

ABSTRACT
Although mitogen-activated protein kinase (MAPK) inhibitors elicit initial regression of BRAF-mutated melanoma, drug resistance is an inevitable and fatal event. We recently reported that in genetically engineered mouse models of BRAF-mutated melanoma, isoform-selective phosphatidylinositol 3-kinase inhibition cooperates with MAPK pathway inhibition to forestall the onset of MAPK pathway inhibitor resistance.
KEYWORDS: Mechanisms of oncogenesis and tumor progression, mechanisms of resistance to therapy, novel therapeutic agents, novel therapeutic targets
The development of therapeutics that inhibit signaling initiated by mutationally activated BRAF (BRAFV600E/K) has dramatically improved the clinical landscape for a significant subset of melanoma patients. However, the majority of patients with BRAF-mutated melanoma who are treated with mitogen-activated protein kinase (MAPK) pathway inhibitors inevitably develop lethal drug-resistant disease.1 Consequently, there is a pressing need to identify new MAPK-independent pathways that can be targeted therapeutically to extend the duration of melanoma responses to inhibitors of BRAFV600E/K signaling.
The phosphatidylinositol 3-kinase (PI3K) signaling pathway has emerged as a compelling melanoma drug target for several reasons. First, the majority of BRAF-mutated melanomas display dysregulation of PI3K signaling, often through silencing of the PI3-lipid phosphatase PTEN.2,3 Second, PI3K pathway activation has been implicated as a mediator of resistance to BRAFV600E-pathway targeted therapeutics.4,5 Genome sequence analysis of matched melanoma samples obtained prior to and after the onset of BRAF inhibitor resistance revealed that 22% of resistant tumors displayed PI3K signaling alterations.5 Thus, there is a compelling rationale for testing whether combined pharmacologic targeting of BRAF and PI3K signaling might prolong the durability of response of BRAF-mutated melanoma patients.
To test this hypothesis in a preclinical setting, we used genetically engineered mouse (GEM) models of BRAF-mutated melanoma in which PI3K signaling was activated either by silencing the expression of Pten or by mutational activation of Pik3ca, encoding PI3Kα.6,7 We previously demonstrated that both of these genetic lesions cooperate with BRAFV600E to elicit melanoma;7 however, BRAFV600E/PTENNull melanomas grew more rapidly than BRAFV600E/PIK3CAH1047R melanomas. Moreover, treatment with a pan-class I PI3K inhibitor (BKM120) lacked single agent efficacy in either GEM melanoma model. Therefore, in our recent paper we sought to further explore PI3K signaling in melanoma progression and maintenance, as well as the therapeutic implications of targeting this pathway using PI3K isoform-selective inhibitors.
Intriguingly, we found that BRAFV600E mice expressing 2 alleles of mutationally activated PI3KCA developed melanoma at a rate that significantly exceeded that of BRAFV600E-expressing melanomas in which PTEN was silenced. These data suggested a correlation between flux through the PI3K pathway and the rate of melanoma growth. As a corollary, these data also suggested that inhibition of PI3K signaling might exert antitumor effects against melanoma. To test this, BRAFV600E/PIK3CAH1047R melanoma cell lines were generated from GEM models and treated with BYL719, a selective PI3Kα inhibitor. BRAFV600E/PIK3CAH1047R melanoma cells displayed a reduction in both cell proliferation and PI3 lipid signaling upon BYL719 treatment, whereas similarly generated BRAFV600E/PTENNull mouse melanoma cells were largely BYL719 insensitive. These data further supported our hypothesis that BRAFV600E/PIK3CAH1047R melanoma cells selectively rely on PI3Kα activity for sustained PI3-lipid signaling and cellular proliferation.
To interrogate the PI3K isoform dependence of BRAFV600E/PTENNull melanoma, the more clinically relevant subset of the disease, we treated BRAFV600E/PTENNull melanoma cell lines with inhibitors of PI3Kβ. Previous reports suggested that PTENNull solid tumors relied on PI3Kβ for PI3-lipid production and sustained proliferation.8,9 However, the PTENNull melanoma cell lines tested were insensitive to treatment with PI3Kβ inhibitors, as well as to combined inhibition of PI3Kα and PI3Kβ. To further interrogate the PI3K isoform dependence of BRAFV600E/PTENNull melanoma cells, we compared the antiproliferative effect of a pan-class I PI3K inhibitor (GDC-0941) to that of a PI3Kβ-sparing inhibitor (GDC-0032). Surprisingly, we noted that the PI3Kβ sparing inhibitor was equipotent to the pan-class I PI3K inhibitor in inhibiting cellular proliferation and PI3-lipid signaling. Together, these results indicate that PI3Kβ has no role in promoting PI3-lipid signaling leading to the proliferation of BRAFV600E/PTENNull melanoma cell lines; instead, this activity is promoted by the combined activities of PI3Kα plus PI3Kδ and/or PI3Kγ.
