

Abstract
A high-density linkage map was constructed using 1,885 newly obtained loci and 3,747 previously published loci, which included 5,152 loci with 4696.03 cM in total length and 0.91 cM in mean distance. Homology analysis in the cotton genome further confirmed the 13 expected homologous chromosome pairs and revealed an obvious inversion on Chr10 or Chr20 and repeated inversions on Chr07 or Chr16. In addition, two reciprocal translocations between Chr02 and Chr03 and between Chr04 and Chr05 were confirmed. Comparative genomics between the tetraploid cotton and the diploid cottons showed that no major structural changes exist between DT and D chromosomes but rather between AT and A chromosomes. Blast analysis between the tetraploid cotton genome and the mixed genome of two diploid cottons showed that most AD chromosomes, regardless of whether it is from the AT or DT genome, preferentially matched with the corresponding homologous chromosome in the diploid A genome, and then the corresponding homologous chromosome in the diploid D genome, indicating that the diploid D genome underwent converted evolution by the diploid A genome to form the DT genome during polyploidization. In addition, the results reflected that a series of chromosomal translocations occurred among Chr01/Chr15, Chr02/Chr14, Chr03/Chr17, Chr04/Chr22, and Chr05/Chr19.
Keywords: tetraploid cotton, genetic linkage map, genome structure, evolution, comparative genomics
1. Introduction
Cotton (Gossypium) is an important cash crop and the uppermost source of textile fibre, and G. hirsutum and G. barbadense account for 90 and 8% of the world cotton production, respectively.1 Thus, genome structure analysis of allotetraploid cotton is important, but the allotetraploid (2n = 4x = 52) species has a large genome size of ∼2,246 Mb,2 indicating that it is especially difficult to generate a reference genome sequence. As a result, a high-density genetic linkage map is an optional tool to reveal genome structure and chromosomal architecture of allotetraploid cotton.
Currently, there are four comparatively dense linkage maps in cotton based on experimental data. Rong et al.3 (2,584 loci, 4447.9 cM in length with an average distance of 1.72 cM) revealed all 13 expected homologous chromosome pairs and reported that there were no major structural changes between DT and D chromosomes, but two reciprocal translocations between AT and A chromosomes and several inversions. Guo et al.4 (2,247 loci, 3440.4 cM in length with an average distance of 1.58 cM) again reported the translocations that occurred between some chromosomes (Chr02 and Chr03, Chr04 and Chr05), providing a glimpse of cotton genome complexity. Based on a genome-wide simple sequence repeat (SSR) genetic map (2,316 loci, 4418.9 cM in length with an average distance of 1.91 cM) constructed in our laboratory,5 21 segregation distortion regions (SDRs) were found, and 3 segregation distorted chromosomes (Chr02, Chr16, and Chr18) were identified with 99.9% of distorted markers segregating towards the heterozygous allele. A genetic linkage map (2,072 loci, 3379.9 cM in length with an average distance of 1.63 cM) constructed by Yu et al.6 showed that the allotetraploid cotton genome produced equivalent recombination frequencies in its two genomes and revealed that the genetically smallest homologous chromosome pair was Chr04 and Chr22, and the largest was Chr05 and Chr19. Just prior to this publication, a sequence-based interspecific genetic map composed of 4,999,048 SNP loci was reported, which contains only 4,049 recombination bins and covers 4,042 cM with an average interbin genetic distance of 1.0 cM.7 First, this map played a role in genomic assembly of allotetraploid cotton. Second, some structural variations not only in the AT genome but also in the DT genome were detected, including 15 first reported simple translocations. Third, centromeric regions of tetraploid cotton were predicted. All of the above genetic maps played fundamental roles in understanding the cotton genome structure and in studying cotton evolutionary genomics.
With the release of G. raimondii genome sequence, genetic relationship analysis showed that G. raimondii and Theobroma cacao belong to a common subclade.8 In addition, the two genomes possess a moderate syntenic relationship, with 463 collinear blocks (with ≥5 genes per block) covering 64.8 and 74.41% of the assembled G. raimondii and T. cacao genomes, respectively.8 Subsequently, G. arboreum genome sequence was released,9 and a close collinear relationship between G. arboreum and T. cacao was also discovered. Besides, collinearity analysis between G. raimondii and G. arboreum revealed that chromosomes 1, 4–6, and 9–13 were highly collinear, whereas large-scale rearrangements were observed on chromosomes 2 and 3 of G. raimondii, and deletions/insertions were observed on chromosomes 7 and 8 of G. arboreum. In 2015, G. hirsutum genome sequence was released,10 which showed a conserved order between the ATDT genome and the already sequenced diploid genomes.8,9 However, another version of the ATDT genome (G. hirsutum)11 showed that such collinearity was not obvious with either G. arboreum9 or G. raimondii.8 Instead, it is largely conserved with another version of the D-progenitor genome (G. raimondii).12 In addition, the overall gene order and collinearity are largely conserved between the AT and DT genomes although at least 9 translocations and 28 inversions were identified.
In the present study, a high-density genetic linkage map including 5,152 loci for tetraploid cotton was constructed mainly based on SSR markers. With this map, we aimed to reveal the cotton genome structure by homologous chromosome comparisons and to explain the polyploidization of tetraploid cotton by comparing this map with two sequenced progenitor diploid cottons.
2. Materials and methods
2.1. Plant materials
Gossypium hirsutum cv. Emian22 and G. barbadense acc. 3–79 were used to detect polymorphisms of all the new markers. Emian22 is a high yield cultivar with moderate fibre quality and no resistance to verticillium wilt, whereas 3–79 is the genetic and cytogenetic standard line for G. barbadense with super fibre quality and high resistance to verticillium wilt. To improve the performance of Emian22 by backcrossing and molecular-assisted selection, a cross between these two materials was performed. Subsequently, the BC1 population [(Emian22 × 3–79) × Emian22] including 141 progeny was used as the mapping population, which had been used to construct a 2,316 loci map.5
2.2. Molecular markers
During map construction in this study, 5,299 new primer pairs were applied, including 4,569 newly published SSRs (2,937 MON-prefixed, 664 NBRI-prefixed, 670 CCRI-prefixed, 200 HAU-prefixed, and 98 NAU-prefixed; http://www.cottongen.org/) and 730 other batches of primers (579 GhirPIP-prefixed,13 115 cg-prefixed,14 and 36 cot-prefixed14).
