

ABSTRACT
PREX2 is a PTEN binding protein that is significantly mutated in melanoma and pancreatic ductal adenocarcinoma. We recently reported the molecular mechanism of tumorigenesis associated with PREX2 mutations: truncating PREX2 mutations activate its RAC1 guanine nucleotide exchanger activity leading to increased PI3K/AKT signaling and enhanced cell proliferation.
KEYWORDS: Epigenetics, Melanoma, Mouse models of cancer, PI3K, PREX2, RAC1
Abbreviations
- PREX2
phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac exchange factor 2
- PTEN
phosphatase and tensin homolog
- RAC1
ras-related C3 botulinum toxin substrate 1
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
Cancer genomic studies have provided tremendous insight into the complexity of somatic mutations and delivered a long list of novel mutated cancer genes.1 It is expected that a wave of functional studies will assign detailed molecular and functional roles for these mutations in tumor development. We identified PREX2 (phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac exchange factor 2) as a gene that is significantly mutated in human melanomas.2 Interestingly, the International Cancer Genomics Consortium (ICGC) recently reported that PREX2 is also significantly mutated in pancreatic ductal adenocarcinoma. 3
PREX2 is a guanine nucleotide exchange factor (GEF) for ras-related C3 botulinm toxin substrate 1 (RAC1) and a known phosphatase and tensin homolog (PTEN) interacting protein.4,5 Functionally, PREX2 has been shown to regulate Rac1-mediated cellular invasion in a manner that crosstalks with PTEN signaling and also regulates insulin signaling and glucose homeostasis through the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway.6,7 Our initial sequencing of the melanoma genome showed various patterns of PREX2 mutations including missense and truncating mutations. Using xenograft models, we were able to show that truncating PREX2 mutations have oncogenic activity;2 however, how these PREX2 mutations contribute to melanoma development was unclear.
To study PREX2 mutations in a spatio-temporally restricted manner, we generated an inducible transgenic mouse model that expresses a truncating PREX2 mutant (TetO-lox-STOP-lox-Prex2E824*) in melanocytes.8 We crossed these transgenic mice with mice that have a melanoma sensitizing background (i.e., they lack the tumor suppressor Ink/Arf and inducibly express a constitutively active NrasQ61K) to generate a genetically engineered mouse (GEM) model of melanoma. Tumor formation was induced by administration of tamoxifen (which induces Cre-mediated recombination and removal of the stopper cassette) and doxycycline (which allows expression of Prex2E824*and NrasQ61K transgenes from tet-responsive promoters). Interestingly, we observed an increased incidence of melanoma formation in mice harboring the inducible Prex2E824*transgene. We also generated xenograft tumors by expressing control GFP, PREX2 wild type, or various PREX2 truncating mutations in primary immortalized human melanocytes. Again, truncating PREX2 mutations induced increased tumor formation. To explore the molecular mechanisms behind the ability of PREX2 mutations to induce increased tumor formation, we performed gene expression profiling of tumors from both xenograft and GEM models. Integrative cross-species analysis revealed regulation of cell cytoskeleton organization, cell cycle, and ribosome biogenesis as key biological pathways that were significantly enriched in tumors with PREX2 truncating mutations. The connection of PREX2 to RAC1 can explain changes in the cell cytoskeleton signaling pathway, whereas the known role of PREX2 in PTEN biology is expected to explain the enrichment in ribosome biogenesis. However, it was not clear why cell cycle regulation is perturbed in PREX2 mutant tumors and we therefore investigated this aspect further.
Histopathologically, we observed that PREX2 mutant tumors are highly proliferative and show increased KI67 staining. Furthermore, we observed reduced expression of key negative cell cycle regulators such as CDKN1C (also known as p57) and CDKN1B (also known as p27) and increased expression of insulin like growth factor 2 (IGF2) in PREX2 mutant tumors. How do truncating mutations in PREX2 result in these biological changes? To answer this question, we next studied the biochemical and signaling consequences of truncating PREX2 mutations. First, we purified recombinant full-length PREX2 or an N-terminal truncated PREX2 from Sf9 cells and performed guanine nucleotide exchange (GEF) activity assays using RAC1 as a substrate. This analysis revealed higher GEF activity of the truncated PREX2 compared to full-length PREX2. In support of these data, we also showed that cells with PREX2 truncating mutation have increased GEF activity as demonstrated by increased GTP-loaded Rac1.
How does truncation of PREX2 increase its GEF activity? Our data suggest the existence of 2 cooperating mechanisms that explain this effect. First, truncated PREX2 mutants lack binding to PTEN, which is known to have a suppressive effect on the GEF activity of PREX2. Hence, by avoiding the inhibitory influence of PTEN, PREX2 mutants have an intrinsically higher GEF activity. Second, using structural modeling of the PREX2:Rac1 interaction, we demonstrated that the GEF domain of PREX2 behaves similarly to the GEF domain of PREX1. It is known that the C-terminus of PREX1 has an auto-inhibitory effect on its N-terminal GEF activity.9,10 Our structural model suggests that a similar mechanism exists in PREX2, and thus PREX2 truncating mutations relieve the auto-inhibition of GEF activity by the C-terminus. Next, we examined the consequences of increased PREX2 GEF activity and RAC1 activation. Using reverse phase protein array (RPPA) and immunoblotting, we observed increased phosphorylation of AKT at Ser473 and Thr308 in PREX2 mutant tumors. Gain-of-function and loss-of-function experiments revealed that the increased activation of Akt by PREX2 mutations was dependent on activation of Rac1. Finally, we provided a mechanism to explain the gene expression changes induced by PREX2 mutations: we showed that PREX2 mutation or PTEN deletion induces DNA hypomethylation and downregulation of expression of p57, a critical cell cycle regulator.
