

Abstract
Stem-like tumor-initiating cells (TICs) are implicated in cancer progression and recurrence, and can be identified by sphere-formation and tumorigenicity assays. Oncolytic viruses infect, replicate in, and kill a variety of cancer cells. In this study, we seek proof of principle that TICs are susceptible to viral infection. HCT8 human colon cancer cells were subjected to serum-free culture to generate TIC tumorspheres. Parent cells and TICs were infected with HSV-1 subtype NV1066. Cytotoxicity, viral replication, and Akt1 expression were assessed. TIC tumorigenicity was confirmed and NV1066 efficacy was assessed in vivo. NV1066 infection was highly cytotoxic to both parent HCT8 cells and TICs. In both populations, cell-kill of >80% was achieved within 3 days of infection at a multiplicity of infection (MOI) of 1.0. However, the parent cells required 2-log greater viral replication to achieve the same cytotoxicity. TICs overexpressed Akt1 in vitro and formed flank tumors from as little as 100 cells, growing earlier, faster, larger, and with greater histologic atypia than tumors from parent cells. Treatment of TIC-induced tumors with NV1066 yielded tumor regression and slowed tumor growth. We conclude that colon TICs are selected for by serum-free culture, overexpress Akt1, and are susceptible to oncolytic viral infection.
Introduction
Cancer stem cell (CSC) theory represents a recent paradigm shift that has challenged previously accepted cancer development processes. In theory, a CSC is a “cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor”.1 CSC theory offers a logical explanation as to why traditional chemoradiotherapies commonly result in transient regression followed by disappointing recurrence. Within the framework of CSC theory, traditional therapy, which targets rapidly dividing cancer cells, spares quiescent CSCs thereby facilitating tumor recurrence months to years after initial therapy.2,3 In addition to demonstrating the need for novel therapies, this theory implies that one cannot cure a cancer without eradicating CSCs.2
Controversy abounds regarding the optimal method for CSC identification.4–7 Authors have used various means, such as cell surface markers, intracellular functional proteins like aldehyde dehydrogenase (ALDH), expulsion of Hoescht dye, formation of tumor spheres in serum-free or soft agar culture, and ATP-binding cassette (ABC) transporters that efflux chemotherapeutic drugs.1,7–12 All of these methods, however, are measured against the functional assay and gold standard of in vivo tumorigenicity from a very small amount of cells.3,8,13 With variations of the above techniques, CSCs have been identified among many different solid tumor types including brain,14 head and neck,15,16 breast,17 lung,18,19 esophageal,20 pancreatic,21 colon,22,23 and ovarian cancers,24 as well as melanoma.25 Several investigators have even established that a single CSC can initiate tumor formation.11,26
The PI3/Akt pathway has been implicated in tumorigenicity and the CSC phenotype. Akt is the primary effector kinase of the lipid kinase PI3K and is constitutively active in many malignant tissues, with promotion of cell survival, proliferation, angiogenesis, and invasion.27 Several authors have shown that upregulated members of the PI3/Akt pathway are crucial to “stemness” in various solid tumors.11,28,29
Oncolytic viral therapy has recently emerged as a forerunner in the race to develop novel adjuvant therapies to treat cancer. Oncolytic viruses infect, replicate in, and kill cancer cells, with specificity that leaves normal cells unharmed.30,31 In terms of CSC treatment, the very aberrations that facilitate CSC resistance to traditional therapies actually make them ideal targets for oncolytic viruses.32 For instance, defective apoptotic signaling has been shown to facilitate p53-independent death in response to viral infection of chemotherapy-resistant cells. This implies that CSCs with strengthened traditional apoptotic pathways would still be susceptible to viral therapy.33 Several authors have demonstrated the potential of various oncolytic viruses against CSCs or stem-like cells using glioblastoma, neuroblastoma, esophageal, and breast cancer models.3,33,34 HSV-1-derived attenuated viruses are unique and potent oncolytic virus that has proven successful in preclinical and clinical models.35–42 NV1066 is an engineered HSV-1 that has shown efficacy against more than 110 cell lines derived from at least 16 cancer cell types, including colorectal cancer.35
We hypothesized that antiapoptotic pathways are upregulated in colorectal CSCs, making them ideal targets for viral therapy. We further theorized that CSCs are susceptible to oncolytic viral infection. As we use a functional tumorigenicity assay to define our stem-like subpopulation of cancer cells in this study, we favor the term “tumor-initiating cells” or TICs.5
Results
Serum-free culture selects for tumor spheres composed of stem-like HCT8 cells that overexpress AKT1 (and underexpress BAD)
Serum-free culture reliably produced tumor spheres. Cells in tumor spheres appeared morphologically different from adherent-growing parent HCT8 cells with rounded shape, with subjectively less-consistent dimensions (Figure 1). Tumorspheres also replicated at a slower rate than their adherent parental counterparts (Table 1). Cell doubling time within a tumorsphere could take up to 48 hours, whereas parent HCT8 cells doubled at least once every 24 hours. Moreover, parent HCT8 cell counts increased 10-fold within 72 hours of plating, versus fivefold in the TIC population (Table 1). These differences in replication speed were consistently observed, and approached but did not achieve statistical significance. TIC upregulation of Akt1 was confirmed with both Western blot and RT-polymerase chain reaction (PCR) (Figure 1c).
Figure 1.