2.3. Genotyping analysis
Polymorphism detection of all the new SSRs was performed as previously described.15 For the remnant monomorphic SSRs, single-strand conformation polymorphism (SSCP) analysis was applied, which is identical to the method described by Li et al.16 For the other three batches of primers (GhirPIP-prefixed, cg-prefixed, and cot-prefixed), SSCP analysis is the only method for genotyping analysis. Subsequently, genotyping of the whole population using polymorphic primers was carried out on the corresponding condition. All DNA fragments were detected with silver staining.
2.4. Map construction
The mapping data for each BC1 individual were scored according to the definition of JoinMap 3.0.17 For each segregating marker, a χ2 analysis was performed to determine whether it is deviated from the expected 1 : 1 segregation ratio. During map construction, the logarithm of odds (LOD) threshold was ≥8.0, and the maximum recombination rate was 0.4. Map distances in centiMorgans (cM) were calculated using the Kosambi mapping function.18 In the resulting linkage map, a region with at least three adjacent loci showing significant segregation distortion (P < 0.05) was defined as the SDR.19
2.5. Collinearity and comparative genomic analysis
Based on the high-density genetic linkage map and sequences corresponding to markers, collinearity analysis between homologous chromosome pairs was performed using a BLASTN search with E ≤ 1e−5, identity ≥80%, and matched length ≥200 bp. Next, the best hit for each marker was chosen, and all the best hits were illustrated intuitively using online drawing tools (http://circos.ca/). Comparative genomic analysis between tetraploid cotton and diploid cottons was performed using a similar method.
3. Results
3.1. Marker information
To construct a high-density genetic linkage map to meet the demands of cotton genetics and breeding, 200 novel EST-SSRs (HAU3599-HAU3798) were developed in this study. In detail, 3,647 unique sequences were obtained from a normalized adversity cDNA library of G. barbadense acc. Hai7124, and then they were searched against the cotton EST database (posted date: 22 March 2009, with a total of 375,349 sequences), with an E-value cut-off of <1e−10. As a result, 255 had no BLAST hits to known sequences. Subsequently, a total of 200 novel EST-SSRs were developed. During map construction, traditional genotyping analysis made 26 (13.00%) polymorphic primer pairs generating 26 polymorphic loci, and SSCP analysis made 14 (8.05%) polymorphic primer pairs generating 16 polymorphic loci from the remnant monomorphic markers. In total, 40 (20.00%) HAU-prefixed EST-SSRs showed polymorphism, and generated 42 polymorphic loci, with an average of 1.05 alleles.
Among the 670 CCRI-prefixed and 98 NAU-prefixed EST-SSRs, traditional genotyping analysis made 75 (11.19%) and 66 (67.35%) polymorphic EST-SSRs generating 78 and 79 polymorphic loci, respectively. For the remnant monomorphic markers, SSCP analysis was applied. As a result, 67 (11.26%) and 11 (34.38%) were polymorphic with 73 and 11 polymorphic loci, respectively. In total, 142 (21.19%) CCRI-prefixed and 77 (78.57%) NAU-prefixed EST-SSRs were polymorphic, and generated 151 and 90 polymorphic loci, with an average of 1.06 and 1.17 alleles, respectively.
The total 2,937 MON-prefixed SSRs consist of 2,521 gSSRs and 416 EST-SSRs. After traditional genotyping analysis, 879 (34.87%) gSSRs and 129 (31.01%) EST-SSRs showed polymorphism, and generated 1,005 and 140 polymorphic loci, respectively. Then, the SSCP analysis was conducted with the remnant monomorphic markers. Subsequently, 126 (7.67%) gSSRs and 12 (4.18%) EST-SSRs showed polymorphism, and generated 138 and 13 polymorphic loci, respectively. In total, 1,005 (39.87%) MON-prefixed gSSRs and 141 (33.89%) MON-prefixed EST-SSRs showed polymorphism, and generated 1,143 and 153 polymorphic loci, with an average of 1.14 and 1.09 alleles, respectively. Taking the 2,937 MON-prefixed SSRs as a whole, 1,146 (39.02%) primers were polymorphic, and generated 1,296 polymorphic loci, with an average of 1.13 alleles.
The total 664 NBRI-prefixed SSRs consist of 263 gSSRs and 401 EST-SSRs. After traditional genotyping analysis, 85 (32.32%) gSSRs and 122 (30.42%) EST-SSRs showed polymorphism, and generated 97 and 136 polymorphic loci, respectively. Then, SSCP analysis was performed with the remnant monomorphic markers. Subsequently, 11 (6.18%) gSSRs and 15 (5.38%) EST-SSRs showed polymorphism, and generated 11 and 17 polymorphic loci, respectively. In total, 96 (36.50%) NBRI-prefixed gSSRs and 137 (34.16%) NBRI-prefixed EST-SSRs showed polymorphism, and generated 108 and 153 polymorphic loci, with an average of 1.13 and 1.12 alleles, respectively. Taking the 664 NBRI-prefixed SSRs as a whole, 233 (35.09%) primers showed polymorphism, and generated 261 polymorphic loci, with an average of 1.12 alleles.
For the 579 GhirPIP-prefixed, 115 cg-prefixed, and 36 cot-prefixed primer pairs, SSCP analysis was directly applied. As a result, 27 (4.66%) GhirPIP-prefixed, 13 (11.30%) cg-prefixed, and 5 (13.89%) cot-prefixed primer pairs showed polymorphism, and each polymorphic primer pair produced one polymorphic locus.
3.2. Map construction and overview
The 1,885 loci obtained in this study were added to the 3,747 loci updated in our laboratory,5,16,20–27 and a total of 5,632 loci were used for map construction. After calculation, a linkage map with 5,152 loci was constructed, and it was 4696.03 cM in total length and 0.91 cM in mean distance (Supplementary Fig. S1 and Table 1). The chromosomes were built with LODs ranging from 8.0 to 15.0 (Table 1). The AT genome contained 2,473 loci with 2359.36 cM in total length and 0.95 cM in mean distance, whereas the DT genome contained 2,679 loci with 2336.67 cM in total length and 0.87 cM in mean distance.
