

Abstract
The role of DnaD in the recruitment of replicative helicase has been identified. However, knowledge of the DNA, PriA, and DnaA binding mechanism of this protein for the DnaA- and PriA-directed replication primosome assemblies is limited. We characterized the DNA-binding properties of DnaD from Staphylococcus aureus (SaDnaD) and analyzed its interactions with SaPriA and SaDnaA. The gel filtration chromatography analysis of purified SaDnaD and its deletion mutant proteins (SaDnaD1-195, SaDnaD1-200 and SaDnaD1-204) showed a stable tetramer in solution. This finding indicates that the C-terminal region aa 196–228 is not crucial for SaDnaD oligomerization. SaDnaD forms distinct complexes with ssDNA of different lengths. In fluorescence titrations, SaDnaD bound to ssDNA with a binding-site size of approximately 32 nt. A stable complex of SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 with ssDNA dT40 was undetectable, indicating that the C-terminal region of SaDnaD (particularly aa 205–228) is crucial for ssDNA binding. The SPR results revealed that SaDnaD1-195 can interact with SaDnaA but not with SaPriA, which may indicate that DnaD has different binding sites for PriA and DnaA. Both SaDnaD and SaDnaDY176A mutant proteins, but not SaDnaD1-195, can significantly stimulate the ATPase activity of SaPriA. Hence, the stimulation effect mainly resulted from direct contact within the protein—protein interaction, not via the DNA—protein interaction. Kinetic studies revealed that the SaDnaD-SaPriA interaction increases the Vmax of the SaPriA ATPase fivefold without significantly affecting the Km. These results indicate that the conserved C-terminal region is crucial for ssDNA and PriA helicase binding, but not for DnaA protein-binding and self-tetramerization.
Introduction
Initiation and re-initiation of chromosomal DNA replication in bacteria is a complex process that depends on divergent multi-protein assembly for loading the replicative DNA helicase at the replication origin [1,2,3,4,5]. DNA damage causes arrest and disassembly of the replication machinery, anywhere along the DNA, leading to replication failure [6,7,8]. Genome integrity should be maintained from generation to generation to ensure proper cell function and survival; thus, collapsed DNA replication forks must be reactivated through origin-independent replisome reloading for genome duplication. The replication restart primosome [9,10,11] is a formidable enzymatic machine; it is a protein—DNA complex that reactivates stalled DNA replication at repaired replication forks after DNA damage [12]. In contrast to the DnaA-directed primosome initiated at the unique oriC site [3,4], the replication restart primosome preferentially recognizes three-way branched DNA structures that possess a leading strand [13,14,15,16].
The replication restart primosome in Escherichia coli includes seven essential proteins, namely, PriA helicase, PriB, PriC, DnaB helicase, DnaC, DnaT, and DnaG primase [10]. The mechanisms of action of DNA replication restart primosome in bacteria have mainly been studied in Gram-negative E. coli, and, to a lesser extent, in Gram-positive bacteria [17]. Different bacteria have different strategies for the functional loading of cellular replicative DNA helicases [18]. In the Gram-positive Bacillus subtilis, the DNA replication initiator protein PriA helicase has a homolog of E. coli [19]. Nevertheless, essential helicase-loading components, such as PriB, PriC, DnaT, and DnaC proteins, are not found in Gram-positive bacteria [20]. Instead, three other proteins, namely, DnaD, DnaB, and DnaI, have been genetically and biochemically proven to be required for the replication restart of the Gram-positive B. subtilis [21,22,23,25,26]. The DnaD and DnaB have no homologs in Gram-negative bacteria, and their functions for DnaA- and PriA-dependent initiation of DNA replication need to be examined.
DnaD interacts with DnaA [26], DnaB [24], and DnaD itself [27]. Following DnaA assembly at oriC in B. subtilis, DnaD and DnaB are sequentially recruited and are required to deposit the protein complex of the helicase loader DnaI and the DNA helicase DnaC onto the unwound DNA duplex [28]. DnaD inhibits the cooperative DNA-binding activity [29], and may regulate DNA replication initiation. DnaD forms scaffolds and enhances duplex melting [30]. DnaD functions as a global regulator of DNA architecture [22] and a potential modulator for global superhelical density [31]. Moreover, this protein interacts with linear DNA and forms a nucleoprotein structure with a round DnaD scaffold in an open circle [32]. DnaD consists of an N-terminal domain with oligomerization activity and a C-terminal domain with single-stranded DNA (ssDNA)-binding activity [27]. The crystal structure of the N-terminal domain of DnaD reveals an extended winged-helix fold [33] and a unique tetramerization motif for the DnaD-mediated scaffold formation [34]. NMR analysis of the structure of the DnaD C-terminal domain reveals the presence of five helices [35]. Although DnaD can bind to DNA and form large nucleoprotein complexes in the absence of the N-terminal domain, it does not exhibit DNA remodeling activity [27].
DnaD is a component of the DNA replication primosome, but information on PriA and DnaA binding of DnaD is limited. Knowledge of stoichiometry, domain function, and binding activity is a prerequisite for formulating any model of protein function in DNA replication. In this study, the DNA-DnaD interactions were analyzed through electrophoretic mobility shift analysis (EMSA) and fluorescence quenching. Furthermore, considering our collective data from surface plasmon resonance (SPR) experiments, ATPase stimulation effects and gold nanoparticle assays on DnaD—PriA and DnaD—DnaA interactions, as well as studies on deletion mutants, we propose and discuss probable domain functions of DnaD.
Materials and Methods
Construction of SaDnaD, SaDnaD1-195, SaDnaD1-200, SaDnaD1-204, SaPriA, and SaDnaA expression plasmids
The gene encoding the putative Staphylococcus aureus PriA (SaPriA), SaDnaA, and SaDnaD was individually amplified by PCR using the genomic DNA of S. aureus subsp. aureus ED98 as template. The forward and reverse primers were designed to introduce unique restriction sites into SaDnaD and its deletion mutants, permitting the insertion of the amplified gene into the pET21e vector for protein expression in E. coli. SaDnaA gene was amplified and inserted into the pET21b vector. Construction of the SaPriA expression plasmid has been reported [36]. The pET21e vector [37] was engineered from the pET21b vector (Novagen Inc., Madison, WI, USA) to avoid N-terminal T7 tag. Primers used for the construction of these plasmids are listed in Table 1.
Table 1. Primers used for construction of plasmids.
| Oligonucleotide | Primer |
|---|---|
| SaDnaD-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| SaDnaD-HindIII-C | GGGAAGCTTCTTACCATCAAGGTTCTC |
| SaDnaD1-204-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| SaDnaD1-204-HindIII-C | GGGAAGCTTTTTATTAAATTTTTCTCT |
| SaDnaD1-200-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| SaDnaD1-200-HindIII-C | GGGAAGCTTTTCTCTTATTTTTCTAGA |
| SaDnaD1-195-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| SaDnaD1-195-HindIII-C | GGGAAGCTTAGAATCGTCAATTGTTTT |
| SaDnaA-NdeI-N | GGGCATATGTCGGAAAAAGAAATTTGG |
| SaDnaA-XhoI-C | GGGCTCGAGTACATTTCTTATTTCTTT |
These plasmids were verified by DNA sequencing. Underlined nucleotides indicate the designated site for the restriction site.
