

ABSTRACT
The c-MYC oncogene is deregulated in virtually all human tumors and therefore constitutes an attractive therapeutic target. We found that the chromatin remodeler BPTF is a c-MYC interactor required for c-MYC chromatin recruitment and transcriptional activity. Moreover, inhibition of BPTF delays tumor development both in vitro and in vivo and its levels positively correlate with c-MYC signatures in human tumors. We propose BPTF as a therapeutic target in c-MYC-addicted tumors.
KEYWORDS: BPTF, c-MYC, chromatin remodeling, transcription
Abbreviations
- BL
Burkitt lymphoma
- BPTF
bromodomain
- PHD
finger transcription factor
- Brd4
bromodomain containing 4
- CDK9
cyclin dependent kinase 9
- KAT2A
K(Lysine) acetyltransferase 2A
- NURF
nucleosome remodeling factor
- SWI/SNF
SWItch/sucrose non-fermentable
- WDR5
WD repeat domain 5
c-MYC expression is deregulated in most, and perhaps all, human tumors through a variety of mechanisms including amplification, translocations, and aberrant activation of upstream signaling pathways. A paradigm of c-MYC–driven tumors is Burkitt lymphoma (BL), characterized by chromosomal translocations leading to c-MYC overexpression under the control of Ig regulatory sequences. Several reports support the relevance of c-MYC as a therapeutic target. Approaches to target c-MYC have remained elusive for a long time, but development of the Omomyc peptide8 and specific inhibitors against co-factors required by c-MYC, such the chromatin reader bromodomain containing 4 (BRD4) inhibitor JQ1,3 have opened new therapeutic possibilities.
c-MYC regulates the expression of thousands of genes in a variety of cell types. To activate transcription, c-MYC dimerizes with its obligate partner MAX and binds to the palindromic E-box motif (CACGTG).2 Binding to non-canonical E-boxes and non-E-box DNA sequences has also been reported upon c-MYC overexpression.2,4 For these reasons, deconvoluting a conserved c-MYC gene signature has proven challenging. Guccione et al. proposed the existence of 2 types of c-MYC target promoters: “high-affinity” and “low-affinity.” c-MYC binds high-affinity sites in multiple cell types regardless of its levels; in contrast, binding to low-affinity targets occurs only when c-MYC is overexpressed. Careful dissection of the epigenetic context of c-MYC targets revealed that high-affinity promoters are characterized by the presence of CpG islands and activating histone marks (H3K4me3, H3K79me, H3K18ac, H3K14ac, H3K9ac, and H2A.Z), whereas low-affinity promoters harbor repressive histone marks (e.g., H3K9me and H4K16Ac).4 The mechanisms whereby c-MYC recognizes transcriptionally active chromatin are poorly understood but likely involve specialized “reader” domains (e.g., bromodomains) that frequently occur in chromatin remodelers and modifiers. Recently Sab∫ et al. proposed a sequential model for c-MYC binding site recognition whereby low-affinity protein–protein interactions between c-MYC:MAX and chromatin-associated complexes and/or transcription factors precede c-MYC binding to DNA.7 Consistent with these findings, Thomas et al. reported that the interaction between c-MYC and the chromatin remodeler WD repeat domain 5 (WDR5, present in multiple chromatin regulatory complexes such as H3K4 methyltransferases) is required for c-MYC recruitment to DNA and biological activity.9 Once bound to its targets, c-MYC recruits co-factors that further modify the chromatin, resulting in increased DNA accessibility and transcriptional activation. Among them are chromatin remodelers (e.g., SWI/SNF), complexes with histone acetyltransferase activity (e.g., KAT2A), and proteins associated with the basal transcriptional machinery (e.g., BRD4 and CDK9) (Fig. 1).10
Figure 1.
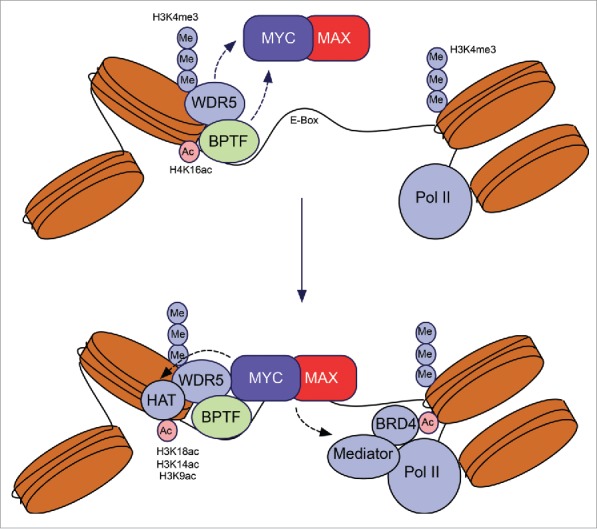
Recruitment of c-MYC on chromatin and transcription. Protein–protein interactions mediated by BPTF and WDR5 facilitate c-MYC chromatin recruitment. BPTF facilitates c-MYC transcriptional activity through remodeling of the surrounding chromatin. It remains to be determined whether BPTF and WDR5 bind concomitantly to c-MYC. BPTF, bromodomain PHD finger transcription factor; WDR5, WD repeat domain 5.