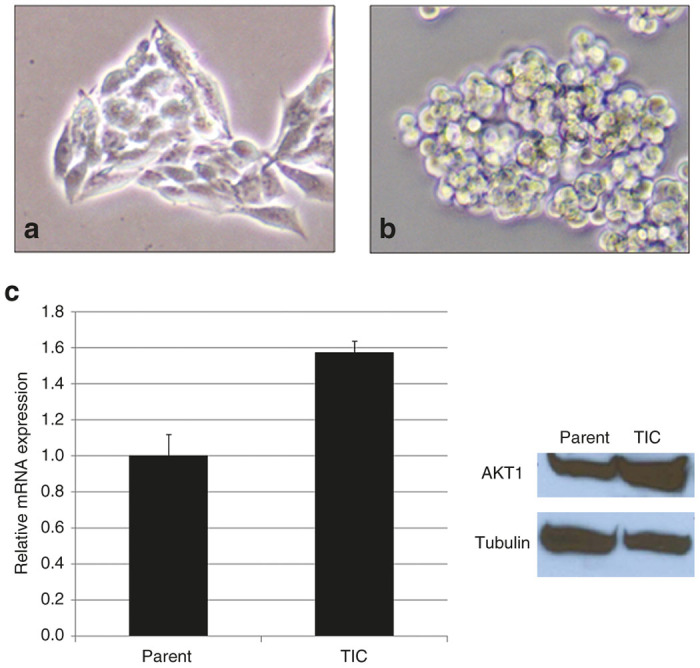
HCT8 parent cells versus tumor-initiating cells (TICs). (a) Parent cells appear morphologically different from their TIC counterparts forming tumorspheres (b). The cells are further distinguishable by varied expression of Akt1 (c).
Table 1. Cell doubling time.
| Parent HCT8 | TIC | P value | |
|---|---|---|---|
| Cells/well | 1 × 105 | 1 × 105 | |
| 24 hours | 2.07 × 105 | 1.62 × 105 | 0.24 |
| 48 hours | 5.44 × 105 | 2.84 × 105 | 0.06 |
| 72 hours | 10.11 × 105 | 5.38 × 105 | 0.15 |
HCT8 TICs are highly tumorigenic compared with parent cells
Injection of the same amount of TICs into the flanks of 7–9-week-old nude athymic mice consistently yielded tumor growth from as few as 100 TICs as soon as 20 days following injection (Figure 2a). Injection of the same quantities of parent HCT8 cells rarely generated tumors. Tumors created by injection with TICs consistently grew faster, larger, sooner, and with greater cellular atypia than those rare tumors formed from parent HCT8 injections (Figure 2b). After 60 days, no tumors resulted from injection of 100 parent HCT8 cells. At 35 days postinjection, tumors began to appear in three of five mice injected with 1,000 parent tumor cells. These tumors grew much later, slower, and with decreased cellular atypia when compared to tumors initiated with 1,000 TICs. Upon sacrifice, no abdominal or thoracic metastases were visible in any of the animals, though ascites was occasionally present.
Figure 2.

Tumor-initiating cells (TICs) are highly tumorigenic when compared to equal number of parent cells injected. (a) Tumors initiated with 1E5 cells of each tumor population were examined histologically. In addition to growing much faster, TIC tumors (c) exhibited much higher levels of histologic atypia compared to tumors initiated by parent cells (b) as noted by H&E staining.
NV1066 infects and expresses GFP in HCT8 parent cells and TICs
In this experiment, NV1066 successfully infected and expressed green fluorescent protein (GFP) in HCT8 parent cells and TICs at all MOIs of 0.01, 0.1, and 1.0. GFP expression was detected by both fluorescent microscopy and by flow cytometry. Table 2 demonstrates sustained dose-dependent increases in GFP expression in both cell populations. Representative experiments at MOI of 0.1 are shown (Figure 3a).
Table 2. Percent green fluorescent protein-expressing cells.
| Day 1 | Day 3 | Day 5 | |
|---|---|---|---|
| Parent | |||
| MOI 1.0 | 31.2 | 100 | 100 |
| MOI 0.1 | 3.09 | 100 | 100 |
| MOI 0.01 | 1.07 | 84.2 | 100 |
| TIC | |||
| MOI 1.0 | 84.8 | 86.8 | 96.7 |
| MOI 0.1 | 40.7 | 88.6 | 94.7 |
| MOI 0.01 | 5.36 | 67.3 | 97.9 |
MOI, multiplicity of infection; TIC, tumor-initiating cells.
Figure 3.

Green fluorescent protein (GFP) expression increases over time after infection. GFP expression reached and sustained close to 100% expression in both HCT8 parent (dashed line, circles) and tumor-initiating cells (TIC) (solid line, squares) populations. A representative experiment at MOI 0.1 is shown (a). HCT8 parent (b) and TIC (c) cells support viral replication at MOIs of 0.01, 0.1, and 1.0, with exponentially increased viral PFUs on day of maximal burst (black bars) compared to initial doses (white bars). However, viral replication does appear to occur on a different scale between populations. In both HCT8 parent (d) and TIC (e) populations, NV1066 was cytotoxic at MOI of 0.01 (circles), 0.1 (squares), and 1.0 (triangles). Cell kill is represented as percentages of live cells compared to control untreated cells grown under identical conditions. MOI: multiplicity of infection, ratio of viral particles to cells; PFU: plaque-forming units, number of viral particles.