Protein expression and purification
The recombinant proteins were expressed and purified using the protocol described previously for PriB [38]. Briefly, E. coli BL21(DE3) cells were transformed with the expression vector and overexpression of the expression plasmids was induced by incubating with 1 mM isopropyl thiogalactopyranoside. The protein was purified from the soluble supernatant by Ni2+-affinity chromatography (HiTrap HP; GE Healthcare Bio-Sciences), eluted with Buffer A (20 mM Tris-HCl, 250 mM imidazole, and 0.5 M NaCl, pH 7.9), and dialyzed against a dialysis buffer (20 mM HEPES and 100 mM NaCl, pH 7.0; Buffer B). Protein purity remained at >97% as determined by SDS-PAGE (Mini-PROTEAN Tetra System; Bio-Rad, CA, USA).
Gel-filtration chromatography
Gel-filtration chromatography was carried out by the AKTA-FPLC system (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). In brief, purified protein (2 mg/mL) in Buffer B was applied to a Superdex 200 HR 10/30 column (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) equilibrated with the same buffer. The column was operated at a flow rate of 0.5 mL/min, and the proteins were detected at 280 nm. The column was calibrated with proteins of known molecular weight: thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), and myoglobin (17 kDa). The Kav values for the standard proteins and the SaDnaD variants were calculated from the equation: Kav = (Ve − Vo)/(Vc − Vo), where Vo is the column void volume, Ve is the elution volume, and Vc is the geometric column volume.
EMSA
EMSA for the SaDnaD variants was conducted using the protocol described previously for PriB [39]. In brief, radiolabeling of various lengths of ssDNA oligonucleotides was carried out with [γ32P]ATP (6000 Ci/mmol; PerkinElmer Life Sciences, Waltham, MA) and T4 polynucleotide kinase (Promega, Madison, WI, USA). Protein (0, 0.06, 0.12, 0.25, 0.5, 1, 2, 4, 8, and 16 μM; monomer) was incubated at 25°C for 30 min with 1.7 nM DNA substrates (dT20–65) in a total volume of 10 μL in 20 mM Tris—HCl (pH 8.0) and 100 mM NaCl. The resulting samples were mixed with gel-loading solution (0.25% bromophenol blue and 40% sucrose; w/v), resolved on a native 8% polyacrylamide gel (8.3 × 7.3 cm) at 4°C in TBE buffer for 1–1.5 h at 100 V, and were visualized by phosphorimaging. The phosphor storage plate was scanned, and the data for complex and free DNA bands were digitized for quantitative analysis. The ssDNA-binding ability for the protein was estimated using linear interpolation from the protein concentration that binds 50% of the input DNA [40]. Each [Protein]50 was calculated as the average of at least three measurements ± S.D.
Preparation of dsDNA substrates
The double-stranded DNA substrates (dsDNA) were prepared with a radiolabeled PS4 strand (3′-GGGCTTAAGCCTATCGAGCCATGGG-5′; 25 mer) and an unlabeled PS3-dT25 strand (5′-CCCGAATTCGGATAGCTCGGTACCC-dT25-3′) at a 1:1 concentration ratio. Each dsDNA substrate was formed in 20 mM HEPES (pH 7.0) and 100 mM NaCl, by brief heating at 95°C for 5 min and then followed by slow cooling to room temperature overnight.
Fluorescence-quenching measurement
Fluorescence titrations were performed in a spectrofluorimeter (Hitachi F-2700; Hitachi High-Technologies, Tokyo, Japan) as described previously [41]. The excitation and emission of tryptophan fluorescence were detected at 295 and 340 nm, respectively. The protein solution (0.1 μM; tetramer) in 2 mL Tris—HCl buffer (20 mM Tris—HCl, and pH 8.0) containing 200 or 500 mM NaCl was titrated with increasing quantities of dT65 oligonucleotide. After the addition of ssDNA, the complex solution was equilibrated for 300 s until no fluorescence change could be observed. Tryptophan fluorescence quenching was used to detect and quantify protein-DNA interactions.
SPR
SPR was conducted using the protocol described previously for DnaC helicase [42]. SaPriA and SaDnaA were individually immobilized on Series S sensor chips CM5 (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The PriA and DnaA-binding experiments were carried out at 293K using a Biacore T200 (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) with running buffer (40 mM Tris, 200 mM NaCl, and 0.05% Tween-20 at pH 8.0). SaDnaD solutions were diluted in the running buffer to final concentrations of 250, 125, 63, 31, 15, and 7.6 nM. Diluted samples were injected in duplicate over each immobilized protein for 120 s at a flow rate of 30 μL/min. The running buffer was then flushed for 300 s at a flow rate of 30 μL/min. Finally, the chip surface was regenerated by injecting 2 M MgCl2 buffer for 60 s at a flow rate of 30 μL/min. Control samples were used to monitor the sensor chip surface stability, demonstrating reproducibility throughout the duration of the experiments. The estimated Kd values were derived by fitting the association and dissociation signals with a 1:1 (Langmuir) model using the Biacore T200 Evaluation Software.
Gold nanoparticle assay
The protein—protein interactions within SaDnaD—SaPriA and SaDnaD—SaDnaA were rapidly analyzed based on the intrinsic optical properties of the gold nanoparticles [43]. The commercialized kit of Ni2+-NTA SAM nanoparticles (SmarticleParticles) was purchased from Minerva Biotechnologies (Waltham, MA, USA). The 75-μL coupling sample 1 contains 50 μL of SaDnaD (1 μM) and 25 μL of Ni2+-NTA SAM nanoparticles, whereas the 75-μL coupling sample 2 contains 50 μL of SaPriA (1 μM) or SaDnaA (1 μM) and 25 μL of Ni2+-NTA SAM nanoparticles. Sample 1 was mixed with sample 2 for 1–10 min. Red/pink indicates a homogeneous suspension, and purple indicates that particles are drawn close together through protein—protein interactions.
ATPase assay
SaPriA ATPase assay was performed with 0.4 mM [γ-32P] ATP and 0.125 μM of SaPriA in reaction buffer containing 40 mM Tris (pH 8.0), 10 mM NaCl, 2 mM DTT, 2.5 mM MgCl2, and 0.1 μM PS4/PS3-dT25 DNA substrate. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a phosphorimager. The kinetic parameters Km and Vmax were determined from a non-linear plot by fitting the hydrolyzing rate from individual experiments to the Michaelis—Menten equation (Enzyme Kinetics module of Sigma-Plot; Systat Software, Chicago, IL, USA) [44].
Cloning, protein expression, and purification of tag-free SaDnaD, SaDnaDY176A, and SaDnaD1-195 proteins
To obtain proteins without a His tag, SaDnaD and SaDnaD1-195 were amplified through PCR and cloned in a pGEX-5X-1 vector (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) by using the restriction sites EcoRI and XhoI. SaDnaDY176A mutant was generated using a QuikChange Site-Directed Mutagenesis kit according to the manufacturer’s protocol (Stratagene, LaJolla, CA). These expression plasmids were verified by DNA sequencing. Table 2 lists the primers utilized to construct these GST plasmids.
Table 2. Primers used for construction of GST plasmids.
| Oligonucleotide | Primer |
|---|---|
| GST-SaDnaD-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| GST-SaDnaD-XhoI-C | GGGCTCGAGTTACTTACCATCAAGGTT |
| GST-SaDnaD1-195-EcoRI-N | GGGGAATTCGATAAATATCAATTAAAA |
| GST-SaDnaD1-195-XhoI-C | GGGCTCGAGTTAAGAATCGTCAATTGT |
| GST-SaDnaDY176A-N | AGCTTTAAAGCTATGGATCGTATTTTA |
| GST-SaDnaDY176A-C | ACGATCCATAGCTTTAAAGCTAAGTTT |
These plasmids were verified by DNA sequencing. Underlined nucleotides indicate the designated site for the restriction site or the mutation site.