Table 1.
Characteristics of the linkage map constructed from the BC1 population
| Chromosome | LOD | Total loci | Size (cM) | Mean distance (cM) | Largest gap (cM) | >10 cM makers |
|---|---|---|---|---|---|---|
| Chr01 | 10.0 | 143 | 186.87 | 1.31 | 21.22 | 1 |
| Chr02 | 12.0 | 130 | 156.03 | 1.20 | 10.67 | 1 |
| Chr03 | 15.0 | 169 | 164.93 | 0.98 | 9.49 | 0 |
| Chr04 | 12.0 | 122 | 149.82 | 1.23 | 7.85 | 0 |
| Chr05 | 13.0 | 285 | 242.76 | 0.85 | 9.16 | 0 |
| Chr06 | 9.0 | 165 | 171.43 | 1.04 | 10.41 | 1 |
| Chr07 | 14.0 | 163 | 105.78 | 0.65 | 3.42 | 0 |
| Chr08 | 11.0 | 213 | 151.02 | 0.71 | 9.38 | 0 |
| Chr09 | 10.0 | 180 | 148.83 | 0.83 | 4.15 | 0 |
| Chr10 | 10.0 | 180 | 200.94 | 1.12 | 17.93 | 1 |
| Chr11 | 13.0 | 271 | 234.77 | 0.87 | 6.09 | 0 |
| Chr12 | 14.0 | 241 | 238.05 | 0.99 | 6.26 | 0 |
| Chr13 | 10.0 | 211 | 208.14 | 0.99 | 6.56 | 0 |
| AT genome | 2,473 | 2,359.36 | 0.95 | 21.22 | 4 | |
| Chr14 | 15.0 | 180 | 164.12 | 0.91 | 9.45 | 0 |
| Chr15 | 11.0 | 215 | 197.10 | 0.92 | 11.05 | 1 |
| Chr16 | 14.0 | 179 | 94.32 | 0.53 | 4.68 | 0 |
| Chr17 | 9.0 | 150 | 162.23 | 1.08 | 22.56 | 2 |
| Chr18 | 10.0 | 188 | 146.95 | 0.78 | 12.18 | 1 |
| Chr19 | 13.0 | 306 | 252.27 | 0.82 | 5.36 | 0 |
| Chr20 | 10.0 | 183 | 117.61 | 0.64 | 8.48 | 0 |
| Chr21 | 13.0 | 265 | 256.03 | 0.97 | 8.85 | 0 |
| Chr22 | 8.0 | 147 | 169.93 | 1.16 | 16.39 | 1 |
| Chr23 | 10.0 | 209 | 193.19 | 0.92 | 12.84 | 2 |
| Chr24 | 10.0 | 225 | 198.85 | 0.88 | 11.81 | 1 |
| Chr25 | 12.0 | 194 | 172.19 | 0.89 | 11.41 | 1 |
| Chr26 | 9.0 | 238 | 211.90 | 0.89 | 14.80 | 2 |
| DT genome | 2,679 | 2,336.67 | 0.87 | 22.56 | 11 | |
| Total | 5,152 | 4,696.03 | 0.91 | 22.56 | 15 |
LOD, logarithm of odds.
The chromosome with the most loci was Chr19 (306 loci), whereas Chr04 had the fewest loci (122 loci), with average loci on each chromosome of 198. The longest chromosome was Chr21 (256.03 cM), whereas the shortest was Chr16 (94.32 cM), with an average chromosome length of 180.62 cM. The largest average distance between markers was on Chr01 (1.31 cM), and the least was on Chr16 (0.53 cM). The largest gap between markers was 22.56 cM on Chr17, and there were a total of 15 gaps >10 cM with 4 on the AT and 11 on the DT genome.
Among the 5,632 polymorphic loci used for map construction, 1,006 loci (17.86%) showed segregation distortion (P < 0.05) and 776 distorted loci, accounting for 15.06% of the mapped loci, were unevenly mapped on cotton chromosomes with 5–125 loci on each chromosome (Supplementary Fig. S1). The most distorted loci were on Chr02 (71), Chr16 (125), and Chr18 (106) (>50% of loci were distorted), accounting for 38.92% of the mapped distorted loci. A total of 62 SDRs were found on 18 cotton chromosomes. More SDRs were found on Chr02 (9), Chr16 (11), and Chr18 (10), which were the chromosomes with the most distorted loci.
3.3. Collinearity in the cotton genome
Based on the 4,807 available marker-derived sequences in the genetic linkage map, homology analysis showed that homologous chromosomes between AT and DT had the highest homology (Fig. 1). In detail, seven homologous chromosome pairs (Chr01 and Chr15, Chr06 and Chr25, Chr08 and Chr24, Chr09 and Chr23, Chr11 and Chr21, Chr12 and Chr26, and Chr13 and Chr18) showed high collinearity. However, not the whole chromosomes showed collinearity between Chr10 and Chr20 (Supplementary Fig. S2A), whereas Chr07 and Chr16 had good homology but hardly any collinearity (Supplementary Fig. S2B). In addition, although most of Chr02 had good collinearity with Chr14, the rest had comparatively good collinearity with Chr17 (Supplementary Fig. S3A). Additionally, most of Chr03 had good collinearity with Chr17, but the rest had comparatively good collinearity with Chr14 (Supplementary Fig. S3B). Moreover, this also appeared among Chr04, Chr05, Chr19, and Chr22 (Supplementary Fig. S3C and D).
Figure 1.

Collinearity among 26 chromosomes in tetraploid cotton. This figure is available in black and white in print and in colour at DNA Research online.
