

ABSTRACT
Alpha thalassemia/mental retardation syndrome X-linked (ATRX) is mutated in nearly a third of pediatric glioblastoma (GBM) patients. We developed an animal model of ATRX-deficient GBM. Using this model combined with analysis of multiple human glioma genome-wide datasets, we determined that ATRX mutation leads to genetic instability, impaired non-homologous end joining, and alternate lengthening of telomeres (ALT).
KEYWORDS: Chromatin, histone mutations, homologous recombination, NHEJ, pediatric GBM
Current therapies for glioblastoma WHO grade IV glioma (GBM) are ineffective and few patients with this diagnosis will survive 2 years.1 Previous efforts have been made to optimize treatments for pediatric and adult GBM based on the molecular attributes of the individual tumor. Such strategies include prioritizing the use of alkylating agents for GBMs that harbor promoter methylation/silencing of the DNA repair gene methylguanine methyltransferase (MGMT).2 However, not all patients with GBMs harboring MGMT promoter methylation will respond to alkylating agents, and despite current molecular stratification nearly all patients with GBM will eventually experience relapse.2
Recurrent mutations in GBM offer additional opportunities for molecular stratification and personalized therapy. In genome-wide sequencing of human GBMs, mutations in the histone chaperone protein α thalassemia/mental retardation syndrome X-linked (ATRX) are seen in approximately 30% of younger patients.3,4 ATRX is mutated only rarely in adult primary GBM, but is commonly found in younger adults with lower grade (WHO grade II/III) glioma.5 ATRX mutation (and loss of immune staining) is strongly associated with the maintenance of telomere length by non-telomerase methods, collectively referred to as alternative lengthening of telomeres (ALT).3 However, no efforts have been made to target ATRX deficiency in glioma, in part due to a lack of adequate animal models.
To this end, we recently developed an animal model of ATRX-deficient GBM using the Sleeping Beauty (SB) transposase system.6 In this model, mice begin to develop brain tumors in the first few weeks of life thus recapitulating the developmental environment of childhood/young adult GBMs, which frequently harbor ATRX mutations. Interestingly, loss of ATRX accelerated mouse GBM growth rate and reduced median survival, uncovering the impact of ATRX loss on glioma progression. We also showed that ATRX loss promoted both ALT and genetic instability in mouse GBM.6 In particular, loss of ATRX resulted in impairment of non-homologous end joining (NHEJ) activity and was correlated with a decrease in the expression of enzymes involved in the NHEJ DNA repair pathway.6
To determine whether the connection between ATRX loss and genetic instability is also seen in human GBM, we downloaded and integrated multiple genome-wide data sets of human GBM available in the European Genome Archive (EGA, accession numbers EGAS00001000192, EGAS00001000572, EGAS00001000575, EGAS00001000720, and EGAS00001001436). We found that ATRX mutation in pediatric GBM is associated with increased mutation rate at the single nucleotide variant (SNV) level but not with alterations at the chromosomal/copy number level. This finding was independent of tumor protein 53 (TP53, best known as p53) mutation, which frequently co-occurs with ATRX mutation in human GBM.6
ATRX encodes a protein with a DNA-binding finger and a SWI2/SNF2-like ATPase motif, making it a member of a family of ATP-dependent chromatin-associated proteins.7 Other members of this class participate in DNA damage repair pathways and previous work has shown that ATRX is recruited to sites of DNA damage.8 However, no previous data had directly linked ATRX to its role in facilitating NHEJ. Based on its known epigenetic role in histone deposition, we hypothesize that reduced ATRX function might lead to conformational changes in heterochromatin that restrict the access of NHEJ proteins to damaged DNA.
Additionally, we demonstrated that ATRX loss in GBM can promote ALT.6 Interestingly, we saw ALT in some of our ATRX-deficient mouse tumors, thus confirming recent work postulating that ATRX loss alone can promote some features of ALT, but that additional genetic changes are needed to fully activate the ALT pathway.9
We did not find any association between ATRX loss and chromosomal/copy number alterations in our mouse or human tumors.6 This was discordant with a previous analysis of pediatric high-grade glioma that showed an association between tumors with concurrent mutations in H3 histone family 3A (H3F3A), TP53, and ATRX and copy number amplifications (CNAs).3 The type of genetic instability observed in tumors can provide a helpful lead in determination of the primary DNA damage repair pathways being used to counteract ongoing genetic insults. Depending on the DNA damage repair pathway used, the repair itself may introduce new somatic mutations and chromosomal rearrangements. The increased frequency of single nucleotide variants at the sequence level raises the possibility that a reduction in NHEJ in ATRX-deficient glioma could lead to an increase in the use of the more error-prone “alternative NHEJ pathway,” which is driven by poly ADP-ribose polymerase (PARP1).10 In the mutator hypothesis of cancer development, mutations in “caretaker genes” can drive further tumor development. Our data show that ATRX mutation may play this role in glioma by impairing NHEJ DNA repair.
Treatments for human cancers are becoming increasingly personalized, and ATRX loss provides a promising target for individualized treatment of children and young adults with GBM. By uncovering the connection between ATRX mutation and impaired NHEJ we provide a mechanism for genetic instability in pediatric GBM and an actionable therapeutic target for ATRX-deficient tumors. Treatment of tumor cells generated from our mouse model has shown that loss of ATRX creates susceptibility to certain DNA-damaging agents that induce double-stranded DNA breaks.6
Based on our animal model, we identified the role of ATRX loss in furthering GBM progression and improving therapy response. We have created a working model of how our results might impact future clinical trials for patients with ATRX-mutated GBM (Fig. 1). One consideration for children or young adults found to have ATRX-deficient GBM would be to select agents that most effectively promote double-strand breaks, such as topoisomerase inhibitors.6 Future work on ATRX-deficient GBM could explore whether the defects in NHEJ can be selectively targeted. For example, it may be feasible to target the relative increase in homologous recombination and/or the increase in tumor mutations seen in tumors with ATRX loss. We are hopeful that this work will continue to highlight areas for improved targeted therapies and precision medicine.
