

ABSTRACT
Gene expression programs are tightly regulated by heritable “epigenetic” information, which is stored as chemical modifications of histones and DNA. With the recent development of sequencing-based epigenome mapping technologies and cancer cellular reprogramming, the tools are now in hand to analyze the epigenetic contribution to human cancer.
Abbreviations
- ABL
Abelson
- AZA
azacytidine
- BCR
breakpoint cluster region
- CML
chronic myeloid leukemia
- DiR
DNMT1-interacting RNA
- Ph+
Philadelphia chromosome
Aberrant DNA methylation is a characteristic feature of several types of cancer including hematological malignancies such as chronic myeloid leukemia (CML).1 Several cancers have been shown to carry many chromosomal abnormalities whereas some, such as CML, show only a single genetic aberration.
A growing body of evidence indicates that cancers can arise from a small numbers of events that affect differentiation, apoptosis, and/or survival processes. Perturbation of these functions (e.g., mutation of a tumor suppressor gene) requires a complete loss of both copies of the involved gene. According to Knudson's “2-hit” hypothesis, cancer results from the accumulation of several insults in the DNA. In some instances the first hit, a somatic or germline mutation, is responsible for the loss of one allele of a tumor suppressor gene,2 and, later on, a second event hitting the other allele causes gene deactivation, inducing cancer. It is conceivable to include among the so-called hits events other than classic genetic mutations, such as DNA methylation. From this viewpoint, aberrant DNA methylation leading to gene silencing can act as a first or second hit, depending on the timeline of its onset, and is therefore able to promote cancer development.
Although most cancers reveal extensive chromosomal aberrations, some of them, such as CML, are classified by very specific genetic mutations. All CML cells are defined by the same cytogenetic lesion: the presence of a specific chromosome named the Philadelphia chromosome (Ph+). The Philadelphia chromosome originates from a reciprocal translocation between chromosomes 9 and 223 that leads to fusion of the Abelson (ABL) oncogene and the breakpoint cluster region (BCR) gene. This translocation creates a chimeric permanently activated kinase, BCR-ABL, that is able to perturb regulation of both cell cycle and apoptosis pathways via activation of several oncoproteins.4 For several years CML has been considered a model of a “one-hit tumor;” consequently, all therapeutic approaches have focused on achieving inhibition of the BCR-ABL oncogene.
Our study5 aimed to elucidate whether and, if relevant, how the BCR-ABL genetic lesion could co-exist and synergize with other epigenetic changes during the leukemogenesis program and whether DNA methylation could function as a second hit in this type of leukemia.5 To that end, we mapped the DNA methylation landscape of primary and laboratory CML cell lines to dissect the mechanistic interplay between genetic versus epigenetic alterations. Interestingly, we found aberrant DNA methylation of genes involved in DNA repair and the cell cycle, such as SALL4 or BRCA1, in both primary and laboratory CML cell lines. To test whether DNA methylation could act as a second hit in CML development, we applied two different methodologic approaches by taking advantage of (1) cellular reprogramming6 and (2) an in vivo CML murine model carrying an inducible BCR-ABL oncogene.7 The first approach allowed us to reset the aberrant methylome leaving intact all the genetic aberrations. Interestingly, the reprogrammed K562 cells (a laboratory CML model cell line) redifferentiated into CD45+ cells and lost their oncogenic potential when transplanted into immunodeficient mice despite the presence of several genetic abnormalities.
The second approach allowed us to test whether induction of BCR-ABL in a wild-type context was, by itself, able to perturb the DNA methylation pattern. Genome-scale DNA methylation analysis of murine stem cells carrying the BCR-ABL oncogene revealed gain of aberrant DNA methylation within CpG islands upon BCR-ABL expression.
To understand the functional contribution of a perturbed DNA methylation pattern to leukemogenesis, different groups of congenic recipient mice were transplanted with murine BCR-ABL leukemic bone marrow cells. The groups were treated with azacytidine (AZA: one of the most commonly used demethylating agents), imatinib (a tyrosine kinase inhibitor used as first-line treatment for CML), or a combination of the 2 drugs. Surprisingly, the group of mice treated with AZA survived even longer than the group treated with imatinib only.
Overall, our data support the hypothesis that DNA methylation changes, promoted by BCR-ABL, function not only as a second hit, but also as a major contributor to leukemia progression.