3.4. Comparative genomics between the tetraploid cotton and diploid cottons
Using the 4,807 available nucleotide sequences of 5,152 mapped markers and the genome sequence of G. arboreum or G. raimondii, we found by comparative genomics that sequences of 3,534 and 3,584 markers were homologous from the physical map of G. arboreum and G. raimondii, with an alignment proportion of 68.59 and 69.57%, respectively. The 3,534 markers blasted on the G. arboreum genome included 1,717 markers from the AT genome with an alignment proportion of 69.43%, and 1,817 markers from the DT genome with an alignment proportion of 67.82%. The 3,584 markers blasted on the G. raimondii genome included 1,631 markers from the AT genome with an alignment proportion of 65.95% and 1,953 markers from the DT genome with an alignment proportion of 72.90%.
In general, most chromosomes had good homology with their corresponding chromosomes in the diploid A or D genome (Figs 2 and 3, and Table 2). Exceptionally, during comparative analysis with the diploid A genome, three homologous chromosome pairs, Chr01/Chr15, Chr02/Chr14, and Chr03/Chr17, also showed good homology with A2, A5, and A7, respectively (Fig. 2 and Table 2). During comparative analysis with the diploid D genome, Chr02, Chr03, Chr04, and Chr05 also showed good homology with D3, D5, D9, and D12, respectively (Fig. 3 and Table 2).
Figure 2.

Intuitive diagram of affinity between the linkage map and G. arboreum. This figure is available in black and white in print and in colour at DNA Research online.
Figure 3.

Intuitive diagram of affinity between the linkage map and G. raimondii. This figure is available in black and white in print and in colour at DNA Research online.
Table 2.
Chromosome collinearity between tetraploid cotton and diploid cotton
| Chromosome in tetraploid cotton | Chromosome of highest/higher homology with G. arboreum | Chromosome of highest/higher homology with G. raimondii | Chromosome in tetraploid cotton | Chromosome of highest/higher homology with G. arboreum | Chromosome of highest/higher homology with G. raimondii |
|---|---|---|---|---|---|
| Chr01 (AT1) | A7/A2 | D2 | Chr15 (DT1) | A7/A2 | D2 |
| Chr02 (AT2) | A5/A2 | D3/D5 | Chr14 (DT2) | A5 | D5 |
| Chr03 (AT3) | A5/A7 | D5/D3 | Chr17 (DT3) | A7/A2 | D3 |
| Chr04 (AT4) | A12 | D12/D9 | Chr22 (DT4) | A12 | D12 |
| Chr05 (AT5) | A10 | D9/D12 | Chr19 (DT5) | A10 | D9 |
| Chr06 (AT6) | A8 | D10 | Chr25 (DT6) | A8 | D10 |
| Chr07 (AT7) | A1 | D1 | Chr16 (DT7) | A1 | D1 |
| Chr08 (AT8) | A3 | D4 | Chr24 (DT8) | A3 | D4 |
| Chr09 (AT9) | A11 | D6 | Chr23 (DT9) | A11 | D6 |
| Chr10 (AT10) | A9 | D11 | Chr20 (DT10) | A9 | D11 |
| Chr11 (AT11) | A4 | D7 | Chr21 (DT11) | A4 | D7 |
| Chr12 (AT12) | A6 | D8 | Chr26 (DT12) | A6 | D8 |
| Chr13 (AT13) | A13 | D13 | Chr18 (DT13) | A13 | D13 |
3.5. Blast analysis between the tetraploid cotton and the mixed genome of two diploid cottons
A blast analysis of markers on the tetraploid cotton genetic linkage map was conducted by taking the G. arboreum and G. raimondii genomes together. Regardless of whether it is from the AT or DT genome, all homologous chromosome pairs, except for Chr01/Chr15, Chr02/Chr14, Chr03/Chr17, and Chr05/Chr19, had the highest homology with the corresponding homologous chromosome in the diploid A genome, and had the second highest homology with the corresponding homologous chromosome in the diploid D genome (Fig. 4 and Table 3). Chr01 was predominantly homologous to A7, followed by A2 and D2; Chr15 had the highest homology with D2, followed by A2 and A7. Chr02 had the highest homology with A5, followed by A2, but very low homology with D5; Chr14 had the highest homology with A5, followed by D5, but very low homology with A2. Chr03 had the highest homology with A5, followed by A7, and low homology with D3; Chr17 had the highest homology with D3, followed by A7, and no homology with A5. Chr04 had the highest homology with A12, followed by D12 and D9; Chr22 had the highest homology with A12, followed by D12. Chr05 had the highest homology with A10, followed by D9 and A12; Chr19 had the highest homology with D9, followed by A10 (Fig. 4, Supplementary Fig. S4, and Table 3).
Figure 4.

Intuitive diagram of blast analysis between tetraploid cotton and the mixed genome of two diploid cottons. This figure is available in black and white in print and in colour at DNA Research online.
Table 3.
Blast analysis between the tetraploid cotton and the mixed genome of two diploid cottons
| Chromosome in tetraploid cotton | Chromosome of highest homology with the mixed genome of two diploid cottons | Chromosome of second highest homology with the mixed genome of two diploid cottons | Chromosome in tetraploid cotton | Chromosome of highest homology with the mixed genome of two diploid cottons | Chromosome of second highest homology with the mixed genome of two diploid cottons |
|---|---|---|---|---|---|
| Chr01 (AT1) | A7 | A2 | Chr15 (DT1) | D2 | A2 |
| Chr02 (AT2) | A5 | A2 | Chr14 (DT2) | A5 | D5 |
| Chr03 (AT3) | A5 | A7 | Chr17 (DT3) | D3 | A7 |
| Chr04 (AT4) | A12 | D12 | Chr22 (DT4) | A12 | D12 |
| Chr05 (AT5) | A10 | D9 | Chr19 (DT5) | D9 | A10 |
| Chr06 (AT6) | A8 | D10 | Chr25 (DT6) | A8 | D10 |
| Chr07 (AT7) | A1 | D1 | Chr16 (DT7) | A1 | D1 |
| Chr08 (AT8) | A3 | D4 | Chr24 (DT8) | A3 | D4 |
| Chr09 (AT9) | A11 | D6 | Chr23 (DT9) | A11 | D6 |
| Chr10 (AT10) | A9 | D11 | Chr20 (DT10) | A9 | D11 |
| Chr11 (AT11) | A4 | D7 | Chr21 (DT11) | A4 | D7 |
| Chr12 (AT12) | A6 | D8 | Chr26 (DT12) | A6 | D8 |
| Chr13 (AT13) | A13 | D13 | Chr18 (DT13) | A13 | D13 |
4. Discussion
4.1. Different polymorphism ratios of markers
High-density genetic linkage maps are becoming increasingly important. Because the polymorphism in cotton is low,28,29 more researchers have paid attention to developing novel markers. Our laboratory also endeavoured to develop more molecular markers to construct high-density linkage maps in cotton to facilitate our cotton genetics and breeding studies. In this study, 200 HAU-prefixed new SSRs were developed from newly released ESTs from an adversity cDNA library of G. barbadense acc. Hai7124, which supplement our previously published markers.