E. coli BL21(DE3) cells were transformed with the expression vector, and overexpression of the proteins was induced through incubation with 1 mM of isopropyl thiogalactopyranoside for 8 h at 25°C. The cells were lysed in a GST loading buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.3). The GST fusion proteins GST-SaDnaD, GST-SaDnaDY176A, and GST-SaDnaD1-195 were purified from the soluble supernatant by GSTrap HP affinity column (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) and were eluted with a GST elution buffer (50 mM Tris-HCl and 10 mM reduced glutathione, pH 8.0). The GST fusion proteins were cleaved by Factor Xa (25 μg/mL; Sigma—Aldrich, St. Louis, MO, USA) for 8 h at 25°C to remove the GST tag. After dialysis against Buffer C (20 mM potassium phosphate and 100mM NaCl, pH 7.0), the protein solution was applied to the Heparin HP column (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) and was eluted with a linear NaCl gradient from 0.1 M to 0.7 M with Buffer C using the AKTA-FPLC system. After the analysis of SDS-PAGE, the peak fractions were collected and concentrated. For further purification and buffer exchange, the resultant protein solution was applied to a Superdex 200 HR 10/30 column equilibrated with Buffer B and purified by the AKTA-FPLC system. The peak fraction for tag-free protein (SaDnaD, SaDnaDY176A, and SaDnaD1-195) was collected and concentrated. Protein purity remained at >97% as determined by SDS-PAGE.
Results
Purification of SaDnaD
The gene SAAV1440 encoding for the putative DnaD was PCR-amplified using the genomic DNA of S. aureus subsp. aureus ED98 as template. This amplified gene was then ligated into the pET21e vector for protein expression. SaDnaD was heterologously overexpressed in E. coli and then purified from the soluble supernatant by Ni2+-affinity chromatography (S1 Fig). Pure protein was obtained in this single chromatographic step with an elution of Buffer B and dialyzed against a dialysis buffer (Buffer C). Approximately >10 mg of purified protein was obtained from 1 L of E. coli cell culture. The truncated SaDnaD proteins were also purified according to the same protocol as that for the wild-type protein, and the purification results were similar.
EMSA of SaDnaD
To investigate the length of nucleotides needed for the formation of the DnaD—ssDNA complex, as well as the ssDNA-binding ability of DnaD, we studied the binding of SaDnaD to dT20, dT25, dT35, dT45, dT55, dT60, and dT65 (Fig 1) with different protein concentrations using EMSA. When we incubated SaDnaD with dT20, no significant band shift was observed. This result indicates that SaDnaD could not form a stable complex with this homopolymer during electrophoresis (Fig 1A). Some smears were observed; thus, SaDnaD appeared to interact with dT20, but the ssDNA may be too short to form a stable complex with SaDnaD. In contrast to dT20, longer dT homopolymers, such as dT25–55 (Fig 1B–1E), produced a significant band shift (C, complex). These findings indicate that the ssDNA-binding activity of SaDnaD is strong enough to form a stable protein—DNA complex in solution. Two different complexes for dT60 (Fig 1F) and dT65 (Fig 1G) were formed by SaDnaD.
Fig 1. EMSA of SaDnaD.

Protein (0, 0.06, 0.12, 0.25, 0.5, 1, 2, 4, 8, and 16 μM; monomer) was incubated at 25°C for 30 min with 1.7 nM of (A) dT20, (B) dT25, (C) dT35, (D) dT45, (E) dT55, (F) dT60, or (G) dT65 in a total volume of 10 μL in 20 mM Tris—HCl (pH 8.0) and 100 mM NaCl. The resulting samples were mixed with gel-loading solution (0.25% bromophenol blue and 40% sucrose; w/v), resolved on a native 8% polyacrylamide gel (8.3 × 7.3 cm) at 4°C in TBE buffer for 1–1.5 h at 100 V, and were visualized by phosphorimaging. The phosphor storage plate was scanned, and the data for complex and free DNA bands were digitized for quantitative analysis. (H) Summary of the [SaDnaD]50 values of SaDnaD. The [SaDnaD]50 values of SaDnaD as a function of the length of the ssDNA determined using EMSA.
Binding constants of SaDnaD—ssDNA complexes determined from EMSA
Binding of SaDnaD to ssDNA of different lengths formed distinct complexes. To compare the binding abilities of SaDnaD to ssDNA of different lengths, the midpoint values for input ssDNA binding that were calculated from the titration curves of EMSA and the [Protein]50 were quantified using linear interpolation from the protein concentration and are summarized in Table 3. As shown in Fig 1H, the DNA binding abilities of SaDnaD were length-dependent. The [SaDnaD]50 for dT65 binding is 0.5 ± 0.1 μM, which is ninefold lower than that for dT25 binding (4.7 ± 0.8 μM). Thus, the protein—DNA contact for each complex of SaDnaD may not be similar.
Table 3. The [SaDnaD]50 values of SaDnaD as analyzed by EMSA.
| DNA | [Protein]50 (μM) |
|---|---|
| dT20 | ND |
| dT25 | 4.7 ± 0.8 |
| dT35 | 2.2 ± 0.3 |
| dT45 | 2.0 ± 0.2 |
| dT55 | 1.4 ± 0.1 |
| dT60 | 1.5 ± 0.1 |
| dT65 | 0.5 ± 0.1 |
[Protein]50 was calculated from the titration curves of EMSA by determining the concentration of the protein (μM) needed to achieve the midpoint value for input DNA binding. For dT60 and dT65, input ssDNA binding was the sum of the intensities from the two separate ssDNA—protein complexes. Errors are standard deviations determined by three independent titration experiments.
C-terminal region of SaDnaD is crucial for ssDNA binding
Given that many conserved positively charged residues are located in the C-terminal domain of DnaD [35], the truncated SaDnaD proteins, namely, SaDnaD1-204 (the C-terminal 24 amino acid residues were removed), SaDnaD1-200, and SaDnaD1-195, were constructed and purified (S1 Fig) to investigate whether the C-terminal region is essential for ssDNA binding and SaDnaD oligomerization. The binding of SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 to dT40 was examined using EMSA. No significant band shift was observed for SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 incubated with dT40, which suggests no complex formation (S2 Fig). Because some smears were observed, SaDnaD1-204 appeared to interact with dT40, but cannot form a stable complex with dT40 during electrophoresis. Thus, the C-terminal region of SaDnaD (the amino acid residues 205–228) is crucial for ssDNA binding.
Oligomeric state of SaDnaD in solution
The analysis of purified SaDnaD protein (4 mg/mL) by gel filtration chromatography revealed a single peak with elution volume of 74.3 mL. Assuming that the shape and partial specific volume of SaDnaD are similar to the standard proteins, the native molecular mass of SaDnaD was estimated to be 107895 Da, which was calculated from a standard linear regression equation, Kav = −0.3684 (logMw) + 2.2707 (Fig 2). The native molecular mass for SaDnaD is approximately four times the molecular mass of a SaDnaD monomer (~27 kDa). Thus, we conclude that SaDnaD in solution is a stable tetramer.
Fig 2. Gel-filtration chromatographic analysis.
