

Abstract
Objective
Neutrophils are involved in the inflammatory responses during atherosclerosis. Human neutrophil peptides (HNPs) released from activated neutrophils exert immune modulating properties. We hypothesized that HNPs play an important role in neutrophil-mediated inflammatory cardiovascular responses in atherosclerosis.
Methods and Results
We examined the role of HNPs in endothelial-leukocyte interaction, platelet activation, and foam cell formation in vitro and in vivo. We demonstrated that stimulation of human coronary artery endothelial cells with clinically relevant concentrations of HNPs resulted in monocyte adhesion and transmigration; induction of oxidative stress in human macrophages, which accelerates foam cell formation; and activation and aggregation of human platelets. The administration of superoxide dismutase or anti-CD36 antibody reduced foam cell formation and cholesterol efflux. Mice deficient in double genes of low-density lipoprotein receptor and low-density lipoprotein receptor–related protein (LRP), and mice deficient in a single gene of LRP8, the only LRP phenotype expressed in platelets, showed reduced leukocyte rolling and decreased platelet aggregation and thrombus formation in response to HNP stimulation.
Conclusion
HNPs exert proatherosclerotic properties that appear to be mediated through LRP8 signaling pathways, suggesting an important role for HNPs in the development of inflammatory cardiovascular diseases.
Keywords: atherosclerosis, leukocytes, transgenic models
Recent studies have revealed that polymorphonuclear neutrophils (PMNs) participate in the development of atherosclerotic lesions.1–5 Indeed, PMNs can be detected in human tissue specimens at sites of plaque rupture and erosion or in thrombi from patients with acute coronary syndrome.6 The number of PMNs in blood and the levels of elastase and myeloperoxidase (MPO) released from PMNs have been reported to be associated with atherosclerosis.6–8 A study in more than 350 000 patients with atherosclerosis demonstrated that the subjects with higher leukocyte counts have higher relative and absolute acute and chronic mortality rates than those with lower leukocyte counts.9 A positive correlation between peripheral PMN count and myocardial infarction has also been reported.10,11 In patients with chronic stable angina, PMN count and activation are an independent predictor of the presence of multiple complex stenoses irrespective of the extent of coronary artery disease.7,12 A recent study demonstrated that peripheral blood PMN counts are higher in patients with rheumatoid arthritis implicated with premature atherosclerosis than in control subjects.13 The increased PMN count is associated with high concentration of inflammatory mediators, including interleukin-6, intercellular adhesion molecule-1 (ICAM-1), E-selectin, and tumor necrosis factor-α in the blood.13
Using apolipoprotein E–deficient mice with fluorescent PMNs to study specifically PMN presence and recruitment in atherosclerotic lesions, Rotzius et al have recently shown that a majority of leukocytes interacting with the endothelium on lesion shoulders are PMNs, suggesting a significant recruitment of these cells to the plaque.14 The functional importance of PMNs in the formation of the atherosclerotic plaque was demonstrated by Zernecke et al,3 who reported that the size of murine atherosclerotic plaques has been shown to be closely correlated with the number of PMNs in the peripheral blood, and depletion of circulating PMNs resulted in reduction of plaque formation.
Evidence has shown that several members of PMN-specific proteins, including human neutrophil peptides (HNPs) and azurocidin (heparin-binding protein) are found at the luminal site of the endothelium,15 suggesting their deposition by PMNs. During inflammation, large amounts of intracellular proteins are released from the activated PMNs into the extracellular milieu as a consequence of PMN degranulation, leakage during phagosome formation, and cell death. The highly homologous HNP-1, -2, and -3, also known as α-defensins, make up more than 50% of the total protein content within the azurophilic granules of PMNs.16 HNPs have the capacity to kill a wide spectrum of Gram-positive and Gram-negative bacteria, fungi, and some enveloped viruses in vitro.17 Recent studies have demonstrated that other leukocytes, including monocytes, macrophages, T cells, and immature dendritic cells, also express HNPs in particular when stimulated with cytokines.18–20 Therefore HNPs appear to participate in both innate and acquired immunity.
In healthy volunteers, plasma levels of HNPs range from undetectable to 50 to 100 ng/mL. However, mean HNP levels are 2- to 4-fold greater at the onset of bacterial infection and during nonbacterial infection compared with healthy volunteers.21 Under septic conditions, plasma HNP levels range from 900 to 170 000 ng/mL, compared with a mean of 42±53 ng/mL in healthy controls.22 An excellent correlation exists between the concentration of HNPs and the number of PMNs in the blood of patients with inflammatory diseases.21 We speculate that these proteins released from PMNs and other leukocytes are not bystanders but play an active role in the development of atherosclerosis.