NV1066 replicates within and is more efficiently cytotoxic to TICs versus parent HCT8 cells
Both HCT8 parent cells and TICs support viral replication. In the parent population, viral titers peaked on day 3 at MOI 1.0 and day 5 at MOI 0.1 and 0.01. In the TIC population, viral titers peaked on day 3 at MOI 1.0 and 0.1, and peaked on day 5 at MOI 0.01. Expectedly, lower viral doses resulted in increased viral replication in order to achieve the same cell kill as higher initial doses. Thus, in both cell populations, an MOI of 0.01 resulted in the maximum observed amplification of viral titers (Figure 3b,c). Interestingly, far less viral replication was needed to achieve similar cytotoxicity in the TIC versus the parent HCT8 cells. For instance, at an MOI of 0.1, peak viral titer in TICs represented a 75-fold increase over initial dose versus a greater-than 3,800-fold increase in the parent HCT8 cells. As shown in Figure 3d, cytotoxicity was achieved in a dose-dependent fashion over a 100-fold range of viral doses. In the TIC population, an MOI of 1.0 resulted in greater than 75% cell kill after 3 versus 4 days in the parent cell population. Furthermore, greater than 80% cell kill was achieved by day 7 at all 3 MOIs in the TIC experiments compared to day 9 in the parent cell experiments. A representative experiment is shown (Figure 3d,e).
NV1066 infection ends in oncolytic cell death, and may induce apoptosis in neighboring noninfected cells
Much debate exists in the literature regarding precise mechanisms and pathways of oncolytic cell death as a result of viral infection. As previously established, viral infection can induce apoptosis in neighboring noninfected cells.43 In order to further characterize the nature and effects of virally-induced cell death in the TIC population, tumorsphere infection was observed using real-time and time-lapsed fluorescent confocal microscopy. Via time-lapsed microscopy, noninfected cells were observed both separating from tumorspheres, and undergoing cell-death (Figure 4a). Infected cells were stained with Cy5-tagged Annexin V and 4’,6-diamidino-2-phenylindole (DAPI) DAPI and examined via fluorescent microscopy. Annexin V is a protein that attaches to phosphatidylserine residues on pre-apoptotic cells. Many non-GFP-expressing, noninfected cells adjacent to GFP-expressing infected cells were positive for Annexin V. These noninfected cells were frequently also positive for DAPI staining, indicating membrane permeability and impending later-stage apoptosis (Figure 4b). Interestingly, very few cells were double-positive for either GFP and Annexin-V-Cy5 or GFP and DAPI, suggesting that viral lytic cell death is a process that likely occurs independent of bystander cell apoptotic pathways. However it should be pointed out that HSV-1 infection can result in phosphatidlyserine translocation, thus there is a possibility that neighboring cells presumed to be noninfected could simply not yet be expressing GFP. Further investigations are needed to establish reproducibility and validity of this possible abscopal effect.
Figure 4.

Fluorescence microscopy demonstrates virus-induced green fluorescent protein (GFP) expression and presumably noninfected bystander cell apoptosis. (a) Images acquired by fluorescent and differential interference contrast confocal microscopy show a tumorsphere on post-infection day 3 following infection with an MOI of 1.0. Cells expressing GFP (green) are infected with NV1066. (b) A merged image of control, uninfected cells expressing only small amounts of Annexin V and DAPI. (c) The control DIC image of the stained tumorsphere. (d) Cy-5 tagged Annexin V-positive cells appear magenta, which indicates phosphatidylserine residues of preapoptotic cells. Cells stained blue have taken up DAPI, indicating increased cellular permeability and a later stage of apoptosis.
NV1066 abrogates tumor growth of TIC flank tumors
In the animal model, injection of virus into both TIC and parent HCT8-generate tumors resulted in cell-swelling, cytoplasmic vacuolization, and intranuclear inclusion bodies with marginalization of chromatin (Figure 5b). Furthermore, in virally-treated tumors, immunohistochemical staining for GFP was positive in areas of necrosis surrounded by areas of tumor regression. Figure 5c shows H&E appearance of treated tumor versus 5 days stained for GFP. On a macroscopic level, among a cohort of 10 mice with 1 × 103 TIC-induced tumors, 4 were treated with PBS and 6 were treated with NV1066. Viral therapy abrogated tumor growth and induced regression in some cases, as indicated by Figure 5a. In fact, phosphate-buffered saline (PBS)-treated mice experienced fourfold greater tumor growth than their virus-treated counterparts.
Figure 5.

Histologic examination of viral treated tumors and in vivo efficacy of viral infection. (a) Viral injection of tumor-initiating cells (TIC)-initiated tumors abates tumor growth and induces regression in some cases. Tumors initiated by 1,000 TICs were treated with two consecutive doses of NV1066 using 5E6 PFU, with the second dose occurring at day 3 on the graph. (b) Microscopic changes in infected tumors show clear signs of tumor cell death. V: viable area of tumor, N: necrotic area, T: transition zone in which cells are undergoing degeneration and necrosis. In this zone note cell swelling, cytoplasmic vacuolation, and numerous cells with intranuclear inclusion bodies with margination of chromatin (arrows) typically seen after viral infection. (c) In virally-treated tumors, areas of necrosis correspond with (d) green fluorescent protein expression.
Discussion
This study demonstrates a distinct sensitivity of colon adenocarcinoma TIC’s to the HSV-1-based oncolytic virus NV1066 in vitro and shows efficacy of this same vector against TIC-derived tumors in vivo. While other authors have shown that HSV-1 kills stem-like glioblastoma, neuroblastoma, and breast cells;44–46 to our knowledge, ours is the first study to investigate the susceptibility of stem-like colonic adenocarcinoma cells to HSV-1 oncolytic viral therapy. Several investigators have previously associated poor colorectal cancer outcomes and increased metastases with expression of cell-surface markers associated with “stemness”.47–50 Thus the sensitivity of colorectal TICs discovered in this study proves a principle that may pave the way for further focus of targeted therapies aimed at TICs.