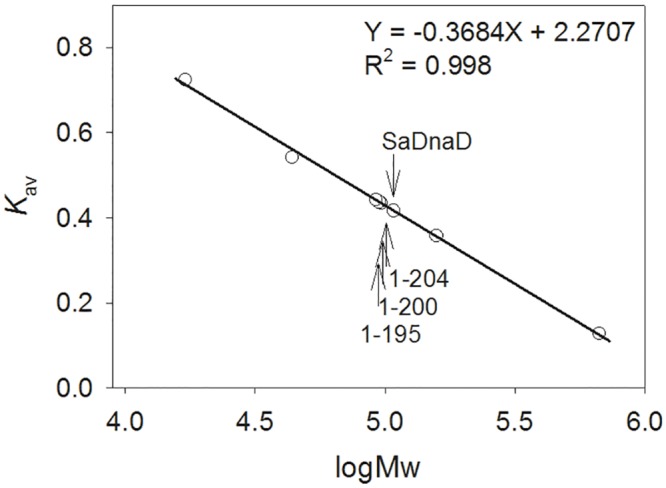
Gel-filtration chromatography was carried out by the AKTA-FPLC system. The column was calibrated with proteins of known molecular weight: thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), and myoglobin (17 kDa). The Kav values for the standard proteins and the SaDnaD variants were calculated from the equation: Kav = (Ve − Vo)/(Vc − Vo), where Vo is the column void volume, Ve is the elution volume, and Vc is the geometric column volume. A standard linear regression curve was generated by plotting the log of the molecular mass of the calibration proteins against their Kav values.
Purified SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 were also analyzed by gel filtration chromatography to investigate the deletion effect in the oligomeric state of SaDnaD. The chromatographic results showed a single peak with elution volumes of 75.6, 75.9, and 76.3 mL for SaDnaD1-204, SaDnaD1-200, and SaDnaD1-195, respectively. The native molecular masses of SaDnaD1-204, SaDnaD1-200, and SaDnaD1-195 were estimated to be 96942, 94872, and 92140 Da, respectively. Thus, SaDnaD1-204, SaDnaD1-200, and SaDnaD1-195 in solution are stable tetramers, which were similar to SaDnaD. Although these deletion mutants of SaDnaD cannot bind to ssDNA (S2 Fig), deletion of the C-terminal region does not cause any change in the oligomeric state of SaDnaD, consistent with what was previously shown for the B. subtilis DnaD [27, 35].
Fluorescence detection of SaDnaD and ssDNA interaction
We examined the quenching fluorescence intensity of tryptophan in SaDnaD upon addition of ssDNA. SaDnaD has three tryptophan residues, namely, Trp148, Trp184, and Trp219, allowing ssDNA binding analysis through tryptophan fluorescence quenching. The protein displayed strong intrinsic fluorescence with a peak wavelength of 340 nm when excited at 295 nm, which is consistent with tryptophan fluorescence. Upon the addition of ssDNA, fluorescence quenching of SaDnaD was observed. The intrinsic fluorescence was progressively quenched as dT65 was titrated into the SaDnaD solution, suggesting an interaction of SaDnaD with dT65 (Fig 3). Upon adding a saturating quantity of ssDNA in the presence of 200 and 500 mM NaCl, the intrinsic fluorescence at 340 nm was quenched by 75% and 65%, respectively. Using these fluorescence data, we calculated the binding stoichiometry between SaDnaD and ssDNA. The saturation curves of SaDnaD in the presence of 200 and 500 mM NaCl suggest that the binding is stoichiometric at 0.5 SaDnaD tetramer per 65-mer ssDNA if enough binding sites were present in the DNA for all SaDnaD molecules to bind. Therefore, the binding-site size of SaDnaD tetramer estimated from fluorescence quenching is approximately 32 nt (65 × 0.5 = 32.5). We also tested the fluorescence quenching of SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 by adding ssDNA. The ssDNA dT65 at the DNA—protein ratio of 1 only quenched <5%, <10%, and ~20% of SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 fluorescence intensities, respectively. Thus, the ssDNA-binding abilities of SaDnaD1-195, SaDnaD1-200, and SaDnaD1-204 were significantly impaired and their ssDNA-binding site sizes were not estimated.
Fig 3. Fluorescence quenching.
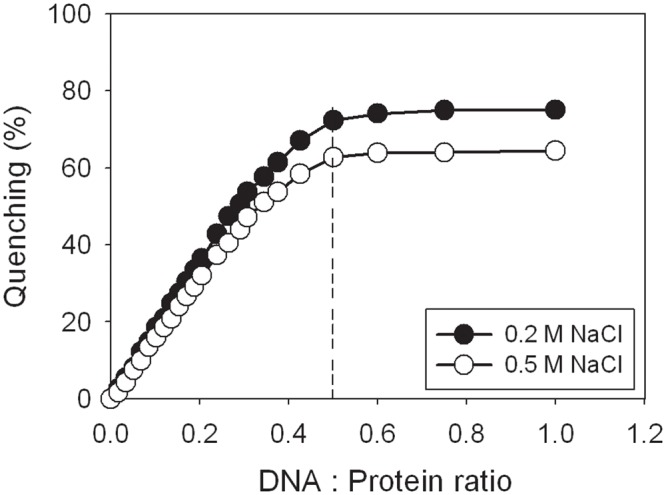
The excitation and emission of tryptophan fluorescence were detected at 295 and 340 nm, respectively. The protein solution (0.1 μM; tetramer) in 2 mL Tris—HCl buffer (20 mM Tris—HCl, and pH 8.0) containing 200 or 500 mM NaCl was titrated with increasing quantities of dT65 oligonucleotide. After the addition of ssDNA, the complex solution was equilibrated for 300 s until no fluorescence change could be observed.
Characterization of SaDnaD bound to SaPriA or SaDnaA
B. subtilis DnaD—DnaA interaction is previously identified through the yeast two-hybrid analysis [26]. To gain insights into direct interactions and further quantitatively characterize whether the C-terminal domain of DnaD was involved in protein—protein interactions within PriA—DnaD or DnaA—DnaD, we provided experimental evidence using SPR to investigate the effect of various fragments on the ability of DnaD to bind to PriA or DnaA. PriA and DnaA were individually immobilized on a sensor chip (as a ligand), and the DnaD solution (as an analyte) was passed over the sensor chip in a microfluidic chamber. Fig 4 shows the SPR results at various DnaD concentrations. The Kd values were calculated from the equilibrium binding isotherms using a simple binding model (a 1:1 Langmuir binding model). As shown in Table 4, C-terminal deletion of DnaD resulted in decreased Kd values for PriA and DnaA binding, indicating a crucial role for protein—protein interactions. DnaD1-195 was injected at increasing concentrations, but the binding response for PriA did not change; this result indicates that aa 196–228 is necessary for PriA binding in DnaD (Fig 4D) but not for DnaA binding (Fig 4F). Therefore, DnaD may bind to PriA in a different manner from that to DnaA.
Fig 4. SPR analysis.

Protein—protein interactions within (A) PriA—DnaD, (B) PriA—DnaD1-204, (C) PriA—DnaD1-200, (D) PriA—DnaD1-195, (E) DnaA—DnaD and (F) DnaA—DnaD1-195 were analyzed using SPR. SaPriA and SaDnaA were individually immobilized on Series S sensor chips CM5, and the binding experiments were carried out using a Biacore T200 (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). Control samples were used to monitor the sensor chip surface stability, demonstrating reproducibility throughout the duration of the experiments. The estimated Kd values were derived by fitting the association and dissociation signals with a 1:1 (Langmuir) model using the Biacore T200 Evaluation Software. The residual plots for SPR fitting were shown in S3 Fig.