Indeed, HNPs are found in the luminal site of human atherosclerotic coronary arteries in both large- and small-vessel endothelium.23 HNPs are also found in intimal smooth muscle cells of atherosclerotic human cerebral arteries.24 Moreover, a significant correlation is observed in men between the amount of HNP deposition in skin and severity of coronary artery disease.25 HNP stimulation increased adhesion of the cultured human endothelial ECV-304 cells in plastic plate coated with gelatin.26 In porcine coronary artery, endothelium-dependent relaxation in response to bradykinin was significantly reduced after treatment with HNPs associated with an increased production of superoxide anion and a decreased expression of endothelial nitric oxide synthase at both levels of mRNA and protein.27 We observed that stimulation of human umbilical vein endothelial cells (HU-VECs) with HNPs resulted in enhanced formation of peroxinitrite that led to endothelin-1 production, which was largely attenuated by the treatment with the reactive oxygen species scavenger N-acetyl-L-cysteine.28 This observation is of interest because endothelin-1 has been found at increased levels in endothelial cells, smooth muscle cells, and macrophages within atherosclerotic plaques.25
Retention of lipoproteins within the vasculature is an important event in the pathogenesis of atherosclerosis. HNPs have been reported to form stable multivalent complexes with lipoprotein(a).29 Whereas HNPs and lipoprotein(a) individually can readily traverse through the endothelial cell membranes, HNP/lipoprotein(a) complexes are lodged onto the cell surface of endothelial and smooth muscle cells, resulting in a marked increase in the total amount of cell-associated lipoprotein.30 Furthermore, HNPs can stimulate the binding of 125I-low-density lipoprotein (LDL) to cultured HUVECs, smooth muscle cells, and fibroblasts in a dose-dependent and saturated manner.30 An antibody against the LDL receptor (LDLR) is able to inhibit the binding of 125I-HNPs to HUVECs.30 Moreover, HNPs bind directly to isolated α2-macroglobulin receptor (LDLR–related protein [LRP]) in a dose-dependent manner, and the binding is inhibited by anti-LRP antibodies and LDLR-associated protein31 to prevent binding of LRP ligands.
Given its biological nature as an inflammatory marker, the ability to promote cellular oxidation, and the interacting capacity with lipoproteins described above, we hypothesized that HNPs function as an effector in the pathogenesis of cardiovascular diseases. Specifically, HNPs increase monocyte-endothelial cell interaction, accelerate foam cell formation, and trigger the activation of platelets through LRP receptors. This hypothesis was tested in vitro with cultured primary human coronary endothelial cells, human acute monocytic leukemia cell line (THP-1)-derived macrophages, and human primary platelets, and in vivo with mice with LDLR and LRP double-gene or LRP8 single-gene deficiency, respectively.
Materials and Methods
The study protocol was approved by the institutional Research Ethics Board for isolation of HNPs from sputum of patients with cystic fibrosis and for human monocyte and platelet isolation from healthy nonsmoking blood donors. The animal experiments were approved by the institutional Animal Care and Use Committee.
HNP Isolation
HNPs were purified from pooled sputum of patients with cystic fibrosis as previously described32 (see supplemental material, available online at http://atvb.ahajournals.org).
Cell Activation, Adhesion, and Transmigration Assays
Primary human coronary artery endothelial cells (HCAECs) (Cell Applications, San Diego, CA) were used within 4 passages. Expression of CD11b was measured to evaluate human monocyte activation after HNP stimulation, and monocyte adhesion and transmigration assays were performed as previously described33 (see supplemental material).
THP-1-Derived Macrophages Expressing CD36 and CD68
The expression of surface markers CD36 and CD68 was used to identify THP-1 monocyte-derived macrophages.34 Briefly, THP-1 cells were stimulated with 5 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich) for 72 hours, and adherent cells were cultured for additional 24 hours. The cells were then incubated with mouse anti-human CD36- (Invitrogen, Carlsbad, CA) or CD68-FITC antibody (AbD Serotec, Oxford, United Kingdom) for 30 minutes followed by flow cytometric analysis (FACSCanto flow cytometer, Becton Dickinson, Franklin Lakes, NJ).