To identify and characterize TICs, this study utilized a serum-free tumorsphere culture of a colorectal cancer cell line, the presence of AKT overexpression, as well as generation of tumors from as few as 100 cells. Despite many studies advocating use of cell surface markers as CSC identifiers, it remains unclear whether stemness and differentiation are mutually exclusive in CSCs. The more likely truth is that cells exist in varying degrees of stemness within a tumor, and that stemness is a flexible characteristic. Thus, several authors advocate a move away from surface markers, or at least the correlation of surface markers with functional cellular assays.51 Most likely, the TIC population induced by our tumorsphere culture was heterogeneous. Inability to produce clonally identical cells is perhaps the foremost limitation of the study. However, we established consistent upregulation of AKT1 in the TICs and by doing so further related them to putative CSCs. Similarly, Vermeulen et al.11 were able to isolate a population of CSCs which could form tumorspheres from a single cell. They then noted that CSC markers like CD133+ and CD24+ are coexpressed on tumorigenic cells, and further demonstrated that inhibition of PI3K pathway actually results in differentiation of stem-like cells, indicating the crucial role of the PI3K/Akt pathway in maintaining stem-like characteristics.11
The susceptibility of human colorectal cancer cell lines to HSV constructs and specifically to NV1066 has been previously established.35,52 This study also established that colorectal TICs are highly sensitive to oncolytic HSV-1. Interestingly, while viral replication occurred efficiently in both parent and TIC populations, several thousand more PFUs were necessary to achieve parent HCT8 cell kill. This finding implies that TICs are more sensitive to viral infection than their parent cell counterparts. It is important to note that infections of parent and TIC populations were performed in parallel, with cells seated and infected at the same time, using the same methods aside from serum-free nonadherent culture required to create the tumor spheres, and same viral stock and repeated in triplicate. Viral titers were also performed in parallel and repeated in triplicate. Thus, we do not feel this difference in replication was merely an issue of cell-plating or difference in viral stock. While a small portion of this difference could be accounted for by the observed increased doubling time of the TIC population, a several thousand fold disparity more likely indicates a fundamental difference in cellular machinery. The finding of distinct efficacy of NV1066 against a TIC cell population holds great clinical promise in that CSC-targeted viral therapy could be administered without generating excessive daughter viral particles. In light of the aforementioned theories, there is reason to suspect that such TIC-targeted therapy may represent a means of preventing the recurrences that plague contemporary treatment regimens.
This study also showed that TICs exhibit upregulated constitutive expression of AKT1 compared to their parent cells. As previously discussed, AKT1 plays a central role in protein synthesis and antiapoptosis processes known to be dysregulated in colorectal cancer. Apoptosis is a protective mechanism employed by infected cells to disable cellular machinery necessary for viral replication. Host cells infected with HSV-1 can undergo phosphorylation of eukaryotic initiation factor-2α which terminates the viral lytic cycle by inhibiting protein synthesis.53 To avert apoptosis, HSV-1 encodes genes like γ134.5, which encodes the ICP45 protein that in turn prevents phosphorylation of eukaryotic initiation factor-2α thereby facilitating continued protein synthesis.54–56 However, many engineered oncolytic HSV-1 strains are attenuated by single deletions of γ134.5, which facilitates oncotropism given that cancer cells usurp antiapoptotic pathways. Thus, even with deficient γ134.5, oncolytic HSV-1 has time and machinery to replicate and propagate. Interestingly, in addition to association with “stemness”, PI3/Akt has been implicated in the upregulation of genes that alter the intracellular environment to facilitate more efficient HSV-1 infection.11,57 Thus, this study opens the door for future research concerning the importance of Akt to viral oncolysis and TIC sensitivity to HSV-1.
This study also observed that while infected cells serve viral purposes by prolonging cell life, neighboring, noninfected cells experience a “bystander effect” and undergo apoptosis. This has been previously demonstrated with HSV-1 infection of gastric cancer cells, theorizing that future inhibition of apoptosis could enhance lateral viral spread by making more cells available to potentially produce viral progeny.43 Using annexin-V and DAPI staining in this study, we were able to visualize a similar phenomenon among infected TICs. Moreover, we found that very few infected cells (e.g., GFP-expressing cells) were also positive for annexin-V or DAPI, indicating that infected cells do not present phosphatidylserine residues, which are thought to result from caspase cascade activation. This is aligned with previous studies indicating that engineered HSV-1 subtypes induce caspase-independent cell death.58 This has several important implications. For instance, inhibition of the protective apoptosis of cells adjacent to infected cells could enhance lateral spread of virus, and thereby facilitate tumor destruction at decreased doses. Even more intriguing is that previous authors have correlated constitutive Akt1 overexpression with cellular protection from apoptosis and membrane phosphatidylserine exposure.59 This again demonstrates the need for further examination of the effects of oncolytic virus on the PI3K/Akt pathway.
Conclusion
Stem-like tumor-initiating colorectal cancer cells overexpress AKT1, and are susceptible to oncolytic HSV-1. Further research is needed to determine if members of the PI3/Akt pathway could yield targets for oncolytic viral therapies aimed at CSC eradication.