Table 4. Protein—protein interactions within PriA—DnaD, PriA—DnaD1-204, PriA—DnaD1-200, PriA—DnaD1-195, DnaA—DnaD, and DnaA—DnaD1-195.
| Ligand | Analyte | Kd (M) |
|---|---|---|
| PriA | DnaD | 2.0 ± 0.3 x 10−9 |
| PriA | DnaD1-204 | 1.1 ± 0.2 x 10−8 |
| PriA | DnaD1-200 | 2.6 ± 0.3 x 10−8 |
| PriA | DnaD1-195 | ND |
| DnaA | DnaD | 1.7 ± 0.2 x 10−8 |
| DnaA | DnaD1-195 | 1.5 ± 0.2 x 10−7 |
The estimated Kd values were derived by fitting the association and dissociation signals with a 1:1 (Langmuir) model using the Biacore T200 Evaluation Software.
To rapidly confirm whether SaDnaD1-195 can interact with SaDnaA and SaPriA, we carried out gold nanoparticle assays. As histidine-tagged proteins, SaDnaD1-195, SaPriA, and SaDnaA can be immobilized on commercialized Ni2+-NTA SAM nanoparticles. Protein-loaded nanoparticles retain the same red color as the unmodified SAM-coated nanoparticles. However, the solution turns purple when gold nanoparticles aggregate, indicating the protein—protein interactions (Fig 5). The gold nanoparticle assay showed that the solution of SaDnaA—SaDnaD1-195 changed from red to purple (Fig 5A), whereas that of SaPriA-SaDnaD1-195 maintained the red color (Fig 5B). Thus, the C-terminal region aa 196–228 in SaDnaD is relatively important for SaPriA binding, which is consistent with the SPR results.
Fig 5. Gold nanoparticle assay.

(A) Analysis of SaDnaA—SaDnaD1-195 interaction using SaDnaA (lane 1), SaDnaD1-195 (lane 2), and mixture of SaDnaA and SaDnaD1-195 (lane 3). (B) Analysis of SaPriA-SaDnaD1-195 interaction using SaPriA (lane 1), SaDnaD1-195 (lane 2), and mixture of SaPriA and SaDnaD1-195 (lane 3). The gold nanoparticle assay showed that the solution of SaDnaA—SaDnaD1-195 changed from red to purple, whereas that of SaPriA-SaDnaD1-195 maintained the red color
SaDnaD can significantly stimulate the ATPase activity of SaPriA but SaDnaD1-195 cannot
Gram-negative bacterial PriA is known as a poor helicase when acting alone in vitro [45]. E. coli PriA activity can be significantly stimulated by PriB and SSB [46,47,48]. Whether SaDnaD and SaDnaD1-195 can enhance SaPriA ATPase is unknown. SaPriA could hydrolyze ATP alone, and this ATPase activity was dramatically stimulated (twelvefold) in the presence of SaDnaD (S4 Fig). However, when acting with SaDnaD1-195, the stimulation of SaPriA ATPase was insignificant (only twofold). Thus, the C-terminal region of SaDnaD is important for the stimulation of SaPriA ATPase activity.
SaDnaDY176A significantly stimulated the ATPase activity of SaPriA as SaDnaD did
In this study, we determined that the ATPase activity of PriA can be significantly stimulated via contact with the C-terminal region of DnaD (Fig 4). We further investigated whether the DNA binding ability of DnaD is related to the stimulation effect. The highly conserved DNA-binding motif of DnaD, YxxxIxxxW, has been identified [35]. The Y180A mutant protein of B. subtilis DnaD cannot bind DNA [35]. The same DNA-binding motif is also found in SaDnaD, and the residue Y180 in B. subtilis DnaD is Y176 in SaDnaD. To assess whether the DNA binding activity of DnaD is also involved in the stimulation of the ATPase activity of PriA, tag-free SaDnaDY176A mutant protein was produced using GST fusion and Factor Xa (S5 Fig). To exclude the possible effect of a His tag, tag-free SaDnaD and SaDnaD1-195 were also constructed and used in the ATPase assay for analysis. The stimulation effect of a tag-free SaDnaD protein was also analyzed according to the same protocol as that for the His-tagged SaDnaD protein, and their results were similar (S6 Fig). SaPriA ATPase activity was dramatically stimulated (elevenfold to twelvefold) in the presence of SaDnaD. However, when acting with SaDnaD1-195, the stimulation of SaPriA ATPase was insignificant (only twofold). We also used tag-free SaDnaDY176A mutant protein for this analysis and obtained a similar result to that of the wild-type protein. Therefore, the DNA-binding ability of DnaD and the His tag fused in the C terminus of DnaD did not play a role in the stimulation of SaPriA ATPase activity. However, SaDnaD1-195 still partially stimulated the PriA ATPase activity (S6 Fig). How SaDnaD1-195 can slightly enhance the ATPase activity of SaPriA remains unclear.
Effect of SaDnaD, SaDnaD1-195, and SaDnaDY176A on the kinetic parameters of SaPriA ATPase
We further determined the kinetic parameters of PriA ATPase in the presence or absence of DnaD. Given that DnaD can bind and may stabilize the dsDNA substrate, the SaDnaDY176A mutant protein was also used for this analysis to rule out any possible role of the DNA-binding involvement of DnaD. As shown in Fig 6, the ATP hydrolyzing reaction was insignificant when only PriA was added into the assay mixture. Under the same ATP concentration assay range, the kinetic parameters of SaPriA ATPase varied when SaDnaD, SaDnaD1-195, and SaDnaDY176A protein was individually added in the hydrolyzing reaction (Table 5). When acting with SaDnaD or SaDnaDY176A, the Vmax values of SaPriA ATPase increased fivefold, whereas the Km values were only slightly affected. The addition of SaDnaD1-195 to the hydrolyzing reaction induced only a minimal effect.
Fig 6. Effect of SaDnaD, SaDnaD1-195, and SaDnaDY176A on the kinetic parameters of the SaPriA ATPase.
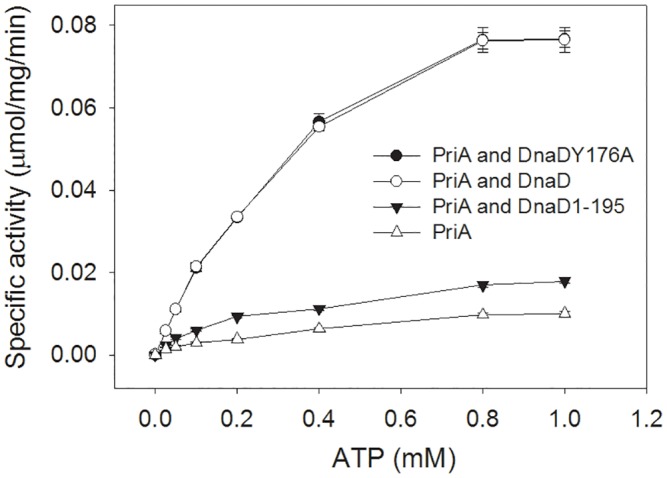
In this experiment, tag-free SaDnaD proteins were used for analysis of SaPriA ATPase activity. SaPriA ATPase assay was performed with 0–1 mM [γ-32P] ATP, 0.125 μM of SaPriA, and 0.1 μM PS4/PS3-dT25 DNA substrate for 1 h. To study the effect, SaDnaD (10 μM), SaDnaDY176A (10 μM), or SaDnaD1-195 (10 μM) was added into the assay solution. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a Phosphorimager. Data points are an average of three determinations and the errors are standard deviation.