Figure 1.
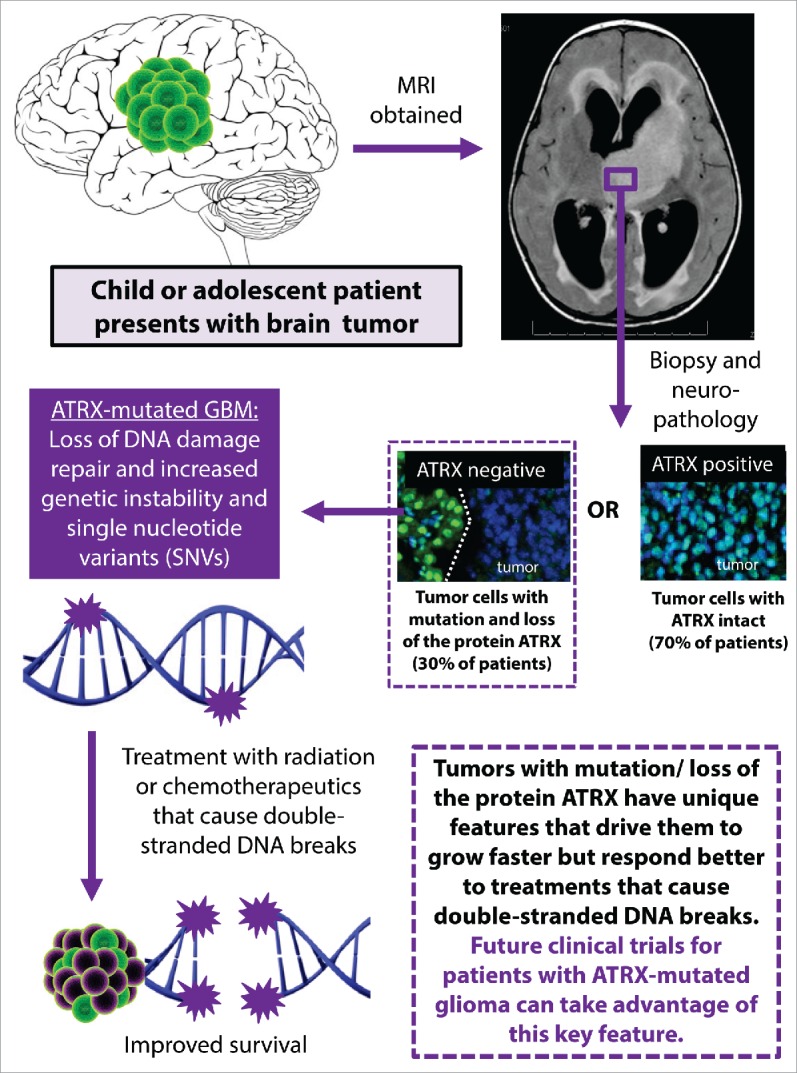
Implication of DNA damage repair deficits in ATRX-deficient glioma for clinical practice. Schematic showing how improved understanding of the role of ATRX mutation in human glioma could be integrated into personalized therapy. ATRX mutation is associated with loss of ATRX by immune staining, which is increasingly used in neuropathological assessment of human gliomas. ATRX, α thalassemia/mental retardation syndrome X-linked; SNV, single nucleotide variant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) grants 1RO1-NS 054193, 1RO1-NS 061107, and 1RO1-NS082311 to PRL; grants 1UO1-NS052465, 1RO1-NS 057711, and 1RO1-NS074387 to MGC. CK was supported by the St. Baldrick's Foundation Fellowship, and the Alex's Lemonade Stand Foundation/Northwestern Mutual Young Investigator Grant.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al.. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Eng J Med 2005; 352(10):987-96; PMID:15758009; http://dx.doi.org/ 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, Mehta MP, Gilbert MR. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol 2008; 26(25):4189-99; PMID:18757334; http://dx.doi.org/ 10.1200/JCO.2007.11.5964 [DOI] [PubMed] [Google Scholar]
- 3.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482(7384):226-31; PMID:22286061; http://dx.doi.org/ 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 4.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, et al.. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012; 3(7):709-22; PMID:22869205; http://dx.doi.org/ 10.18632/oncotarget.588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, et al.. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genetics 2015; 47(5):458-68; PMID:25848751; http://dx.doi.org/ 10.1038/ng.3273 [DOI] [PubMed] [Google Scholar]
- 6.Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M, Mulpuri L, et al.. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med 2016; 8(328):328ra328; PMID:26936505; http://dx.doi.org/12953102 10.1126/scitranslmed.aac8228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A 2003; 100(19):10635-40; PMID:12953102; http://dx.doi.org/ 10.1073/pnas.1937626100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung JW, Ghosal G, Wang W, Shen X, Wang J, Li L, Chen J. Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem 2013; 288(9):6342-50; PMID:23329831; http://dx.doi.org/ 10.1074/jbc.M112.411603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suvà ML, Benes CH, et al.. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015; 347(6219):273-7; PMID:25593184; http://dx.doi.org/ 10.1126/science.1257216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betermier M, Bertrand P, Lopez BS. Is non-homologous end-joining really an inherently error-prone process? PLoS Genetics 2014; 10(1):e1004086; PMID:24453986; http://dx.doi.org/ 10.1371/journal.pgen.1004086 [DOI] [PMC free article] [PubMed] [Google Scholar]