Materials and Methods
Cell lines
The human colorectal cancer cell line HCT8 (American Type Culture Collection (ATCC), Rockville, MD) was studied. HCT8s were grown in Roswell Park Memorial Institute (RPMI) medium supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal calf serum (FCS). Vero cells (ATCC) were grown in minimum essential medium (MEM) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% FCS. Cells were maintained in a 5% CO2 humidified incubator at 37 °C.
Sphere isolation and characterization
After a minimum of three passages, HCT8 (ATCC, Manassas, VA) cells were washed in PBS and harvested with 0.25% trypsin in 0.02% ethylenediaminetetraacetic acid and centrifuged for 5 minutes at 800 rpm. They were then resuspended in PBS, recentrifuged, and finally resuspended in serum-free Roswell Park Memorial Institute medium (RPMI) supplemented with 20 µg/l human recombinant epidermal growth factor (hrEGF), 10 μg/l fibroblast growth factor (FGF), and 5 ml/l insulin-transferrin-selenium (ITS) liquid media supplement 100× (Sigma-Aldrich, St. Louis, MO). Cells were then placed overnight in a regular adherence flask. The next day, cells growing in suspension were removed from the flask, centrifuged, resuspended in serum-free RPMI, and cultured in ultra-low attachment flasks (Corning Incorporated, Corning, NY).
Spheres were characterized using Western blot and PCR for Akt1 expression (techniques described below). Hemocytometer standard counting methods were employed to discern cell quantities. To determine doubling times, TICs and parent HCT8 cells were seated in six-well plates and counted at 24-hour intervals. Six measurements from each well were taken and the total calculated cells per well of at least three wells were averaged to yield daily values.
Virus
NV1066 is a replication-competent, attenuated herpes simplex type-1 virus (HSV-1) whose construction has been described in detail previously.60 This virus is derived from the wild-type HSV-1 F-strain backbone, and has single copy deletions of ICP-4, ICP-0, and γ1–34.5, which increase tumor selectivity and decrease virulence (Figure 6). NV1066 also contains a transgene for enhanced GFP under the control of a constitutively active cytomegalovirus promoter. Infected cells thus express GFP that can be detected in vitro by both fluorescent microscopy and flow cytometry.35 Viral stocks were propagated on Vero cells and titered by standard plaque assay.52
Figure 6.
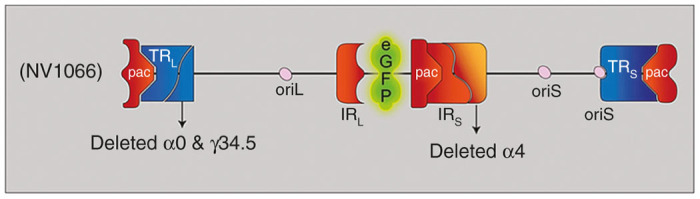
NV1066 viral construct. Herpes simplex virus type 1 is a double-stranded DNA virus with a 152 kb DNA genome comprised of unique long and short segments, each flanked by inverted (IRL and IRS) and terminal repeats (TRL and TRS). The genome further contains three DNA packaging (pac) signals, which enable construction of virions. There are two different origins of replication, one in the unique long segment (oriL), and one in the unique short segment (oriS). Several genes are duplicated as a result of the inverted repeats. These include oriS, γ34.5, α0, and α4. Deletion of these genes enhances viral specificity and decreased neurovirulence. An enhanced green fluorescent protein (eGFP) is encoded on a constitutively active cytomegalovirus (CMV) promoter. Infected cells thus express eGFP.
Cytotoxicity assay
HCT8 cells (1.0 × 104) were plated and grown overnight in standard 24-well flat-bottom plates, while suspended HCT8 tumor spheres were grown in Corning ultra-low attachment 24-well plates. Cells were infected with NV1066 at multiplicities of infection (MOIs) of 1.0, 0.1, and 0.01, and incubated at 37 °C in a 5% CO2 humidified incubator for 1 to 9 days. Each day after infection, adherent HCT8 supernatant was removed and cells were lysed in 24-well plates with a 1.5% Triton-X solution (% volume/PBS). HCT8 tumorsphere suspensions were collected and centrifuged at 1,200 rpm for 5 minutes and then lysed with the same Triton-X solution to release intracellular lactate dehydrogenase. The cytotoxic effect was determined by comparing release of intracellular lactate dehydrogenase from virally infected tumor cells to untreated control cells grown under identical conditions. Lactate dehydrogenase was quantified using a Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) that measures the conversion of a tetrazolium salt into a formazan product. Absorbance was measured at 490 nm using a SpectraMAX 190 microplate reader with the assistance of SoftMax Pro software v5.0.1 (Molecular Devices, Sunnyvale, CA). Results are expressed as the surviving fraction of treated cells compared to untreated control cells. All samples were tested in triplicate and the experiment was repeated at least three times.
