

ABSTRACT
Dicer has been well studied in cancer; however, deciphering its exact function in tumorigenesis continues to be a challenge. While partial suppression or truncation of Dicer promotes tumorigenesis, its complete deletion inhibits tumor growth. Here, we discuss this Dicer cancer conundrum in the context of its recently discovered role in the DNA damage response.
Dicer has a well-established function in the biogenesis of microRNAs (miRNAs). However, emerging evidence indicates that Dicer is also important for processing other small noncoding RNAs (ncRNAs). Among these RNAs, which include mirtrons and transcriptional start site miRNAs (TSS-miRNAs), are ncRNAs that are important for the DNA damage response (DDR). These recently discovered DNA damage response ncRNAs (DNA damage-response RNAs [DDRNAs] or double-strand break [DSB]-induced RNAs [diRNAs]) are exciting because they highlight novel physiological functions of Dicer that are independent of miRNAs. Importantly, these DDRNAs/diRNAs also provide insight into the pathologic consequences of Dicer deficiency that may not readily be explained by the lack of miRNAs alone. Here, we focus on the consequences of Dicer deficiency in the context of cancer and propose a model that could potentially explain an apparent paradox.
Although mutations in DICER (often resulting in truncations or a reduction in DICER expression) are commonly seen in many cancers, loss of heterozygosity (LOH) resulting in complete deficiency of DICER function is extremely rare.1 Mutational analyses of human cancers indicate that, while partial loss of DICER is associated with many cancers, its complete loss is not well tolerated. In that sense, DICER does not fit the classical definition of either a tumor suppressor gene or an oncogene. Interestingly, further insight into the function of Dicer in cancer comes from studies in which the consequence of heterozygous or homozygous deletion of Dicer was examined in mouse models. A pivotal study by Tyler Jacks's group demonstrated that shRNA-mediated downregulation of Dicer resulted in enhanced cellular transformation and tumorigenesis in mouse lung adenocarcinoma cells.2 Subsequently, Dicer heterozygous mice were shown to exhibit increased tumor burden and reduced survival in K-Ras–driven mouse models of lung cancer and sarcoma.3 Most interestingly, while loss of a single allele of Dicer enhanced tumorigenesis, loss of both copies of Dicer had the opposite effect. Indeed, it was rare to find tumors that had lost both copies of Dicer, and enforced complete deletion of Dicer led to inhibition of tumorigenesis.
What could be the mechanism by which partial loss of Dicer is pro-tumorigenic whereas its complete loss is anti-tumorigenic? One possible explanation is that partial deletion of Dicer results in increased amounts of oncogenic versus tumor suppressive miRNAs, whereas the complete loss of all miRNAs is detrimental for cellular growth. However, the recently discovered miRNA-independent function of Dicer in DNA damage repair presents an attractive alternative possibility. In 2012, two independent studies reported that Dicer-mediated processing of ncRNAs (referred to as DDRNAs or diRNAs) is essential for the repair of damaged DNA.4,5 Specifically, these DDRNAs/diRNAs correspond to the sites of DNA double-strand breaks and are thought to act as templates for efficient DNA repair.
In our recent paper, we examined this function of Dicer and found marked accumulation of DNA damage in cells deficient for Dicer.6 This was particularly evident in rapidly dividing cells that experience replication stress-associated DNA damage. Such cells included embryonic stem cells as well as neural precursor cells in the developing brain, where the absence of Dicer alone resulted in the accumulation of DNA damage and tumor protein p53 (TP53 or p53)-dependent cell death. Importantly, we also examined this DNA repair function of Dicer in the context of brain tumors. In the absence of Dicer, medulloblastoma cells accumulated significant DNA damage, resulting in extensive cell death and decreased tumor burden. Together, these results highlight the importance of Dicer function in DDR and show that a major consequence of Dicer deficiency is the accumulation of DNA damage and cell death in rapidly dividing cells during development and tumorigenesis.
We propose that this role of Dicer in DDR at least in part provides a plausible explanation for why partial loss of Dicer is pro-tumorigenic whereas its complete loss is anti-tumorigenic (Fig. 1). While tumors with normal levels of Dicer are able to repair the DNA damage that is caused by oncogenic stress, tumors completely lacking Dicer would accumulate significant DNA damage resulting in cell death. In contrast, partial reduction of Dicer (e.g., Dicer heterozygosity) could result in increased but sublethal levels of DNA damage that actually promotes tumorigenicity. Low levels of mutagenesis are well recognized to stimulate cancer development and progression. Indeed, recent studies have shown that sublethal DNA damage caused by limited caspase activation potentiates cancer growth.7
Figure 1.