Foam Cell Assay
The macrophages described above were incubated with 10 μg/mL HNPs, 50 μg/mL native LDL (Calbiochem, La Jolla, CA), or HNPs and LDL together in serum-free medium for 16 hours. As for the positive control condition, macrophages were incubated with 50 μg/mL oxidized LDL (oxLDL) (Kalen Biomedical, Savage, MD) or in combination with HNPs for 16 hours. To investigate the effects of HNPs on LDL oxidation in macrophages, additional cells were treated with the O2− scavenger superoxide dismutase (200 U/mL, Sigma-Aldrich) 5 minutes before the incubation with HNPs and LDL. The foam cell formation was evaluated as previously described.35
Cholesterol Efflux Assay
To label the intracellular cholesterol pool, the THP-1 monocyte-derived macrophages were loaded with HNPs, LDL, oxLDL, anti-CD36 antibody and fluorescent Pennsylvania Green/N-alkyl-3β-cholesterylamine-derived molecular probe (F-Ch, 10 μmol/L from a 10 mmol/L dimethyl sulfoxide stock solution, provided by Dr B. Peterson, University of Kansas). After 24 hours, cells were washed and incubated in 1% FBS medium for another 12 hours to allow the labeled cholesterol to be distributed to various intracellular compartments. After washing, efflux was induced by addition of high-density lipoprotein (100 μg/mL) for 1 hour. The medium was collected, and cells were lysed and centrifuged at 13 200g for 10 minutes. The F-Ch in the medium and cell lysates was quantified relative to individual standard curves using excitation and emission wavelengths of 485 and 535 nm, respectively.36
Murine Platelet Isolation
Male B6129SF2-Lrp8tm1Her/J mice (6 to 8 weeks, Jackson Laboratory, Bar Harbor, ME) and heterozygotes were mated to generate LRP8+/+ and LRP8−/− littermates. LRP8−/− mice have 2 exons deleted from the cytoplasmic domain of the LRP8 receptor, resulting in a DNA sequence that is 100 base pairs shorter than full-length gene sequence in wild-type mice. Platelets from mice were isolated as previously described to obtain platelet-rich plasma (PRP) and platelet-poor plasma.37 Gel filtration of PRP on columns of Sepharose 2B was performed, and 2×108/mL of platelets was used in experiments.
Competitive Binding Assay Between HNPs and Recombinant Human LRP8
HNPs were preincubated with recombinant human LRP8 (R&D Systems Inc, Minneapolis, MN) at a 10:1 molar ratio for 30 minutes at 37°C. Murine and human platelets were stimulated at room temperature for 5 minutes with HNPs (10 μg/mL), vehicle, or the mixture of HNPs and recombinant human LRP8. Platelets were stained with anti-mouse CD62P-FITC antibody (BD Biosciences, Mississauga, ON) or anti-human CD62P-FITC antibody (R&D Systems), respectively, for 25 minutes and fixed for analysis by flow cytometry.
Platelet Aggregation Assay
Human and murine gel-filtered platelets and PRP were stimulated with HNPs, and aggregation was assessed at 37°C using an aggregometer. For priming experiments, murine and human PRP were incubated with HNPs or vehicle for 2 minutes before the addition of ADP. At the end of the aggregation experiment, an aliquot of 30 μL of PRP from each condition was placed on coverslips for examination of thrombus formation under a confocal microscope.
Murine Pulmonary Endothelial Cell Isolation
Male LRP8−/− and age (2 to 3 weeks) and background strain-matched mice (Jackson Laboratory) were used for endothelial cell isolation by using rat anti-mouse CD31 antibody (PECAM-1, Bio-legend, San Diego, CA) followed by a magnetic separation (Dynal MPC-S, Invitrogen Dynal AS, Oslo, Norway) (see supplemental material for details). FITC rat anti-mouse CD54/ICAM-1 monoclonal antibody (1:50, Biolegend), PE rat anti-mouse CD106/vascular cell adhesion molecule-1 monoclonal antibody (1:50, GenWay Biotech, Inc, San Diego, CA), and DAPI (Molecular Probe, Molecular Probes, Inc, Eugene, OR) were used for immunostaining, followed by quantitative analysis using ImageJ software (NIH, http://imagej.nih.gov/ij/download.html).
Mouse Monocyte Isolation and Flow Cytometry
Male LRP8−/− and age (2 to 3 weeks) and background strain-matched mice (Jackson Laboratory) were euthanized by overdose anesthesia for monocyte isolation from bone marrow (Mouse Monocyte Enrichment Kit, Stemcell Technologies Inc, Vancouver, British Columbia, Canada). Monocytes (2×105) were suspended in RPMI-1640 in 24-well plates for 2 hours and then incubated without or with HNPs 10 μg/mL for 1 hour. The cells were stained with unlabeled or PE rat anti-mouse CD11b monoclonal antibody (Biolegend) for 30 minutes at room temperature and then were analyzed using a BD FACSCanto with BD FACSDiva software (BD Biosciences, Mountain View, CA).
Measurements
Concentrations of reactive nitrogen species, HNPs, MPO, chemokines, and adhesion molecules were measured (see supplemental material).