Among all the SSR markers applied in this study, NAU-prefixed EST-SSRs showed the highest polymorphism ratio (78.57%), mainly owing to the artificial selection of polymorphic SSRs based on published results.30 Polymorphisms of HAU (20.00%), CCRI (21.19%), MON (33.89%), and NBRI (34.16%)-prefixed EST-SSRs were universally lower than that of MON (39.87%) and NBRI (36.50%)-prefixed gSSRs, which is in accordance with previous reports that ESTs are more conservative because of more selective pressure.16,31 Polymorphisms of MON- and NBRI-prefixed EST-SSRs were much higher than that of HAU- and CCRI-prefixed EST-SSRs for the following reasons. MON-prefixed SSRs reliably generated two or more amplicons,32 which raised the possibility of producing polymorphisms in each primer pair. NBRI-prefixed SSRs were developed from G. herbaceum (AA);33 when they were used in tetraploid cottons (AADD), high polymorphism between G. hirsutum and G. barbadense may be detected. Generally speaking, the SSR markers in this study were comparatively highly polymorphic. One cause may be the great difference between the two parents, and the other would be the application of the SSCP method during genotyping analysis.
GhirPIP-prefixed markers13 (containing intron single-nucleotide polymorphisms and intron length polymorphisms with a predicted ratio of 3 : 1), cg-prefixed SNP/InDel markers14 (developed from 3′ end, 5′ end, or intron sequences), and cot-prefixed SNP/InDel markers14 (developed from conserved orthologous sets) all showed low polymorphism (4.66, 11.30, and 13.89%, respectively), mainly because the SSCP method applied in this study has its recognized limits in reflecting SNPs/InDels. The reason why GhirPIP-prefixed markers showed such a low polymorphism rate (4.66%) may be that they were developed from the predicted introns of G. hirsutum based on the complete genome sequence of model plants, which existed deviation undoubtedly.
Thus, together with previous reports that SSR markers are highly reproducible across species,34 and could provide more convenient assays of collinearity between different genomes,35 SSRs are ideal markers for map construction.
4.2. Characteristics of genome structure in tetraploid cotton
As previously reported, a high-density genetic linkage map could accelerate genome structure analysis. The present map (5,152 loci, 4696.03 cM in length with an average distance of 0.91 cM; Supplementary Fig. S1 and Table 1) revealed that (i) more loci were found on the DT genome than on the AT genome, consistent with the results of Guo et al.4 and Yu et al.,5 but inconsistent with those of Rong et al.3 and Yu et al.;6 (ii) the DT genome was shorter than the AT genome, consistent with the results of Rong et al.,3 Yu et al.,5 and Yu et al.,6 but inconsistent with those of Guo et al.;4 (iii) the average marker distance of the DT genome is shorter than that of the AT genome, consistent with the results of Rong et al.,3 Guo et al.,4 and Yu et al.,5 but inconsistent with those of Yu et al.;6 and (iv) there were more gaps (>10 cM) on the DT genome than on the AT genome, consistent with the results of Yu et al.,5 but inconsistent with those of Rong et al.,3 Guo et al.,4 and Yu et al.6 Variations among the five genetic linkage maps are likely the results of differences in the mapping population types and sizes, as well as in the numbers and sources of molecular markers.
In this study, 776 of the total 1,006 distorted loci were mapped on the 26 cotton chromosomes, and 62 SDRs were discovered. Further analysis showed that three chromosomes (Chr02, Chr16, and Chr18) had extreme segregation distortion (>50% of loci showing distortion, accounting for 38.92% of the mapped distorted loci, and containing 30 SDRs), which has been identified by our previous study.5 Because segregation distortion is increasingly recognized as a potentially powerful evolutionary force,36 the present results suggest the prospect for a wider application. In addition, segregation distortion mechanisms of these three chromosomes are being researched using eight reciprocal backcrossing populations, which were financially supported by the National Science Foundation of China (Grant No. 31171593).