Table 5. Apparent Michaelis—Menten constants for SaPriA in the presence of the SaDnaD protein.
| PriA | ||||
|---|---|---|---|---|
| DnaD | Vmax | Km | Vmax/Km | Fold |
| PriA | 0.023 ± 0.001 | 0.47 ± 0.07 | 0.049 | 1.0 |
| PriA and DnaD | 0.114 ± 0.005 | 0.44 ± 0.04 | 0.259 | 5.3 |
| PriA and DnaD1-195 | 0.026 ± 0.003 | 0.42 ± 0.09 | 0.062 | 1.2 |
| PriA and DnaDY176A | 0.115 ± 0.005 | 0.46 ± 0.04 | 0.250 | 5.1 |
The kinetic parameters Km (mM) and Vmax (μmol/mg/min) were determined by fitting the hydrolyzing rate from individual experiments to the Michaelis—Menten equation (Enzyme Kinetics module of Sigma-Plot; Systat Software, Chicago, IL, USA), and then the standard errors were given. The enzyme activity of PriA for kinetic fitting is an average determined at three measurements.
Discussion
S. aureus is a Gram-positive pathogen that exhibits a remarkable ability to develop antibiotic resistance [49,50,51,52,53,54]. Considering that PriA and DnaA-directed primosomes are required for bacterial DNA replication restart processes, these primosomes may be suitable targets for antibiotic development. Because PriA, DnaA, and DnaD are not found in mammals, inhibitors based on these proteins are potentially safe for human use. We found that SaDnaD1-195 can bind to DnaA and can form a tetramer, but it cannot interact with ssDNA and PriA. This binding information on SaDnaD is a prerequisite for new antibacterial drug development and for formulating any model of protein function in DNA replication and DNA replication restart.
In this study, we discovered that the primosomal DnaD protein, which is only found in the Gram-positive bacteria, can bind PriA helicase via the C-terminal region, and this interaction significantly stimulated the activity of PriA. PriA activity can also be stimulated by the Gram-negative E. coli PriB and SSB proteins [47,48]. For SSB, the 15 C-terminal amino acids are absolutely needed for PriA binding and stimulation. Moreover, the stimulation of PriA by the SSB C-terminus requires binding of SSB to ssDNA within the substrate [48]. For PriB, the stimulation of PriA by PriB needs to form a stable PriA-PriB-DNA complex. In addition, the ssDNA-containing DNA substrate is required to simulate PriA by PriB [47]. These two stimulatory cases of PriA by PriB and SSB indicate that both protein—protein interaction and DNA-binding ability are important factors in simulating PriA. However, several lines of evidence indicate that the stimulatory mechanism of PriA by DnaD, PriB or SSB may differ. First, unlike PriB and SSB, DnaD is not an OB-fold protein, thus binding ssDNA differently [38,55]. Second, DnaD is found to bind dsDNA (but with lower affinity than ssDNA) [35], whereas PriB and SSB are not. Third, the DNA-binding mutant SaDnaDY176A still stimulated PriA activity as the wild-type SaDnaD did, whereas the DNA-binding mutants of PriB and SSB do not exhibit a stimulation effect for PriA. Fourth, although the C-terminal region of DnaD was also crucial for the PriA stimulation as the case in SSB, the amino acid residues in SSB and DnaD C-terminal region are quite different and the sequences are not similar. Fifth, the C-terminal region of DnaD is important both for DNA- and PriA-binding, whereas the ssDNA binding region is located at the N-terminal domain of SSB, not the C terminus [55]. Occupying of partially unwound intermediates of DNA unwinding reactions by SSB is known to prevent ssDNA reannealing and enhance the apparent processivity of more distributive helicases. If the DNA binding activity of DnaD plays no role in the activity stimulation of PriA, how can the protein—protein interactions within PriA-DnaD can conduct such effect? The results from our kinetic studies further reveal that PriA binding to DnaD did not significantly change the Km value. Thus, DnaD binding to PriA may not alter the ATP binding environment of PriA. Whether the conformation of PriA changes upon DnaD binding and whether the increased Vmax value of PriA ATPase for DnaD binding results from facilitating the product (ADP) release remains undiscovered.
Roles of the N-terminal domain of DnaD have been well established. The structure and detailed functions of the C-terminal domain of DnaD are still poorly understood. The C-terminal region of DnaD was found to be crucial for both PriA and ssDNA binding, suggesting that these binding sites are overlapped on DnaD. Future studies should focus on whether the binding site for ssDNA on DnaD is necessary to overlap the binding site for PriA, because PriA (a monomer) and DnaD (a tetramer) have different oligomerization states. For example, only one monomer of the PriB dimer can interact with the DNA and its partner proteins [38,56]. The complex structures of DnaD are useful in helping us to understand the primosome assembly mechanisms.
Most DNA helicases of superfamily I and superfamily II are almost non-hexameric and have poor dsDNA unwinding activities when acting alone in vitro [45]. E. coli PriA helicase activity can be significantly stimulated by PriB and SSB. Determining whether DnaD can enhance the Gram-positive PriA helicase or the Gram-negative PriA helicase is important to understand deeply the role of DnaD in the primosome assembly. SPR results indicate that the binding sites on DnaD for PriA and ssDNA do not overlap with the binding site for DnaA (Fig 4), suggesting that the DnaD expression in cells may simultaneously work with DnaA in replication and with PriA in replication restart.
Supporting Information
Coomassie Blue-stained SDS-PAGE (12%) of the purified SaDnaD (lane 1), SaDnaD1-204 (lane 2), SaDnaD1-200 (lane 3), SaDnaD1-195 (lane 4) and molecular mass standards (M) are shown. The sizes of the standard proteins, from the top down, are as follows: 170, 130, 100, 70, 55, 40, 35, 25, 15, and 10 kDa.
(TIF)
(A) SaDnaD1-195, (B) SaDnaD1-200, or (C) SaDnaD1-204, was incubated at 25°C for 30 min with 1.7 nM dT40 in a total volume of 10 μL in 20 mM Tris—HCl (pH 8.0) and 100 mM NaCl. The resulting samples were mixed with gel-loading solution (0.25% bromophenol blue and 40% sucrose; w/v), resolved on a native 8% polyacrylamide gel (8.3 × 7.3 cm) at 4°C in TBE buffer for 1–1.5 h at 100 V, and were visualized by phosphorimaging. The phosphor storage plate was scanned, and the data for complex and free DNA bands were digitized for quantitative analysis.
(TIF)
The estimated Kd values were derived by fitting the association and dissociation signals with a 1:1 (Langmuir) model using the Biacore T200 Evaluation Software. These residual plots were the calculated difference between the experimental and fitted data [57] for PriA—DnaD, PriA—DnaD1-204, PriA-DnaD1-200, DnaA—DnaD, and DnaA—DnaD1-195, respectively.
(TIF)
SaPriA ATPase assay was performed with 0.4 mM [γ-32P] ATP, 0.125 μM of SaPriA, and 0.1 μM PS4/PS3-dT25 DNA substrate for 1 h. To study the effect, SaDnaD (10 μM) or SaDnaD1-195 (10 μM) was added into the assay solution. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a Phosphorimager.
(TIF)
To obtain proteins without a His tag, tag-free SaDnaD, SaDnaDY176A, and SaDnaD1-195 proteins were produced using GST fusion and Factor Xa and were used in succeeding analysis. Coomassie Blue-stained SDS-PAGE (12%) of the purified SaDnaD (lane 1), GST-SaDnaD (lane 2), SaDnaD1-195 (lane 3), GST-SaDnaD1-195 (lane 4), GST-SaDnaDY176A (lane 5), SaDnaDY176A (lane 6), and molecular mass standards (M) are shown. The sizes of the standard proteins, from the top down, are as follows: 170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa.