In summary, our study demonstrated the oncogenic capacity of truncating PREX2 mutations in vivo. We also identified a direct link to an established oncogenic signaling pathway—the PI3K/AKT pathway—and provided insights into the downstream regulation of a tumor suppressor—p57—in melanoma pathogenesis (Fig. 1). However, a number of important outstanding questions remain. High-resolution X-ray structures of full-length and truncated PREX2 protein in complex with its substrate will be of great help in elucidating the molecular basis of PREX2 GEF activation. Our understanding of the role of Rac1 in activation of the PI3K/AKT pathway is still rudimentary and must be explored in greater depth using biochemical, genetic, and pharmacologic tools. Finally, the link between changes in the PI3K/AKT pathway and downstream epigenetic and gene expression changes caused by PREX2 mutations is not known and deserves further investigation.
Figure 1.
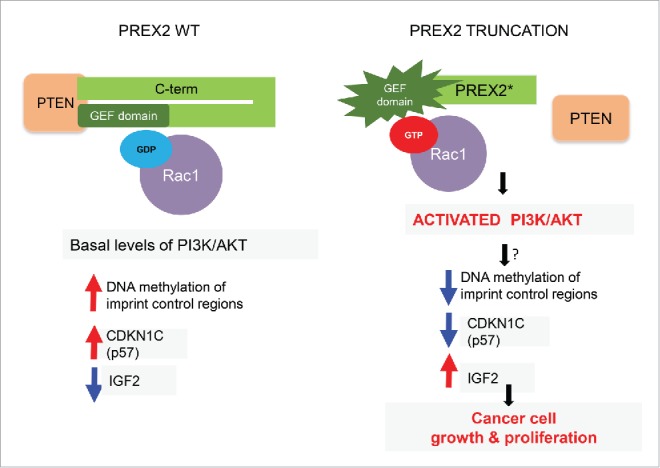
Mechanistic consequences of PREX2 truncating mutations. In wild-type cells PREX2 is bound to PTEN and its GEF domain is kept in an inactive state through association with its C-terminus (C-term), with resultant basal levels of downstream signaling events such as GDP-loaded RAC1. In cells and tumors with truncating PREX2 mutations, PREX2 is unable to bind to PTEN and its intramolecular inhibitory conformation is lost, resulting in activation of its GEF activity. This results in GTP loading of RAC1, which in turn leads to direct activation of the PI3K/AKT pathway. These alterations (through a still unknown link) perturb DNA methylation at a critical genomic imprint control region leading to downregulation of a key cell cycle regulator CDKN1C (also known as p57) and upregulation of IGF2, which favors accelerated cell growth and proliferation. GDP, guanosine diphosphate; GEF, guanine nucleotide exchange factor; GTP, guanosine triphosphate; IGF2, insulin like growth factor 2; PREX2, phosphatidylinositol-3, 4, 5-triphosphate-dependent Rac exchange factor 2; PTEN, phosphatase and tensin homolog; RAC1, ras-related C3 botulinum toxin substrate 1.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013; 153:17-37; PMID:23540688; http://dx.doi.org/ 10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al.. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012; 485:502-6; PMID:22622578; http://dx.doi.org/ 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al.. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518:495-501; PMID:25719666; http://dx.doi.org/ 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S, et al.. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 2009; 325:1261-5; PMID:19729658; http://dx.doi.org/ 10.1126/science.1173569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donald S, Hill K, Lecureuil C, Barnouin R, Krugmann S, John Coadwell W, Andrews SR, Walker SA, Hawkins PT, Stephens LR, et al.. P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Lett 2004; 572:172-6; PMID:15304343; http://dx.doi.org/ 10.1016/j.febslet.2004.06.096 [DOI] [PubMed] [Google Scholar]
- 6.Mense SM, Barrows D, Hodakoski C, Steinbach N, Schoenfeld D, Su W, Hopkins BD, Su T, Fine B, Hibshoosh H, et al.. PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci Signal 2015; 8:ra32; PMID:25829446; http://dx.doi.org/ 10.1126/scisignal.2005840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, Kern PA, Hawkins PT, Stephens LR, Parsons R. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc Natl Acad Sci U S A 2014; 111:155-60; PMID:24367090; http://dx.doi.org/ 10.1073/pnas.1213773111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lissanu Deribe Y, Shi Y, Rai K, Nezi L, Amin SB, Wu CC, Akdemir KC, Mahdavi M, Peng Q, Chang QE, et al.. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc Natl Acad Sci U S A 2016.. Mar 1; 113(9):E1296-305; PMID:26884185; http://dx.doi.org/ 10.1073/pnas.1513801113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucato CM, Halls ML, Ooms LM, Liu HJ, Mitchell CA, Whisstock JC, Ellisdon AM. The Phosphatidylinositol (3,4,5)-Trisphosphate-dependent Rac Exchanger 1.Ras-related C3 Botulinum Toxin Substrate 1 (P-Rex1.Rac1) Complex Reveals the Basis of Rac1 Activation in Breast Cancer Cells. J Biol Chem 2015; 290:20827-40; PMID:26112412; http://dx.doi.org/ 10.1074/jbc.M115.660456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urano D, Nakata A, Mizuno N, Tago K, Itoh H. Domain-domain interaction of P-Rex1 is essential for the activation and inhibition by G protein betagamma subunits and PKA. Cell Signal 2008; 20:1545-54; PMID:18514484; http://dx.doi.org/ 10.1016/j.cellsig.2008.04.009 [DOI] [PubMed] [Google Scholar]