We explored the relevance of these findings in vivo using our GEM models (Fig. 1). First, mice bearing BRAFV600E/PIK3CAH1047R melanomas were treated with a BRAF inhibitor (LGX818) and a PI3Kα inhibitor (BYL719), either alone or in combination. Whereas the BRAFV600E inhibitor promoted profound melanoma regression, the PI3Kα selective inhibitor elicited only a cytostatic effect. Further, although PI3Kα inhibition augmented melanoma regression elicited by BRAFV600E inhibition, the overall effect was modest. Concordant results were obtained when BRAFV600E/PTENNull melanomas were treated with a MEK1/2 inhibitor (GDC-0973) and the PI3Kβ-sparing inhibitor (GDC-00032) either alone or in combination.
Figure 1.
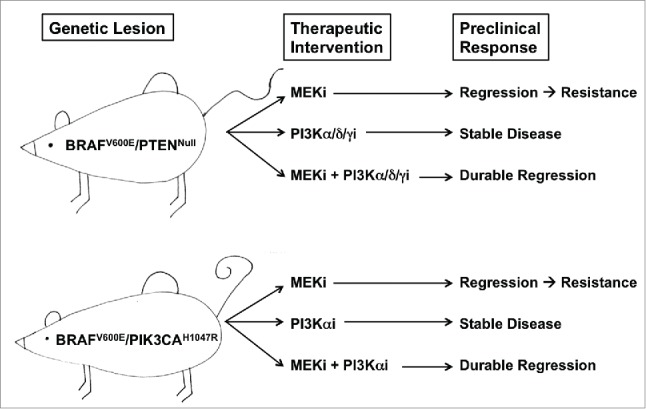
Genetically engineered mouse (GEM) models of BRAF-mutated melanoma display differential responses to inhibition of the mitogen activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K) pathway. GEM models of the indicated genotype were treated with inhibitors of either the MAPK or the PI3K pathway, and the indicated tumor responses were noted.
Finally, we explored whether PI3K inhibition would influence the development of resistance to MAPK pathway-targeted therapy. Cohorts of mice bearing either BRAFV600E/PIK3CAH1047R or BRAFV600E/PTENNull melanomas were treated with single agent MEK1/2 inhibitor therapy in the absence or presence of the relevant PI3K inhibitor (Fig. 1). Over the course of more than 100 days of treatment, we found that the vast majority of mice receiving MEK1/2 inhibitor monotherapy developed drug-resistant melanoma. In contrast, all of the mice receiving MEK1/2 inhibitor plus PI3K inhibitor therapy remained in a durable state of melanoma regression. These results suggest that the combined deployment of MAPK pathway inhibitors plus PI3K inhibitors may forestall the onset of drug resistance for a subset of patients with BRAF-mutated melanoma.
The implications of these studies remain to be tested clinically. The fact that blockade of PI3Kα in a PIK3CAH1047R addicted melanoma elicited only cytostatic effects may indicate that initial clinical expectations for PI3K inhibitors were overly enthusiastic. The capacity for PI3-lipid signaling to shift PI3K isoform dependency in the face of pharmacologic blockade demonstrates the complex dynamics of this signaling system.10 Finally, PI3K signaling is crucial for a range of normal physiologic functions, such that there may be unacceptable toxicity associated with combined inhibition of both MAPK and PI3K signaling in patients. Therefore, translating preclinical studies into the world of clinical trials remains a challenge.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Dr. McMahon has received grant support from Novartis Inc. Financial support was also received from the National Cancer Institute (grant number CA176839).
References
- 1.Ascierto PA, Minor D, Ribas A, Lebbe C, O'Hagan A, Arya N, Guckert M, Schadendorf D, Kefford RF, Grob JJ, et al.. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 2013; 31:3205-11; PMID:23918947; http://dx.doi.org/ 10.1200/JCO.2013.49.8691 [DOI] [PubMed] [Google Scholar]
- 2.Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol 2006; 126:154-60; PMID:16417231; http://dx.doi.org/ 10.1038/sj.jid.5700026 [DOI] [PubMed] [Google Scholar]
- 3.Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res 2011; 24:879-97; PMID:21707960; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00880.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al.. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18:683-95; PMID:21156289; http://dx.doi.org/ 10.1016/j.ccr.2010.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al.. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014; 4:80-93; PMID:24265155; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544-52; PMID:19282848; http://dx.doi.org/ 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marsh Durban V, Deuker MM, Bosenberg MW, Phillips W, McMahon M. Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J Clin Invest 2013; 123:5104-18; PMID:24200692; http://dx.doi.org/ 10.1172/JCI69619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 2008; 105:13057-62; PMID:18755892; http://dx.doi.org/ 10.1073/pnas.0802655105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al.. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008; 454:776-9; PMID:18594509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, et al.. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-Specific inhibitors in luminal breast cancer. Cancer Cell 2015; 27:97-108; PMID:25544637; http://dx.doi.org/ 10.1016/j.ccell.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]