Microscopic examination of infected cells
All infected cells were visualized under fluorescent microscopy. All samples were evaluated with an inverted microscope (Nikon Eclipse TE300, Nikon, Tokyo, Japan) using phase-contrast and fluorescence microscopy. A GFP emission filter was used to detect green fluorescence, and images were obtained using NIS software. Infected tumorspheres were further examined with a Leica TCS AOBS SP2 spectral confocal microscope on an upright stand (Leica Microsystems, Wetzlar, Germany) following staining with Annexin V- Cy5 Apoptosis Detection Kit (BioVision, Mountain View, CA) and 4′, 6-diamidino-2-phenylindole dihydrochloride DAPI for measures of early and late apoptosis. Time-lapse images of sphere formation and cellular infection were obtained using a Zeiss LSM 5 Live confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Viral replication assay
Standard plaque assays were performed to quantify viral replication in HCT8 cells following infection with NV1066. Cells (1 × 103) were plated in six-well flat-bottom assay plates in 2 ml media. After 12 to 24 hours, cells were treated with NV1066 at MOIs of 0.01, 0.1, and 1.0 in 100 µl media. Cells maintained at 37 °C served as controls. Supernatants and cells were collected from culture wells each day after infection for 7 days. Serial dilutions of supernatants and cell lysates were cultured on confluent layers of Vero cells and viral titers were determined by counting viral plaques after 72 hours. All samples were tested, and experiments were replicated in triplicate.
Flow cytometry for GFP
5 × 104 HCT8 cells were grown in standard six-well plates and the same number of cells from HCT8 tumorspheres were grown in ultra-low attachment six-well plates (Corning Incorporated). Cells were infected at MOIs of 1.0, 0.1, and 0.01. Adherent HCT8 cells were harvested with 0.25% trypsin in 0.02% ethylenediaminetetraacetic acid, washed in PBS, and then resuspended in 200 µl PBS. 5 µl of 7-amino-actinomycin (7-AAD, BD Pharmingen, San Diego, CA) was added as an exclusion dye for cell viability. Data for GFP expression was acquired on a FACSCalibur machine (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (Tree Star, Ashland, OR).
Flank tumor model
Eight-to-ten-week-old athymic nude female mice obtained from Harlan Sprague Dawley laboratories were provided with food and water ad libitum. All animal work occurred with the approval and strict guidance of the Memorial Sloan-Kettering Institutional Animal Care and Use Committee. Mouse flanks were injected with 1 × 101 to 1 × 105 HCT8 human colorectal cancer cells or an equal number of cells from tumor spheres produced by serum-free culture. Cells were injected as a 1:1 mixture with Growth-Factor-Reduced BD Matrigel (BD Biosciences). Animals were weighed and tumors measured every 3–5 days. Tumor development occurred reliably 2–5 weeks following injection depending upon amount and type of cells injected. Tumor volume was calculated using the following formula: Volume (cm3) = (length × width2) × (π ÷ 6). Animals in which either excessive weight loss or tumor growth occurred were sacrificed prior to experimental completion. In one cohort of ten 1 × 103 TIC-induced tumor-bearing mice, when visible tumor growth was achieved, five mice were treated with 5 × 106 PFU NV1066 in 100 µl PBS tumor injection whereas five received 100 µl PBS alone for two consecutive doses administered on days 1 and 3.
Histologic analysis
Following sacrifice, flank tumors were removed en bloc. Tumor and carcass weights were recorded. Tumors were fixed in 4% paraformaldehyde overnight, then rinsed with PBS, and placed in 70% ethanol. Samples were then embedded in paraffin blocks, routinely sectioned at 4 µm, and stained for hematoxylin and eosin, as well as GFP. All pathology slides were reviewed by a veterinary histopathologist, blinded to tumor treatment modalities.
Western blot
Protein from total cell lysates were resolved by sodium dodecyl sulfate -polyacrylamide gel electrophoresis (SDS-PAGE) SDS-PAGE, transferred onto Immuno-Blot polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA), and probed with antibodies including AKT1, β-actin, and tubulin (Cell Signaling Technology, Danvers, MA). Detection was performed with enhanced chemiluminescence reagents (Amersham Biosciences, England).
Real-time PCR (RT-PCR)
RNA was isolated using an RNeasy Mini-Kit (Qiagen, Valencia, CA) as described by the manufacturer. Reverse transcription was performed using random hexamer priming and TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA). RNA was quantified by spectrophotometry with the Nanodrop 1000 spectrophotometer (Thermo Scientific, Waltham, MA). RT-PCR was performed on a ThermoHybrid thermocycler (Thermo Scientific). For each reaction, 2 µg of RNA was amplified in a 100 µl reaction containing 20 µl of deoxynucleotide triphosphate (2.5 mmol/l each dNTP), 22 µl MgCL2 (25 mmol/l), 10 µl 10×-RT buffer, 5 µl random hexamers (50 µmol/l), 2 µl RNase inhibitor (20 U/µl), and a variable amount of free water to total 100 µl depending on RNA sample concentration. Samples were transferred to the Thermo Hybrid thermocycler at 25 °C for 10 minutes, 48 °C for 30 minutes, and 95 °C for 5 minutes. The resulting cDNA samples were stored at −20 °C. Quantitative real-time PCR (RT-PCR) using TaqMan Assay-on-Demand Gene Expression Assays (Applied Biosystems) was performed on the ABI Prism 7900HT Sequence Detection System. DNA was amplified in a 20 µl reaction containing 1 µmol/l of the desired primer, 2 µl cDNA, and TaqMan Universal PCR Master Mix (Applied Biosystems). Each PCR reaction was subjected to 30 minutes at 48 °C and 10 minutes at 95 °C followed by 40 cycles at 95 °C for 15 seconds and 60 °C for 60 seconds. Each sample was assayed at least in triplicate and experiments were repeated at least three times. Results were analyzed using SDS version 2.3 software (Applied Biosystems). The relative expressions of described genes were determined relative to the endogenous control 18s rRNA using ΔCT analysis.