(TIF)
In this experiment, tag-free SaDnaD proteins were used for analysis of SaPriA ATPase activity. SaPriA ATPase assay was performed with 0.4 mM [γ-32P] ATP, 0.125 μM of SaPriA, and 0.1 μM PS4/PS3-dT25 DNA substrate for 1 h. To study the effect, SaDnaD (10 μM), SaDnaDY176A (10 μM), or SaDnaD1-195 (10 μM) was added into the assay solution. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a Phosphorimager.
(TIF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by a grant from the Ministry of Science and Technology, Taiwan (MOST 103-2320-B-040-018-MY2 to C.Y. Huang). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Merrikh H, Machon C, Grainger WH, Grossman AD, Soultanas P (2011) Co-directional replication-transcription conflicts lead to replication restart. Nature 470: 554–557. 10.1038/nature09758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McHenry CS (2011) DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80: 403–436. 10.1146/annurev-biochem-061208-091655 [DOI] [PubMed] [Google Scholar]
- 3.Leonard AC, Grimwade JE (2011) Regulation of DnaA assembly and activity: taking directions from the genome. Annu. Rev. Microbiol. 65: 19–35. 10.1146/annurev-micro-090110-102934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mott ML, Berger JM (2007) DNA replication initiation: mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 5: 343–354. [DOI] [PubMed] [Google Scholar]
- 5.Heller RC, Marians KJ (2006) Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell. Biol. 7: 932–943. [DOI] [PubMed] [Google Scholar]
- 6.McGlynn P, Lloyd RG (2002) Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell. Biol. 3: 859–870. [DOI] [PubMed] [Google Scholar]
- 7.Cox MM (2001) Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu. Rev. Genet. 35: 53–82. [DOI] [PubMed] [Google Scholar]
- 8.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ (2000) The importance of repairing stalled replication forks. Nature 404: 37–41. [DOI] [PubMed] [Google Scholar]
- 9.Sandler SJ (2000) Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155: 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marians KJ (2000) PriA-directed replication fork restart in Escherichia coli. Trends Biochem. Sci. 25: 185–189. [DOI] [PubMed] [Google Scholar]
- 11.Schekman R, Weiner A, Kornberg A (1974) Multienzyme systems of DNA replication. Science 186: 987–993. [DOI] [PubMed] [Google Scholar]
- 12.Masai H, Tanaka T, Kohda D (2010) Stalled replication forks: making ends meet for recognition and stabilization. Bioessays 32: 687–697. 10.1002/bies.200900196 [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T, Mizukoshi T, Sasaki K, Kohda D, Masai H (2007) Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J. Biol. Chem. 282: 19917–19927. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki K, Ose T, Okamoto N, Maenaka K, Tanaka T, Masai H, et al. (2007) Structural basis of the 3'-end recognition of a leading strand in stalled replication forks by PriA. EMBO J. 26: 2584–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizukoshi T, Tanaka T, Arai K, Kohda D, Masai H (2003) A critical role of the 3' terminus of nascent DNA chains in recognition of stalled replication forks. J. Biol. Chem. 278: 42234–42239. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T, Mizukoshi T, Taniyama C, Kohda D, Arai K, Masai H (2002) DNA binding of PriA protein requires cooperation of the N-terminal D-loop/arrested-fork binding and C-terminal helicase domains. J. Biol. Chem. 277: 38062–38071. [DOI] [PubMed] [Google Scholar]
- 17.Briggs GS, Smits WK, Soultanas P (2012) Chromosomal replication initiation machinery of low-G+C-content Firmicutes. J. Bacteriol. 194: 5162–5170. 10.1128/JB.00865-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soultanas P (2012) Loading mechanisms of ring helicases at replication origins. Mol. Microbiol. 84: 6–16. 10.1111/j.1365-2958.2012.08012.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruand C, Ehrlich SD, Janniere L (1995) Primosome assembly site in Bacillus subtilis. EMBO J. 14: 2642–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang YH, Huang CY (2014) Structural insight into the DNA-binding mode of the primosomal proteins PriA, PriB, and DnaT. Biomed. Res. Int. 2014: 195162 10.1155/2014/195162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruand C, Velten M, McGovern S, Marsin S, Serena C, Ehrlich SD, et al. (2005) Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol. Microbiol. 55: 1138–1150. [DOI] [PubMed] [Google Scholar]
- 22.Turner IJ, Scott DJ, Allen S, Roberts CJ, Soultanas P (2004) The Bacillus subtilis DnaD protein: a putative link between DNA remodeling and initiation of DNA replication. FEBS Lett. 577: 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soultanas P (2002) A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res. 30: 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marsin S, McGovern S, Ehrlich SD, Bruand C, Polard P (2001) Early steps of Bacillus subtilis primosome assembly. J. Biol. Chem. 276: 45818–45825. [DOI] [PubMed] [Google Scholar]
- 25.Bruand C, Farache M, McGovern S, Ehrlich SD, Polard P (2001) DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol. Microbiol. 42: 245–255. [DOI] [PubMed] [Google Scholar]
- 26.Ishigo-Oka D, Ogasawara N, Moriya S (2001) DnaD protein of Bacillus subtilis interacts with DnaA, the initiator protein of replication. J. Bacteriol. 183: 2148–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carneiro MJ, Zhang W, Ioannou C, Scott DJ, Allen S, Roberts CJ, et al. (2006) The DNA-remodelling activity of DnaD is the sum of oligomerization and DNA-binding activities on separate domains. Mol. Microbiol. 60: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smits WK, Goranov AI, Grossman AD (2010) Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol. Microbiol. 75: 452–461. 10.1111/j.1365-2958.2009.06999.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonilla CY, Grossman AD (2012) The primosomal protein DnaD inhibits cooperative DNA binding by the replication initiator DnaA in Bacillus subtilis. J. Bacteriol. 194: 5110–5117. 10.1128/JB.00958-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Machon C, Orta A, Phillips N, Roberts CJ, Allen S, et al. (2008) Single-molecule atomic force spectroscopy reveals that DnaD forms scaffolds and enhances duplex melting. J. Mol. Biol. 377: 706–714. 10.1016/j.jmb.2008.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Allen S, Roberts CJ, Soultanas P (2006) The Bacillus subtilis primosomal protein DnaD untwists supercoiled DNA. J. Bacteriol. 188: 5487–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Carneiro MJ, Turner IJ, Allen S, Roberts CJ, Soultanas P (2005) The Bacillus subtilis DnaD and DnaB proteins exhibit different DNA remodelling activities. J. Mol. Biol. 351: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang CY, Chang YW, Chen WT (2008) Crystal structure of the N-terminal domain of Geobacillus kaustophilus HTA426 DnaD protein. Biochem. Biophys. Res. Commun. 375: 220–224. 10.1016/j.bbrc.2008.07.160 [DOI] [PubMed] [Google Scholar]