Tetraploid cotton, containing AT and DT genomes, was formed from an interspecific hybridization event between diploid A and diploid D cotton species that may have evolved from a common ancestor.37,38 As a result, 13 expected homologous AT/DT chromosome pairs were further confirmed by the present results that the highest homology or collinearity existed between them (Fig. 1), which is in accordance with previous reports.3 These results may be useful in identifying candidate genes. For example, if one quantitative trait locus (QTL) was detected on the AT chromosome, a candidate gene controlling the same trait could be predicted on the corresponding collinear segment of the DT chromosome, so that we can study the functional divergence of homologous genes. However, it is notable that an obvious inversion appeared on Chr10 or Chr20, and repeated inversions appeared on Chr07 or Chr16 (Fig. 1 and Supplementary Fig. S2). In addition, two reciprocal translocations between Chr02 and Chr03 and between Chr04 and Chr05 were confirmed in this study (Fig. 1 and Supplementary Fig. S3), corresponding to previous reports.3,6,28,39 All of the above may result from genome rearrangements during or after the polyploidization process of the two ancestral diploid genomes, indicating complex but linear features of the tetraploid cotton genome.6 Another possibility is that the conformation of Chr02, Chr03, Chr04, and Chr05 in the AT genome may result from fracture and fusion of ancient chromosomes, since diploid cotton is a paleohexaploid with a radix of seven.12
4.3. Good collinearity between the AT/DT genome in tetraploid and diploid A/D genomes
Comparative genomic analysis between tetraploid cotton and diploid cottons showed that 12 chromosomes in the AT genome, except for Chr02 (AT2), showed good collinearity with chromosomes in the diploid A genome (Fig. 2 and Table 2); and 13 chromosomes in the DT genome showed good collinearity with chromosomes in the diploid D genome (Fig. 3 and Table 2), which is consistent with the results of Li et al.9 and Wang et al.,8 respectively, and is consistent with the report that no major structural changes exist between the DT and D chromosomes, but rather between the AT and A chromosomes.3,40 Moreover, homology between the AT genome and the diploid D genome (Fig. 3 and Table 2), as well as between the DT genome and the diploid A genome (Fig. 2 and Table 2), supplied further evidence to define 13 homologous chromosome pairs in tetraploid cotton. In addition, it also provided evidence for collinearity between the diploid A and D genomes, as well as homology between chromosomes from different diploid genomes. The present results are a complete unification of the reports that are entirely based on the genome sequences of two diploid species.9
4.4. The diploid A genome dominates the tetraploid cotton genome
It is well known that tetraploid cotton was formed from an interspecific hybridization event between an A-genome species and a D-genome species,4 and the diploid A genome is nearly 2-fold larger than the D genome, although they diverged from a common ancestor ∼5–10 million years ago.38 However, previous studies,3–6 as well as the present study, reported that there is a minor difference between the AT and DT genomes in tetraploid cotton. Previous reports that the AT genome is more stable than the DT genome,28 and the present fact that regardless of an AT or DT genome origin, most chromosomes had the highest homology with the corresponding homologous chromosomes in the diploid A genome (Fig. 4 and Table 3), support the inference that during the hybridization evolution of tetraploid cotton, the DT genome underwent invasion of the diploid A genome (Fig. 5). This result may partly explain why the diploid D-genome species do not produce spinnable fibre,41 while many QTLs for fibre-related traits have been detected in the DT genome of tetraploid cotton.42–44
Figure 5.
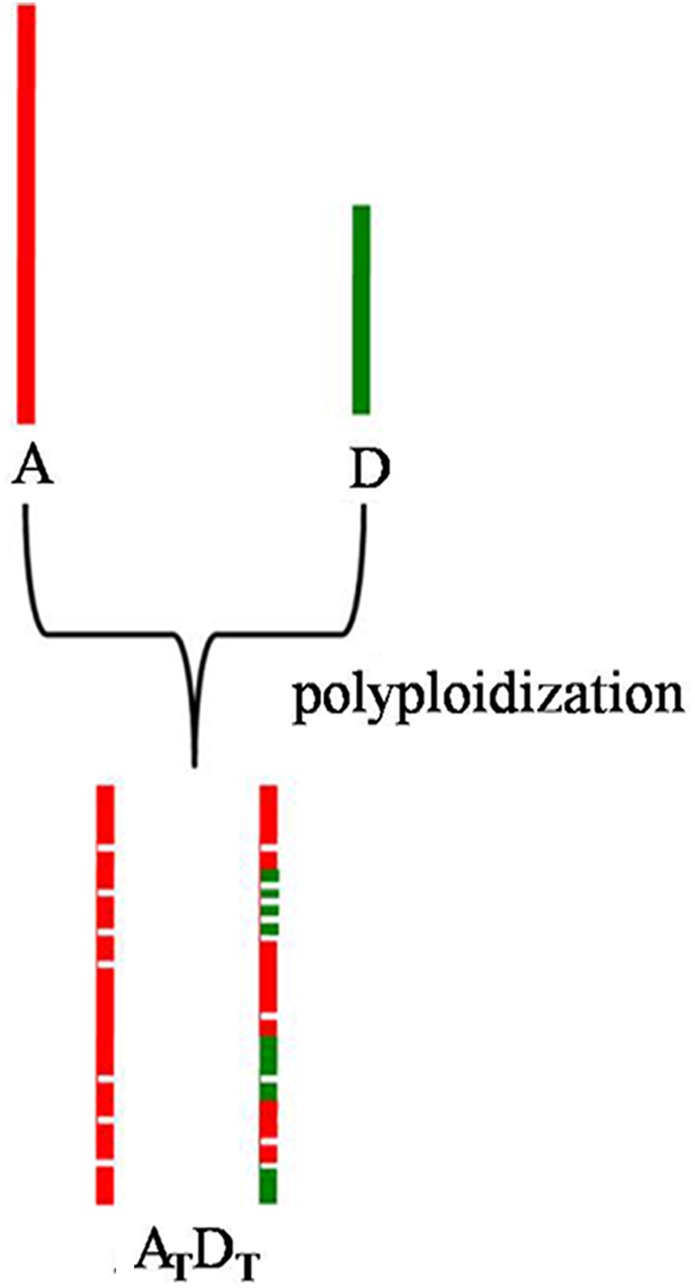
Schematic drawing that illustrates the evolution of most chromosomes. This figure is available in black and white in print and in colour at DNA Research online.
If the formation of Chr04/Chr22 and Chr05/Chr19 was similar to other homologous chromosome pairs, their structure should be as shown in Fig. 6A. However, blast analysis between the tetraploid cotton genome and the mixed genome of two diploid cottons showed that Chr04 had the highest homology with A12, followed by D12 and D9 that were equal to each other. Additionally, Chr05 had the highest homology with A10, followed by D9 and A12 that were equal to each other (Fig. 4 and Supplementary Fig. S4). Thus, it is predicted that Chr04 should have a segment of D9, and Chr05 should have a segment of A12 (as shown in Fig. 6C). This prediction could also be confirmed by comparative genomics between the tetraploid cotton and diploid cottons (Figs 2 and 3, and Table 2). In that way, chromosomal translocations must occur during interspecific hybridization events as shown in Fig. 6B. In detail, one chromosome translocation occurred between Chr04 and Chr05, and another one occurred between Chr04 and Chr19. Similarly, considering the results of comparative genomics between the tetraploid cotton and diploid cottons, and the results of blast analysis between the tetraploid cotton genome and the mixed genome of two diploid cottons, a series of chromosomal translocations among Chr01/Chr15, Chr02/Chr14, and Chr03/Chr17 could also be predicted (Fig. 7). All of the above demonstrate that these five homologous chromosome pairs changed in a complex and dramatic manner in that they coevolved during polyploidization, while other chromosomes in the AT genome just happened minor variations and those in the DT genome just underwent invasion of the corresponding homologous chromosome in the diploid A genome.