Statistical analysis
All data were expressed as mean ± standard error of the mean. A two-tailed Student t-test was used to determine significance between treatment groups in both in vitro and in vivo experiments. A P value of <0.05 was considered statistically significant.
Acknowledgments
The authors would like to thank Tony Riley, of the Medical Graphics and Photography Department at the Memorial Sloan-Kettering Cancer Center for his invaluable illustration of Figure 1. The authors further thank Jackie Candelier of the laboratory of comparative pathology for assisting in the processing of tumor samples. The authors thank Viola Weeda of Bhuvanesh Singh’s laboratory for her invaluable assistance with various laboratory techniques. The authors thank Sho Fujisawa, Yevgeniy Romin, and Tao Tong of the Molecular Cytology Core Facility at Memorial Sloan-Kettering Cancer Center for assistance with the microscopic aspects of this project. Finally, the authors especially thank Meryl Greenblatt for her editorial assistance, and also Scott Tuorto of the Department of Surgery for his research assistance.
The authors declare no conflict of interest.
References
- Clarke, MF, Dick, JE, Dirks, PB, Eaves, CJ, Jamieson, CH, Jones, DL et al. (2006). Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 66: 9339–9344. [DOI] [PubMed] [Google Scholar]
- Dingli, D and Michor, F (2006). Successful therapy must eradicate cancer stem cells. Stem Cells 24: 2603–2610. [DOI] [PubMed] [Google Scholar]
- Ribacka, C and Hemminki, A (2008). Virotherapy as an approach against cancer stem cells. Curr Gene Ther 8: 88–96. [DOI] [PubMed] [Google Scholar]
- Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res 2006;66:1891–1895; discussion 1890. [DOI] [PubMed] [Google Scholar]
- Woodward, WA and Sulman, EP (2008). Cancer stem cells: markers or biomarkers? Cancer Metastasis Rev 27: 459–470. [DOI] [PubMed] [Google Scholar]
- Croker, AK and Allan, AL (2008). Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med 12: 374–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, P, Clanton, DJ, Snipas, TS, Lee, J, Mitchell, E, Nguyen, ML et al. (2009). Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int J Cancer 124: 1312–1321. [DOI] [PubMed] [Google Scholar]
- Dou, J and Gu, N (2010). Emerging strategies for the identification and targeting of cancer stem cells. Tumour Biol 31: 243–253. [DOI] [PubMed] [Google Scholar]
- Dean, M (2009). ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 14: 3–9. [DOI] [PubMed] [Google Scholar]
- Gou, S, Liu, T, Wang, C, Yin, T, Li, K, Yang, M et al. (2007). Establishment of clonal colony-forming assay for propagation of pancreatic cancer cells with stem cell properties. Pancreas 34: 429–435. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Todaro M, de Sousa Mello F, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci USA 2008;105:13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger, AW and Salmon, SE (1977). Primary bioassay of human tumor stem cells. Science 197: 461–463. [DOI] [PubMed] [Google Scholar]
- Spillane, JB and Henderson, MA (2007). Cancer stem cells: a review. ANZ J Surg 77: 464–468. [DOI] [PubMed] [Google Scholar]
- Singh, SK, Hawkins, C, Clarke, ID, Squire, JA, Bayani, J, Hide, T et al. (2004). Identification of human brain tumour initiating cells. Nature 432: 396–401. [DOI] [PubMed] [Google Scholar]
- Zito, G, Richiusa, P, Bommarito, A, Carissimi, E, Russo, L, Coppola, A et al. (2008). In vitro identification and characterization of CD133(pos) cancer stem-like cells in anaplastic thyroid carcinoma cell lines. PLoS One 3: e3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007;104:973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, MM, Ng, AV, Lam, S and Hung, JY (2007). Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res 67: 4827–4833. [DOI] [PubMed] [Google Scholar]
- Kim, CF, Jackson, EL, Woolfenden, AE, Lawrence, S, Babar, I, Vogel, S et al. (2005). Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121: 823–835. [DOI] [PubMed] [Google Scholar]
- Huang, D, Gao, Q, Guo, L, Zhang, C, Jiang, W, Li, H et al. (2009). Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev 18: 465–473. [DOI] [PubMed] [Google Scholar]
- Hermann, PC, Huber, SL, Herrler, T, Aicher, A, Ellwart, JW, Guba, M et al. (2007). Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1: 313–323. [DOI] [PubMed] [Google Scholar]
- O’Brien, CA, Pollett, A, Gallinger, S and Dick, JE (2007). A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106–110. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani, L, Lombardi, DG, Pilozzi, E, Biffoni, M, Todaro, M, Peschle, C et al. (2007). Identification and expansion of human colon-cancer-initiating cells. Nature 445: 111–115. [DOI] [PubMed] [Google Scholar]
- Bapat, SA, Mali, AM, Koppikar, CB and Kurrey, NK (2005). Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res 65: 3025–3029. [DOI] [PubMed] [Google Scholar]
- Schatton, T, Murphy, GF, Frank, NY, Yamaura, K, Waaga-Gasser, AM, Gasser, M et al. (2008). Identification of cells initiating human melanomas. Nature 451: 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi I, Sanzone S, Astigiano S, et al. The properties of a mammary gland cancer stem cell. Proc Natl Acad Sci USA 2007;104:10476–10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo, A and Pandolfi, PP (2008). The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27: 5527–5541. [DOI] [PubMed] [Google Scholar]
- Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA 2007;104:618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler, CE, Foo, WC, LaFiura, KM, McLendon, RE, Hjelmeland, AB and Rich, JN (2008). Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26: 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki, K, Ebert, O and Woo, SL (2005). Eradication of advanced hepatocellular carcinoma in rats via repeated hepatic arterial infusions of recombinant VSV. Hepatology 41: 196–203. [DOI] [PubMed] [Google Scholar]