- 34.Schneider S, Zhang W, Soultanas P, Paoli M (2008) Structure of the N-terminal oligomerization domain of DnaD reveals a unique tetramerization motif and provides insights into scaffold formation. J. Mol. Biol. 376: 1237–1250. 10.1016/j.jmb.2007.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marston FY, Grainger WH, Smits WK, Hopcroft NH, Green M, Hounslow AM, et al. (2010) When simple sequence comparison fails: the cryptic case of the shared domains of the bacterial replication initiation proteins DnaB and DnaD. Nucleic Acids Res. 38: 6930–6942. 10.1093/nar/gkq465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang YH, Huang CC, Chen CC, Yang KJ, Huang CY (2015) Inhibition of Staphylococcus aureus PriA helicase by flavonol kaempferol. Protein J. 34: 169–172. 10.1007/s10930-015-9609-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang CC, Tsau HW, Chen WT, Huang CY (2010) Identification and characterization of a putative dihydroorotase, KPN01074, from Klebsiella pneumoniae. Protein J. 29: 445–452. 10.1007/s10930-010-9272-2 [DOI] [PubMed] [Google Scholar]
- 38.Huang CY, Hsu CH, Sun YJ, Wu HN, Hsiao CD (2006) Complexed crystal structure of replication restart primosome protein PriB reveals a novel single-stranded DNA-binding mode. Nucleic Acids Res. 34: 3878–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang YH, Lo YH, Huang W, Huang CY (2012) Crystal structure and DNA-binding mode of Klebsiella pneumoniae primosomal PriB protein. Genes Cells 17: 837–849. 10.1111/gtc.12001 [DOI] [PubMed] [Google Scholar]
- 40.Huang YH, Lin MJ, Huang CY (2013) DnaT is a single-stranded DNA binding protein. Genes Cells 18: 1007–1019. 10.1111/gtc.12095 [DOI] [PubMed] [Google Scholar]
- 41.Huang CY (2015) Inhibition of a putative dihydropyrimidinase from Pseudomonas aeruginosa PAO1 by flavonoids and substrates of cyclic amidohydrolases. PLoS One 10: e0127634 10.1371/journal.pone.0127634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo YH, Tsai KL, Sun YJ, Chen WT, Huang CY, Hsiao CD (2009) The crystal structure of a replicative hexameric helicase DnaC and its complex with single-stranded DNA. Nucleic Acids Res. 37: 804–814. 10.1093/nar/gkn999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson AB, Calhoun AK, Smagghe BJ, Stevens MD, Wotkowicz MT, Hatziioannou VM, et al. (2011) A gold nanoparticle platform for protein-protein interactions and drug discovery. ACS Appl. Mater. Interfaces 3: 2979–2987. 10.1021/am200459a [DOI] [PubMed] [Google Scholar]
- 44.Peng WF, Huang CY (2014) Allantoinase and dihydroorotase binding and inhibition by flavonols and the substrates of cyclic amidohydrolases. Biochimie 101: 113–122. 10.1016/j.biochi.2014.01.001 [DOI] [PubMed] [Google Scholar]
- 45.Lohman TM, Tomko EJ, Wu CG (2008) Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell. Biol. 9: 391–401. 10.1038/nrm2394 [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharyya B, George NP, Thurmes TM, Zhou R, Jani N, Wessel SR, et al. (2014) Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. U. S. A. 111: 1373–1378. 10.1073/pnas.1318001111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cadman CJ, Lopper M, Moon PB, Keck JL, McGlynn P (2005) PriB stimulates PriA helicase via an interaction with single-stranded DNA. J. Biol. Chem. 280: 39693–39700. [DOI] [PubMed] [Google Scholar]
- 48.Cadman CJ, McGlynn P (2004) PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 32: 6378–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyama N, Inokoshi J, Tomoda H (2012) Anti-infectious agents against MRSA. Molecules 18: 204–224. 10.3390/molecules18010204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otto M (2010) Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 64: 143–162. 10.1146/annurev.micro.112408.134309 [DOI] [PubMed] [Google Scholar]
- 51.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML (2010) Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23: 99–139. 10.1128/CMR.00042-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, et al. (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466: 935–940. 10.1038/nature09197 [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi SD, DeLeo FR (2009) An update on community-associated MRSA virulence. Curr. Opin. Pharmacol. 9: 545–551. 10.1016/j.coph.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 54.Fischbach MA, Walsh CT (2009) Antibiotics for emerging pathogens. Science 325: 1089–1093. 10.1126/science.1176667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan KW, Lee YJ, Wang CH, Huang H, Sun YJ (2009) Single-stranded DNA-binding protein complex from Helicobacter pylori suggests an ssDNA-binding surface. J. Mol. Biol. 388: 508–519. 10.1016/j.jmb.2009.03.022 [DOI] [PubMed] [Google Scholar]
- 56.Szymanski MR, Jezewska MJ, Bujalowski W (2010) Interactions of the Escherichia coli primosomal PriB protein with the single-stranded DNA. Stoichiometries, intrinsic affinities, cooperativities, and base specificities. J. Mol. Biol. 398: 8–25. 10.1016/j.jmb.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drescher DG, Ramakrishnan NA, Drescher MJ (2009) Surface plasmon resonance (SPR) analysis of binding interactions of proteins in inner-ear sensory epithelia. Methods Mol. Biol. 493: 323–343. 10.1007/978-1-59745-523-7_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coomassie Blue-stained SDS-PAGE (12%) of the purified SaDnaD (lane 1), SaDnaD1-204 (lane 2), SaDnaD1-200 (lane 3), SaDnaD1-195 (lane 4) and molecular mass standards (M) are shown. The sizes of the standard proteins, from the top down, are as follows: 170, 130, 100, 70, 55, 40, 35, 25, 15, and 10 kDa.
(TIF)
(A) SaDnaD1-195, (B) SaDnaD1-200, or (C) SaDnaD1-204, was incubated at 25°C for 30 min with 1.7 nM dT40 in a total volume of 10 μL in 20 mM Tris—HCl (pH 8.0) and 100 mM NaCl. The resulting samples were mixed with gel-loading solution (0.25% bromophenol blue and 40% sucrose; w/v), resolved on a native 8% polyacrylamide gel (8.3 × 7.3 cm) at 4°C in TBE buffer for 1–1.5 h at 100 V, and were visualized by phosphorimaging. The phosphor storage plate was scanned, and the data for complex and free DNA bands were digitized for quantitative analysis.
(TIF)
The estimated Kd values were derived by fitting the association and dissociation signals with a 1:1 (Langmuir) model using the Biacore T200 Evaluation Software. These residual plots were the calculated difference between the experimental and fitted data [57] for PriA—DnaD, PriA—DnaD1-204, PriA-DnaD1-200, DnaA—DnaD, and DnaA—DnaD1-195, respectively.
(TIF)
SaPriA ATPase assay was performed with 0.4 mM [γ-32P] ATP, 0.125 μM of SaPriA, and 0.1 μM PS4/PS3-dT25 DNA substrate for 1 h. To study the effect, SaDnaD (10 μM) or SaDnaD1-195 (10 μM) was added into the assay solution. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a Phosphorimager.
(TIF)
To obtain proteins without a His tag, tag-free SaDnaD, SaDnaDY176A, and SaDnaD1-195 proteins were produced using GST fusion and Factor Xa and were used in succeeding analysis. Coomassie Blue-stained SDS-PAGE (12%) of the purified SaDnaD (lane 1), GST-SaDnaD (lane 2), SaDnaD1-195 (lane 3), GST-SaDnaD1-195 (lane 4), GST-SaDnaDY176A (lane 5), SaDnaDY176A (lane 6), and molecular mass standards (M) are shown. The sizes of the standard proteins, from the top down, are as follows: 170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa.
(TIF)
In this experiment, tag-free SaDnaD proteins were used for analysis of SaPriA ATPase activity. SaPriA ATPase assay was performed with 0.4 mM [γ-32P] ATP, 0.125 μM of SaPriA, and 0.1 μM PS4/PS3-dT25 DNA substrate for 1 h. To study the effect, SaDnaD (10 μM), SaDnaDY176A (10 μM), or SaDnaD1-195 (10 μM) was added into the assay solution. Aliquots (5 μL) were taken and spotted onto a polyethyleneimine cellulose thin-layer chromatography plate, which was subsequently developed in 0.5 M formic acid and 0.25 M LiCl for 30 m. Reaction products were visualized by autoradiography and quantified with a Phosphorimager.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.