Figure 6.
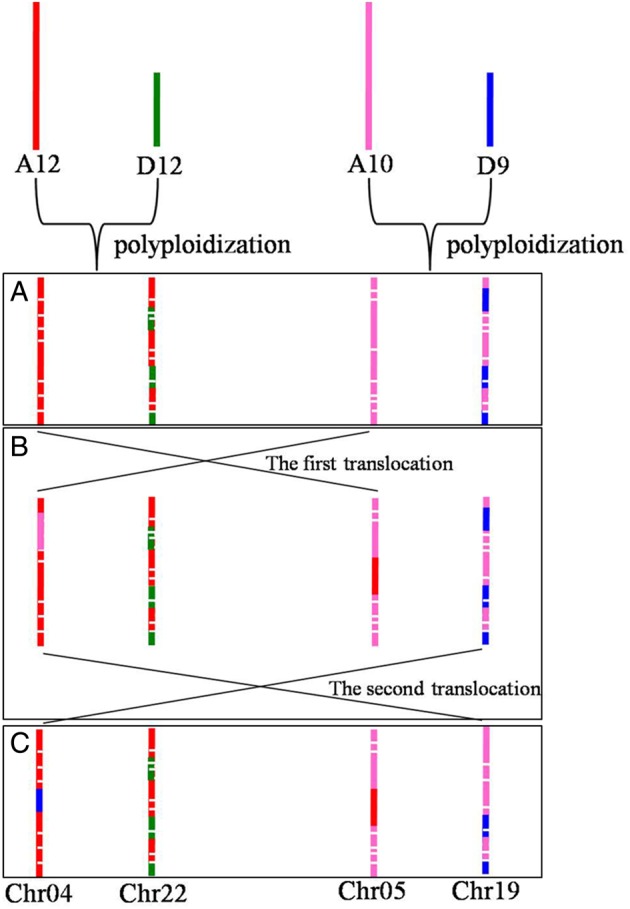
Schematic drawing that illustrates the evolution of Chr04/Chr22 and Chr05/Chr19. This figure is available in black and white in print and in colour at DNA Research online.
Figure 7.

Schematic drawing that illustrates the evolution of Chr01/Chr15, Chr02/Chr14, and Chr03/Chr17. This figure is available in black and white in print and in colour at DNA Research online.
4.5. Applications of the high-density linkage map in tetraploid cotton genomics, genetics, and breeding
The present high-density genetic linkage map, with an average interval of 0.91 × 400 kb between genetic markers on the basis of a consensus estimate of genome size of ∼2,246Mb, provides a foundation to facilitate genome sequencing and sequence assembly.45 Besides, it well revealed the genome structure of tetraploid cotton and provided preliminary hypotheses of tetraploid cotton formation from two diploid cotton species. Until now, cotton breeders have been working to transfer excellent genes controlling fibre quality from G. barbadense to G. hirsutum. And, introgression lines have proved to be an optional way to solve high sterility and crazed segregation of interspecific hybrid progeny. Excellent introgression lines are usually constructed by marker-assisted selection, which is inevitably linked to a genetic linkage map. The present high-density linkage map could also provide markers that are tightly linked with target traits, which is undoubtedly very useful to molecular breeding and genetic improvement.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was financially supported by the National Science Foundation of China (grant no. 31171593) and Genetically Modified Organisms Breeding Major Project of China (no. 2014ZX08009). Funding to pay the Open Access publication charges for this article was provided by the National Science Foundation of China (grant no. 31171593).
Supplementary Material
References
- 1.Zhang H.B., Li Y., Wang B., Chee P.W.. 2008, Recent advances in cotton genomics, Int. J. Plant Genomics, 2008, 742304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinisch A.J., Dong J.M., Brubaker C.L., Stelly D.M., Wendel J.F., Paterson A.H.. 1994, A detailed RFLP map of cotton Gossypium hirsutum× Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome, Genetics, 138, 829–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rong J., Abbey C., Bowers J.E. et al. 2004, A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium), Genetics, 166, 389–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo W., Cai C., Wang C., Zhao L., Wang L., Zhang T.. 2008, A preliminary analysis of genome structure and composition in Gossypium hirsutum, BMC Genomics, 9, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y., Yuan D., Liang S. et al. 2011, Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense, BMC Genomics, 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu J.Z., Kohel R.J., Fang D.D. et al. 2012, A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome, G3 (Bethesda), 2, 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S., Chen J., Zhang W. et al. 2015, Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes, Genome Biol., 16, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K., Wang Z., Li F. et al. 2012, The draft genome of a diploid cotton Gossypium raimondii, Nat. Genet., 44, 1098–103. [DOI] [PubMed] [Google Scholar]
- 9.Li F., Fan G., Wang K. et al. 2014, Genome sequence of the cultivated cotton Gossypium arboreum, Nat. Genet., 46, 567–72. [DOI] [PubMed] [Google Scholar]
- 10.Li F., Fan G., Lu C. et al. 2015, Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution, Nat. Biotechnol., 33, 524–30. [DOI] [PubMed] [Google Scholar]
- 11.Zhang T., Hu Y., Jiang W. et al. 2015, Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement, Nat. Biotechnol., 33, 531–7. [DOI] [PubMed] [Google Scholar]
- 12.Paterson A.H., Wendel J.F., Gundlach H. et al. 2012, Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres, Nature, 492, 423–7. [DOI] [PubMed] [Google Scholar]
- 13.Yang L., Jin G., Zhao X., Zheng Y., Xu Z., Wu W.. 2007, PIP: a database of potential intron polymorphism markers, Bioinformatics, 23, 2174–7. [DOI] [PubMed] [Google Scholar]
- 14.Van Deynze A., Stoffel K., Lee M. et al. 2009, Sampling nucleotide diversity in cotton, BMC Plant. Biol., 9, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Z., He D., Zhang X. et al. 2005, Linkage map construction and mapping QTL for cotton fibre quality using SRAP, SSR and RAPD, Plant. Breed, 124, 180–7. [Google Scholar]
- 16.Li X., Yuan D., Wang H. et al. 2012, Increasing cotton genome coverage with polymorphic SSRs as revealed by SSCP, Genome, 55, 459–70. [DOI] [PubMed] [Google Scholar]
- 17.Stam P. 1993, Construction of integrated genetic linkage maps by means of a new computer package: Join Map, Plant J., 3, 739–44. [Google Scholar]
- 18.Kosambi D.D. 1944, The estimation of map distance from recombination values, Ann. Eugen., 12, 172–5. [Google Scholar]
- 19.Paillard S., Schnurbusch T., Winzeler M. et al. 2003, An integrative genetic linkage map of winter wheat (Triticum aestivum L.), Theor. Appl. Genet., 107, 1235–42. [DOI] [PubMed] [Google Scholar]
