

ABSTRACT
Patients with inflammatory bowel disease are often deficient in micronutrients such as selenium and have an increased risk of colon cancer. We tested whether the selenium transport protein, selenoprotein P, could modify colitis-associated cancer. Our results indicate that global SEPP1 haploinsufficiency augments tumorigenesis and mediates oxidative damage in the intestine.
KEYWORDS: Cancer, IBD, selenoprotein P, SEPP1, ulcerative colitis
The macronutrient selenium, via its incorporation into selenoproteins, has antioxidant roles, influences immune activity, and is inversely correlated with inflammatory bowel disease (IBD) and cancer risk in numerous epidemiologic studies.1-3 Indeed, previous studies from our laboratory have demonstrated that dietary selenium influences the severity of colitis and colitis-associated cancer (CAC) in mouse models.4 The most abundant plasma selenoprotein is selenoprotein P (SEPP1). SEPP1 contains 10 selenocysteine residues and is essential for selenium transport and the production of other selenoproteins throughout the body.5 However, SEPP1 is also thought to possess an endogenous antioxidant function, suggesting that it could play a role in cancer prevention, particularly in the context of inflammatory cancers characterized by increased oxidative stress. Furthermore, at baseline the gastrointestinal tract is susceptible to oxidative damage resulting from direct contact of the colonic epithelium with microbial and food-derived reactive oxygen species (ROS). Thus, it was our hypothesis that SEPP1 may regulate intestinal homeostasis and protect against colitis and CAC.
To specifically investigate the contribution of SEPP1 to colonic epithelial biology and how its loss affects CAC we used a global SEPP1 knockout model. Sepp1 wild-type (WT, Sepp1+/+), heterozygous (Sepp1+/−), and null (Sepp1−/−) mice were subjected to an azoxymethane (AOM) and chronic dextran sodium sulfate (DSS) protocol. A key finding of our study was that SEPP1 functions as a haploinsufficient tumor suppressor in the AOM/DSS CAC model.6 Mice with reduced SEPP1 (i.e., Sepp1+/−) had greater tumor multiplicity and these tumors were larger, had a higher degree of dysplasia, and were more proliferative than those observed in SEPP1 WT mice. These results phenocopy our prior observations in selenium-deficient mice and suggest that SEPP1 is a major mediator of the effects we observed with selenium reduction.
However, specifically studying the role of SEPP1 is confounded by its role in selenium transport, as loss of SEPP1 decreases the selenium available for total selenoprotein synthesis. Indeed, loss of other selenoproteins, namely the glutathione peroxidases, can directly cause colitis in genetic knockout (KO) models.7 Thus, although SEPP1 is the most abundant plasma selenoprotein, many ill effects observed in the setting of selenium deficiency have instead been attributed to other antioxidant selenoproteins rather than SEPP1. In addition to the global Sepp1 KO mice, we were fortunate to have access to mice deficient in either the selenium-rich transport domain or the N-terminal redox motif of SEPP1. Surprisingly, both mouse models showed almost identical increases in tumor formation in the AOM/DSS model. Thus, our results solidify SEPP1 as an important antioxidant in the setting of intestinal inflammation while illustrating that both functions of SEPP1 (i.e., selenium transport and antioxidant activity) contribute to tumorigenesis and likely act synergistically in the AOM/DSS model.
The role of increased oxidative damage in the development of malignancy is well characterized, and ROS are a well-known contributor to a chronic inflammatory microenvironment.8 Thus, while striking, it may not be surprising that loss of the antioxidant SEPP1 potentiates tumor formation. On the other hand, our AOM/DSS studies also found that complete loss of SEPP1 was relatively protective in AOM/DSS models of CAC. Tumors in these mice were small and scarce, and exhibited high genomic instability, decreased proliferation, and increased apoptosis. Although paradoxical, this observation is likely due to the “double-edged sword” of ROS-induced injury by which instead of promoting malignancy, critically high levels of oxidative injury lead to the clearance of initiated Sepp1−/− cells (Fig. 1A). This hypothesis was further investigated using 3D ex vivo enteroid cultures from SEPP1 WT and null mice in the presence of hydrogen peroxide (H2O2) to stimulate ROS-mediated injury. Not only did Sepp1−/− cultures have increased baseline levels of ROS and oxidative damage, they were also extremely sensitive to ROS-mediated injury. In contrast to WT cultures that were able to persist in the presence of H2O2, SEPP1-deficient enteroids were completely lost by 24 hours. Together, these studies suggest that while the modestly increased ROS induced by Sepp1 heterozygosity may increase disease severity, complete loss of SEPP1 (and likely other selenoproteins) leads to continuous oxidative assault that cells are unable to overcome.
Figure 1.