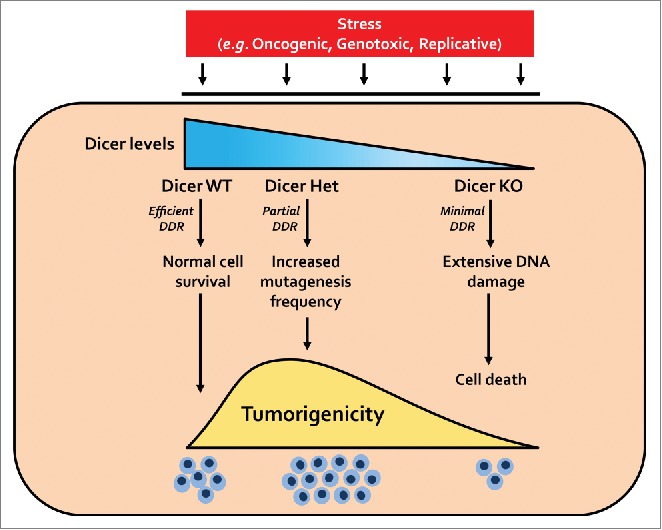
The paradox of Dicer in cancer. Tumor cells with normal levels of Dicer (Dicer WT) are able to efficiently repair the DNA damage that occurs with cellular stress. Tumors completely lacking Dicer (Dicer KO) have extensive DNA damage that results in cell death and reduced tumor burden. In contrast, tumors with partial reduction in Dicer (Dicer Het) may accumulate sublethal levels of DNA damage that results in enhanced tumorigenesis. DDR, DNA damage response: Het, heterozygous; KO, knockout; WT, wild type.
There are several predictions of this model that can be readily tested. For example, in the SmoM2 mouse model of medulloblastoma where we have observed accumulation of DNA damage and reduced tumorigenesis with complete Dicer deletion, we expect Dicer heterozygous mice to exhibit limited DNA damage and enhanced tumorigenesis. Consistent with this, using a different medulloblastoma model another group demonstrated that Dicer heterozygous tumors were indeed more aggressive than WT tumors.8 Also, since the model predicts that the inability of complete Dicer deletion to support tumor growth is due to lethal accumulation of DNA damage and activation of p53-mediated apoptosis, p53 deficiency is anticipated to permit the growth of Dicer-deficient tumors. Indeed, co-deletion of Dicer and p53 has been reported to induce highly aggressive skin carcinomas.9 Likewise, although it has been challenging to generate cancer cells completely deficient in Dicer, we note that the recent report of the generation of Dicer-deficient sarcoma cells was achieved in the context of p53 inactivation.10 However, we acknowledge that, in other contexts, the rescue of Dicer loss with p53 deficiency may only be partial since persistent DNA damage can also activate p53-independent pathways of cell death.
Lastly, it is important to consider that since the threshold of DNA damage that is permissive for cell survival is likely to be different in different cell types, the exact extent of Dicer reduction and DNA damage that potentiates versus inhibits tumorigenesis could vary according to the type of tumor. Nevertheless, partial loss of Dicer under conditions of oncogenic stress is anticipated to have an outcome on cancer progression that is completely opposite to complete loss of Dicer in multiple tumor types.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 2014; 14:662-72; PMID:25176334; http://dx.doi.org/ 10.1038/nrc3802 [DOI] [PubMed] [Google Scholar]
- 2.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007; 39:673-7; PMID:17401365; http://dx.doi.org/ 10.1038/ng2003 [DOI] [PubMed] [Google Scholar]
- 3.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009; 23:2700-4; PMID:19903759; http://dx.doi.org/ 10.1101/gad.1848209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d'Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012; 488:231-5; PMID:22722852; http://dx.doi.org/ 10.1038/nature11179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A Role for Small RNAs in DNA Double-Strand Break Repair. Cell 2012; 149:101-12; PMID:22445173; http://dx.doi.org/ 10.1016/j.cell.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 6.Swahari V, Nakamura A, Baran-Gale J, Garcia 4, Crowther AJ, Sons R, Gershon TR, Hammond S, Sethupathy P, Deshmukh M. Essential Function of Dicer in Resolving DNA Damage in the Rapidly Dividing Cells of the Developing and Malignant Cerebellum. Cell Reports 2016; 14:216-24; PMID:26748703; http://dx.doi.org/ 10.1016/j.celrep.2015.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gama V, Deshmukh M. Life after MOMP. Mol Cell 2015; 58:199-201; PMID:25884366; http://dx.doi.org/ 10.1016/j.molcel.2015.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zindy F, Lee Y, Kawauchi D, Ayrault O, Merzoug LB, Li Y, McKinnon PJ, Roussel MF. Dicer Is Required for Normal Cerebellar Development and to Restrain Medulloblastoma Formation. PLoS ONE 2015; 10:e0129642; PMID:26091048; http://dx.doi.org/ 10.1371/journal.pone.0129642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyle S, Hoover K, Colpan C, Zhu Z, Matijasevic Z, Jones SN. Dicer Cooperates with p53 to Suppress DNA Damage and Skin Carcinogenesis in Mice. PLoS ONE 2014; 9:e100920; PMID:24979267; http://dx.doi.org/ 10.1371/journal.pone.0100920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravi A, Gurtan AM, Kumar MS, Bhutkar A, Chin C, Lu V, Lees JA, Jacks T, Sharp PA. Proliferation and Tumorigenesis of a Murine Sarcoma Cell Line in the Absence of DICER1. Cancer Cell 2012; 21:848-855; PMID:22698408; http://dx.doi.org/ 10.1016/j.ccr.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]