We recently reported that c-MYC interacts with the bromodomain PHD finger transcription factor (BPTF), the largest and essential subunit of the ATP-dependent nucleosome remodeling factor (NURF). BPTF provides specificity to NURF through its interaction with transcription factors and histone modifications (H3K4me3 and H4K16ac) (Alkhatib et al. 2011). We showed that BPTF silencing impaired activation of the full c-MYC transcriptional program and was associated with decreased c-MYC recruitment to low-affinity promoters and a reduction in DNA accessibility at c-MYC target chromatin. As a result, BPTF silencing impaired several biological processes regulated by c-MYC; we observed a selective effect on cell proliferation and replication stress, but not on apoptosis. To induce cell proliferation, c-MYC directly binds to genes participating in DNA replication and cell cycle control and activates their transcription.6 In contrast, c-MYC-driven apoptosis is mostly indirect: it involves the inhibition of MIZ1 without direct binding of c-MYC to DNA and stabilization of the cyclin dependent kinase inhibitor p19ARF (CDKN2A, best known as p19ARF) and the tumor suppressor p53 (TP53, best known as p53).5 Based on these findings, we propose that BPTF is only required for c-MYC functions that involve direct binding to chromatin (Fig. 1).
Analysis of public Omics data sets revealed that BPTF levels positively correlate with c-MYC signatures in c-MYC-driven tumors (BL, colorectal, prostate, and pancreatic cancer) but not in N-MYC– or L-MYC–driven tumors (medulloblastoma and ovarian carcinoma). We extended these results using Ela1-Myc mice in which c-MYC is overexpressed in the acinar compartment of the pancreas, leading to the development of aggressive mixed acinar/ductal pancreatic tumors with a high penetrance. In this model, elimination of a single Bptf allele was sufficient to delay tumor initiation and progression. Further work is required to determine whether BPTF is also required for tumors driven by N- and L-MYC.
In conclusion, our work suggests that BPTF is an important protein involved in the chromatin remodeling required for the action of c-MYC (Richart et al. 2016). The broad pattern of expression of BPTF in tissues and the correlation between BPTF and c-MYC signatures in human tumors suggest that a wide range of tumor types might be amenable to therapeutic disruption of the c-MYC-BPTF axis. We therefore propose that the development of small interfering peptides that disrupt the c-MYC–BPTF interaction or specific BPTF inhibitors might represent valuable approaches to the treatment of c-MYC–addicted tumors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Alkhatib SG, Landry JW. The nucleosome remodeling factor. FEBS lett 2011; 585:3197-207; PMID:21920360; http://dx.doi.org/ 10.1016/j.febslet.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H. Binding of Myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol 1993; 13:5216-24; PMID:8395000; http://dx.doi.org/ 10.1128/MCB.13.9.5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson M, Prinjha R, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM et al.. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529-33; PMID:21964340; http://dx.doi.org/ 10.1038/nature10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, Zardo G, Nervi C, Bernard L, Amati B. Myc binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol 2006; 8:764-70; PMID:16767079; http://dx.doi.org/ 10.1038/ncb1434 [DOI] [PubMed] [Google Scholar]
- 5.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene 2008; 27:6462-72; PMID: 18955973; http://dx.doi.org/ 10.1038/onc.2008.312 [DOI] [PubMed] [Google Scholar]
- 6.Perna D, Fagà G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R et al.. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblast. Oncogene 2012; 31:1695-1709; PMID: 21860422; http://dx.doi.org/ 10.1038/onc.2011.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabò A, Amati B. Genome recognition by MYC. Cold Spring Harb Perspect Med 2014; 4:pii: a014191; PMID: 24492846; http://dx.doi.org/ 10.1101/cshperspect.a014191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679-83; PMID: 18716624; http://dx.doi.org/ 10.1038/nature07260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas L, Wang Q, Grieb B, Phan J. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol Cell 2015; 58:440-52; PMID: 25818646; http://dx.doi.org/ 10.1016/j.molcel.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635-45; PMID: 16064138; http://dx.doi.org/ 10.1038/nrm1703 [DOI] [PubMed] [Google Scholar]