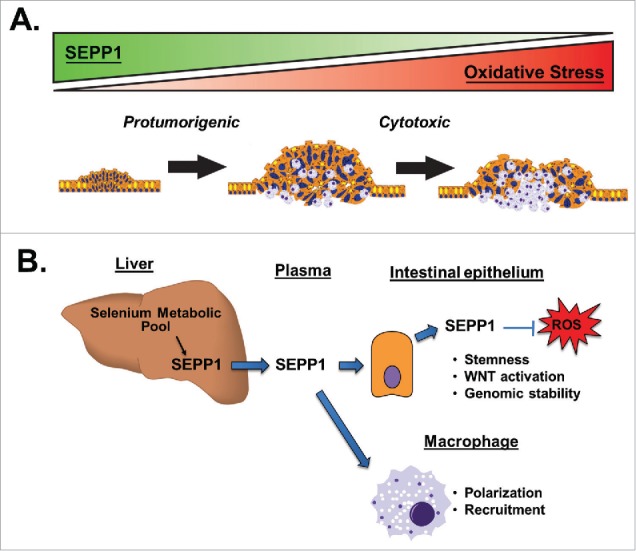
Roles for selenoprotein P (SEPP1) in intestinal homeostasis and tumorigenesis. Our results from azoxymethane and repeated dextran sodium sulfate (AOM/DSS) experiments demonstrated increased tumor numbers in mice heterozygous for Sepp1 (Sepp1+/−), which parallels the protumorigenic effects of increased oxidative injury (Fig. 1A). In contrast, we found relatively few tumors in global Sepp1 knockout mice, most likely because of critically high levels of oxidative damage and cytotoxicity. These studies also identified tissue-specific effects of SEPP1 that may independently contribute to tumorigenesis (Fig. 1B). We determined that epithelial-derived SEPP1 inhibits reactive oxygen species (ROS) production and contributes to genome stability, stemness, and WNT activity in ex vivo cultures. SEPP1 expression also affected macrophage populations, as we identified altered recruitment and skewed polarization in the setting of Sepp1 heterozygosity. Together, these results illustrate a significant role for SEPP1 in both intestinal homeostasis and disease development.
To add to the complexity of SEPP1's role in CAC, we further identified tissue-specific effects of SEPP1 that likely influence tumorigenesis (Fig. 1B). SEPP1 is highly expressed in macrophage populations and is one of the most upregulated genes in alternatively activated (M2) macrophages.9 Indeed, macrophages from Sepp+/− mice were preferentially skewed to the protumorigenic M2 phenotype and naïve macrophages displayed a heightened response to M2 stimuli. A separate SEPP1 pool in intestinal epithelial cells, however, modulated epithelial stem cell behavior and increased “stemness.” This was likely due to activation of the WNT signaling pathway, which is a crucial pathway for both stem cell maintenance and intestinal tumorigenesis.10 Both of these cell type-specific effects can promote tumor development and progression, and further suggest that SEPP1 contributes to cell homeostasis in a variety of tissues.
Our study analyzed the specific contribution of SEPP1 to intestinal homeostasis and disease. Interestingly, while our observations firmly establish the importance of SEPP1 in colitis and intestinal disease, most likely through its role in mediating oxidative damage, they also greatly add to the complexity of SEPP1 biology. Whether the numerous functional and tissue-specific effects of SEPP1 contribute independently to colitis and intestinal tumorigenesis is still unknown. Our studies establish a critical role for SEPP1 in intestinal biology, homeostasis, injury response, preservation of genomic integrity, and inflammatory carcinogenesis and suggest that SEPP1 could serve as a therapeutic target in the prevention of CAC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was funded by grants from National Institute of Diabetes and Digestive and Kidney Diseases (R01DK099204), Crohn's and Colitis Foundation of America (290326), and U.S. Department of Veteran Affairs (1I01BX001426).
References
- 1.Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr 2005; 25:215-35; PMID:16011466; http://dx.doi.org/ 10.1146/annurev.nutr.24.012003.132120 [DOI] [PubMed] [Google Scholar]
- 2.Shamberger RJ, Willis CE. Selenium distribution and human cancer mortality. CRC Crit Rev Clin Lab Sci 1971; 2:211-21; PMID:4950948; http://dx.doi.org/ 10.3109/10408367109151308 [DOI] [PubMed] [Google Scholar]
- 3.Speckmann B, Steinbrenner H. Selenium and selenoproteins in inflammatory bowel diseases and experimental colitis. Inflamm Bowel Dis 2014; 20:1110-9; PMID:24694793 [DOI] [PubMed] [Google Scholar]
- 4.Barrett CW, Singh K, Motley AK, Lintel MK, Matafonova E, Bradley AM, Ning W, Poindexter SV, Parang B, Reddy VK, et al.. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One 2013; 8:e67845; PMID:23861820; http://dx.doi.org/ 10.1371/journal.pone.0067845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burk RF, Hill KE. Regulation of selenium metabolism and transport. Annu Rev Nutr 2015; 35:109-34; PMID:25974694; http://dx.doi.org/ 10.1146/annurev-nutr-071714-034250 [DOI] [PubMed] [Google Scholar]
- 6.Barrett CW, Reddy VK, Short SP, Motley AK, Lintel MK, Bradley AM, Freeman T, Vallance J, Ning W, Parang B, et al.. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J Clin Invest 2015; 125:2646-60; PMID:26053663; http://dx.doi.org/ 10.1172/JCI76099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esworthy RS, Aranda R, Martin MG, Doroshow JH, Binder SW, Chu FF. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol 2001; 281:G848-55; PMID:11518697 [DOI] [PubMed] [Google Scholar]
- 8.Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract 2008; 204:511-24; PMID:18571874; http://dx.doi.org/ 10.1016/j.prp.2008.04.011 [DOI] [PubMed] [Google Scholar]
- 9.Bosschaerts T, Guilliams M, Noel W, Herin M, Burk RF, Hill KE, Brys L, Raes G, Ghassabeh GH, De Baetselier P, et al.. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol 2008; 180:6168-75; PMID:18424738; http://dx.doi.org/ 10.4049/jimmunol.180.9.6168 [DOI] [PubMed] [Google Scholar]
- 10.de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci 2007; 12:471-91; PMID:17127311; http://dx.doi.org/ 10.2741/2076 [DOI] [PubMed] [Google Scholar]