- 20.Wang X., Ren G., Li X., Tu J., Lin Z., Zhang X.. 2012, Development and evaluation of intron and insertion-deletion markers for Gossypium barbadense, Plant. Mol. Biol. Rep., 30, 605–13. [Google Scholar]
- 21.Liu C., Lin Z., Zhang X.. 2012, Unbiased genomic distribution of genes related to cell morphogenesis in cotton by chromosome mapping, Plant Cell Tissue Org. Cult., 108, 529–34. [Google Scholar]
- 22.Li X., Yuan D., Zhang J., Lin Z., Zhang X.. 2013, Genetic mapping and characteristics of genes specifically or preferentially expressed during fiber development in cotton, PLoS One, 8, e54444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X., Gao W., Zhang J., Zhang X., Lin Z.. 2013, Linkage mapping and expression analysis of miRNAs and their target genes during fiber development in cotton, BMC Genomics, 14, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren G., Li X., Lin Z.. 2014, Mining, genetic mapping and expression analysis of EST-derived resistance gene homologs (RGHs) in cotton, BMC Plant. Biol., 14, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X., Gao W., Guo H., Zhang X., Fang D.D., Lin Z.. 2014, Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping, BMC Genomics, 15, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Li X., Gao W., Jin X., Zhang X., Lin Z.. 2014, Comparison and development of EST–SSRs from two 454 sequencing libraries of Gossypium barbadense, Euphytica, 198, 277–88. [Google Scholar]
- 27.Chen X., Jin X., Li X., Lin Z.. 2015, Genetic mapping and comparative expression analysis of transcription factors in cotton, PLoS One, 10, e0126150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo W., Cai C., Wang C. et al. 2007, A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium, Genetics, 176, 527–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Z., Zhang Y., Zhang X., Guo X.. 2009, A high-density integrative linkage map for Gossypium hirsutum, Euphytica, 166, 35–45. [Google Scholar]
- 30.Zhao L., Yuanda L., Caiping C. et al. 2012, Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information, BMC Genomics, 13, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sourdille P., Tavaud M., Charmet G., Bernard M.. 2001, Transferability of wheat microsatellites to diploid Triticeae species carrying the A, B and D genomes, Theor. Appl. Genet., 103, 346–52. [Google Scholar]
- 32.Xiao J., Wu K., Fang D.D., Stelly D.M., Yu J., Cantrell R.G.. 2009, New SSR markers for use in cotton (Gossypium spp.) improvement, J. Cotton Sci., 13, 75–157. [Google Scholar]
- 33.Jena S.N., Srivastava A., Rai K.M. et al. 2012, Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.), Theor. Appl. Genet., 124, 565–76. [DOI] [PubMed] [Google Scholar]
- 34.Eujayl I., Sledge M.K., Wang L. et al. 2004, Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp., Theor. Appl. Genet., 108, 414–22. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y., Sledge M.K., Bouton J.H.. 2007, Genome mapping of white clover (Trifolium repens L.) and comparative analysis within the Trifolieae using cross-species SSR markers, Theor. Appl. Genet., 114, 1367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor D.R., Ingvarsson P.K.. 2003, Common features of segregation distortion in plants and animals, Genetica, 117, 27–35. [DOI] [PubMed] [Google Scholar]
- 37.Wendel J.F. 1989, New World tetraploid cottons contain old world cytoplasm, Proc. Natl Acad. Sci. USA, 86, 4132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wendel J.F., Cronn R.C.. 2003, Polyploidy and the evolutionary history of cotton, Adv. Agron., 78, 139–86. [Google Scholar]
- 39.Blenda A., Fang D.D., Rami J.F., Garsmeur O., Luo F., Lacape J.M.. 2012, A high density consensus genetic map of tetraploid cotton that integrates multiple component maps through molecular marker redundancy check, PLoS One, 7, e45739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H., Jin X., Zhang B., Shen C., Lin Z.. 2015, Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing, DNA Res., 22, 147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Applequist W.L., Cronn R., Wendel J.F.. 2001, Comparative development of fiber in wild and cultivated cotton, Evol. Dev., 3, 3–17. [DOI] [PubMed] [Google Scholar]
- 42.Jiang C., Wright R.J., El-Zik K.M., Paterson A.H.. 1998, Polyploid formation created unique avenues for response to selection in Gossypium (cotton), Proc. Natl Acad. Sci. USA, 95, 4419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park Y.H., Alabady M.S., Ulloa M. et al. 2005, Genetic mapping of new cotton fiber loci using EST-derived microsatellites in an interspecific recombinant inbred line (RIL) cotton population, Mol. Genet. Genomics, 274, 428–41. [DOI] [PubMed] [Google Scholar]
- 44.Paterson A.H., Saranga Y., Menz M., Jiang C.X., Wright R.J.. 2003, QTL analysis of genotype × environmental interactions affecting cotton fiber quality, Theor. Appl. Genet., 106, 384–96. [DOI] [PubMed] [Google Scholar]
- 45.Yuan D., Tang Z., Wang M. et al. 2015, The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres, Sci. Rep., 5, 17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.