Further studies are now needed to elucidate how BCR-ABL, and oncogenes in general, function mechanistically to induce epigenetic changes. We can envisage two potential pathways involved in a similar mechanism. Recently, our group demonstrated the existence of a specific class of RNAs called DNMT1-interacting RNAs (DiRs).8 DiRs originate from transcriptionally active loci and, by binding with high affinity to DNA (cytosine-5-)-methyltransferase 1 (DNMT1), prevent DNA methylation of the corresponding gene loci. Consequently, any change occurring at the DiR transcriptional level could result in aberrant DNA methylation at specific gene loci Additionally, oncogenes such as BCR-ABL might negatively regulate DiR expression directly or indirectly, causing specific gene silencing (Fig. 1). Another possibility implicates increased expression of DNMTs induced by the oncogene.9 Although unlikely, oncogenes might also be able to increase DNMT enzymatic activity thus disrupting the balance that keeps specific gene loci active (Fig. 1).
Figure 1.
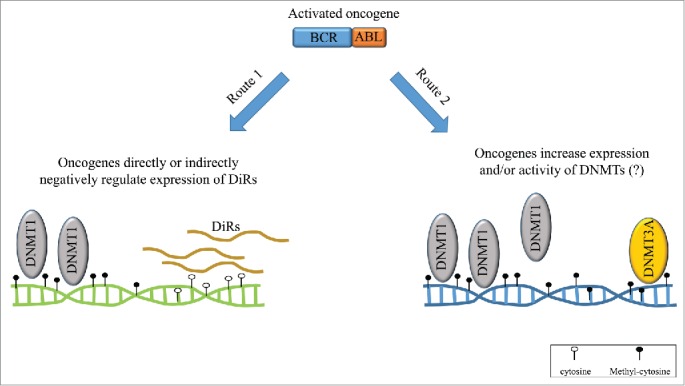
Independent routes to aberrant DNA methylation. Following oncogene activation, 2 independent potential paths leading to aberrant DNA methylation can be hypothesized that would represent the “second hit” during cancer development.
Overall, the results obtained in our study open a new prospective on the role of aberrant DNA methylation in the pathogenesis of a cancer historically considered as a single-hit tumor and, more generally, on the synergistic effect played by both genetic and epigenetic dysregulation.
We believe that this study sheds light on our still limited understanding on the role of epigenetic dysregulation in human malignancies and paves the way for novel therapeutic approaches based on pathogenetic insight.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the K99/R00 Career Development Award (1K99CA188595–01), the Career Development Award from the Giovanni Armenise-Harvard Foundation and partially the Progetto Start-up 2014 (N.15347) to ADR.
References
- 1.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042-54; PMID:14627790; http://dx.doi.org/ 10.1056/NEJMra023075 [DOI] [PubMed] [Google Scholar]
- 2.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer 2001; 1:157-62; PMID:11905807; http://dx.doi.org/ 10.1038/35101031 [DOI] [PubMed] [Google Scholar]
- 3.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243:290-3; PMID:4126434; http://dx.doi.org/ 10.1038/243290a0 [DOI] [PubMed] [Google Scholar]
- 4.Stam K, Heisterkamp N, Grosveld G, de Klein A, Verma RS, Coleman M, Dosik H, Groffen J. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med 1985; 313:1429-33; PMID:3864009; http://dx.doi.org/ 10.1056/NEJM198512053132301 [DOI] [PubMed] [Google Scholar]
- 5.Amabile G, Di Ruscio A, Müller F, Welner RS, Yang H, Ebralidze AK, Zhang H, Levantini E, Qi L, Martinelli G, et al.. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat Commun 2015; 6:7091; PMID:25997600; http://dx.doi.org/ 10.1038/ncomms8091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med 2009; 15:59-68; PMID:19162546; http://dx.doi.org/ 10.1016/j.molmed.2008.12.003 [DOI] [PubMed] [Google Scholar]
- 7.Koschmieder S, Göttgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, Dayaram T, Geary K, Green AR, Tenen DG, et al.. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 2005; 105:324-34; PMID:15331442; http://dx.doi.org/ 10.1182/blood-2003-12-4369 [DOI] [PubMed] [Google Scholar]
- 8.Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich-Jorda M, Zhang P, et al.. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013; 503:371-6; PMID:24107992; http://dx.doi.org/ 10.1038/nature12598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya H, et al.. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol 2009; 186:393-407; PMID:19667129; http://dx.doi.org/ 10.1083/jcb.200904105 [DOI] [PMC free article] [PubMed] [Google Scholar]