- Fong, Y, Kim, T, Bhargava, A, Schwartz, L, Brown, K, Brody, L et al. (2009). A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectal cancer. Mol Ther 17: 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripe, TP, Wang, PY, Marcato, P, Mahller, YY and Lee, PW (2009). Targeting cancer-initiating cells with oncolytic viruses. Mol Ther 17: 1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coukos, G, Makrigiannakis, A, Kang, EH, Rubin, SC, Albelda, SM and Molnar-Kimber, KL (2000). Oncolytic herpes simplex virus-1 lacking ICP34.5 induces p53-independent death and is efficacious against chemotherapy-resistant ovarian cancer. Clin Cancer Res 6: 3342–3353. [PubMed] [Google Scholar]
- Zhang, X, Komaki, R, Wang, L, Fang, B and Chang, JY (2008). Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin Cancer Res 14: 2813–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumilli, PS, Stiles, BM, Chan, MK, Eisenberg, DP, Yu, Z, Stanziale, SF et al. (2006). Real-time diagnostic imaging of tumors and metastases by use of a replication-competent herpes vector to facilitate minimally invasive oncological surgery. FASEB J 20: 726–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinblatt, M, Pin, RH and Fong, Y (2007). Herpes viral oncolysis: a novel cancer therapy. J Am Coll Surg 205(4 Suppl): S69–S75. [DOI] [PubMed] [Google Scholar]
- Kemeny, N, Brown, K, Covey, A, Kim, T, Bhargava, A, Brody, L et al. (2006). Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther 17: 1214–1224. [DOI] [PubMed] [Google Scholar]
- Markert, JM, Medlock, MD, Rabkin, SD, Gillespie, GY, Todo, T, Hunter, WD et al. (2000). Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 7: 867–874. [DOI] [PubMed] [Google Scholar]
- Rampling, R, Cruickshank, G, Papanastassiou, V, Nicoll, J, Hadley, D, Brennan, D et al. (2000). Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther 7: 859–866. [DOI] [PubMed] [Google Scholar]
- Senzer, NN, Kaufman, HL, Amatruda, T, Nemunaitis, M, Reid, T, Daniels, G et al. (2009). Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27: 5763–5771. [DOI] [PubMed] [Google Scholar]
- Nagano, S, Perentes, JY, Jain, RK and Boucher, Y (2008). Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res 68: 3795–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow, S, Papanastassiou, V, Harland, J, Mabbs, R, Petty, R, Fraser, M et al. (2004). HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther 11: 1648–1658. [DOI] [PubMed] [Google Scholar]
- Stanziale, SF, Petrowsky, H, Adusumilli, PS, Ben-Porat, L, Gonen, M and Fong, Y (2004). Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clin Cancer Res 10: 3225–3232. [DOI] [PubMed] [Google Scholar]
- Mahller, YY, Williams, JP, Baird, WH, Mitton, B, Grossheim, J, Saeki, Y et al. (2009). Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One 4: e4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto, H, Kesari, S, Farrell, CJ, Curry, WT Jr, Zaupa, C, Aghi, M et al. (2009). Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res 69: 3472–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, TT, Roth, JC, Friedman, GK and Gillespie, GY (2014). Oncolytic viral therapy: targeting cancer stem cells. Oncolytic Virother 2014: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeelenberg, IS, Ruuls-Van Stalle, L and Roos, E (2003). The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res 63: 3833–3839. [PubMed] [Google Scholar]
- Gassmann, P, Haier, J, Schlüter, K, Domikowsky, B, Wendel, C, Wiesner, U et al. (2009). CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia 11: 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigusa, S, Tanaka, K, Toiyama, Y, Yokoe, T, Okugawa, Y, Ioue, Y et al. (2009). Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann Surg Oncol 16: 3488–3498. [DOI] [PubMed] [Google Scholar]
- Horst, D, Scheel, SK, Liebmann, S, Neumann, J, Maatz, S, Kirchner, T et al. (2009). The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J Pathol 219: 427–434. [DOI] [PubMed] [Google Scholar]
- Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci USA 2003;100:3547–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooby, DA, Carew, JF, Halterman, MW, Mack, JE, Bertino, JR, Blumgart, LH et al. (1999). Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207). FASEB J 13: 1325–1334. [DOI] [PubMed] [Google Scholar]
- Cassady, KA, Gross, M and Roizman, B (1998). The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol 72: 7005–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Roizman B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci USA 1992;89:3266–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Chen JJ, Gross M, Roizman B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci USA 1995;92:10516–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, B, Chou, J, Brandimarti, R, Mohr, I, Gluzman, Y and Roizman, B (1997). Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol 71: 6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod, IJ and Minson, T (2010). Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One 5: e9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan, V, Brandimarti, R and Roizman, B (1999). Herpes simplex virus 1 blocks caspase-3-independent and caspase-dependent pathways to cell death. J Virol 73: 3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, JQ, Chong, ZZ and Maiese, K (2003). Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol 64: 557–569. [DOI] [PubMed] [Google Scholar]
- Wong, RJ, Joe, JK, Kim, SH, Shah, JP, Horsburgh, B and Fong, Y (2002). Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Hum Gene Ther 13: 1213–1223. [DOI] [PubMed] [Google Scholar]