Intravital Microscopy
Leukocyte Adhesion
Male B6129SF2 and age-matched (6 to 8 weeks) and background strain–matched Ldlrtm1Her/Lrpap1tm1Her/J, and LRP8−/− mice (Jackson Laboratory) were anesthetized and received intravenously HNPs (10 mg/kg body weight corresponding to approximately 10 μg/mL)38 or vehicle control solution. In additional experiments, platelets were depleted in mice who received intraperitoneal injection of rat anti-mouse CD41 monoclonal antibody at 2 μg per mouse (from a 500 μg/mL PBS stock solution provided by Dr A. Lazarus, the University of Toronto) 24 hours before HNP injection. This dose and timing of application of anti-CD41 resulted in 80% platelet depletion in the whole blood. An equal volume of PBS was used as for vehicle control.
To label circulating leukocytes,39 rhodamine 6G at 200 μL (20 μg/μL, Sigma-Aldrich, St. Louis, MO) was injected intravenously. The carotid artery was exposed and monitored under an upright microscope (Axiotechvario 100HD, Zeiss, Jena, Germany) on a custom-built, computer-controlled warmed stage. The percentage of rolling leukocytes was calculated as previously described.39
Platelet Aggregation
Male LRP8−/− and age (6 to 8 weeks) and background strain-matched mice (Jackson Laboratory) were anesthetized and received intravenously HNPs (10 mg/kg body weight)38 or vehicle control solution. Ex vivo–labeled platelets (250×106 to 300×106) were infused intravenously into the tail vein for real-time visualization by using the intravital fluorescence microscope setup described above. The thrombus formation was recorded during the first 15 minutes after FeCl3 administration to determine the time required for reaching maximal vessel occlusion.
Statistical Analysis
Data are presented as a means±SE. Statistical significance was determined using a 1-way ANOVA followed by a Dunnett comparison. Analyses were performed using GraphPad Prism, version 5.00c (GraphPad Software Inc, San Diego, CA). A probability value <0.05 was considered statistically significant.
Results
HNPs Induce Monocyte-Endothelial Interaction and Chemokine Production
When the HCAECs were incubated with HNPs, monocyte adhesion to HCAECs increased at 10 μg/mL compared with the vehicle control group (Figure 1A). In addition, HNPs induced significant monocyte transmigration through the HCAEC monolayer in a dose-dependent manner (Figure 1B). The expression of ICAM-1 but not vascular cell adhesion molecule-1 increased in HCAECs, and CD11b increased in human monocytes after stimulation with HNPs, compared with the vehicle control groups (Figure 1C and 1D). An increase in the production of interleukin-8 was observed in the cultural supernatants of both HCAECs and monocytes after HNP stimulation at 10 μg/mL (Figure 1E) and monocyte chemotactic protein-1 in HCAECs at both 1 μg/mL and 10 μg/mL (Figure 1F).
Figure 1.

Human neutrophil peptides (HNPs) induce leukocyte-monocyte interaction. A, Human monocyte adhesion on human coronary artery endothelial cells (HCAECs) after stimulation with HNPs for 4 hours. V indicates vehicle. B, Monocyte (defined by CD33 as surface marker) transmigration through HCAECs after stimulation with HNPs. C, Expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in HCAECs after stimulation with HNPs for 4 hours. D, CD11 expression 1 hour after HNP stimulation in primary human monocytes. E, Interleukin-8 (IL-8) production in coculture supernatants of HCAECs and monocytes stimulated with HNPs for 4 hours (n=5 experiments). F, Monocyte chemotactic protein-1 (MCP-1) production in HCAECs stimulated with HNPs for 4 hours (n=5 experiments). *P<0.05 vs V under identical conditions.
HNPs Increase Oxidative Stress and Accelerate Foam Cell Formation
The macrophage scavenger receptors CD36 and CD68 are major receptors responsible for uptake of modified LDL contributing to foam cell formation. We observed an increase in macrophage surface expression of CD36 and CD68 after stimulation with HNPs, as compared with the vehicle control group (Figure 2A and 2B). An enhanced oxidative stress as reflected by nitrotyrosine formation was observed in macrophages after HNP stimulation at 10 μg/mL (Figure 2C).
Figure 2.

Human neutrophil peptides (HNPs) increase oxidative stress and foam cell formation. A and B, Surface expression of CD36 and CD68 on human acute monocytic leukemia cell line (THP-1) derived macrophages stimulated with HNPs or vehicle (V) control for 8 hours. C, Concentration of nitrotyrosine measured in human THP-1 derived macrophage lysates after HNP stimulation. n=5 experiments. D, Foam cell formation, quantified by Oil Red O (ORO) staining. Human macrophages were incubated with HNPs (10 μg/mL) in the presence of low-density lipoprotein (LDL) or oxidized LDL (oxLDL) for 16 hours. In additional experiments, superoxide dismutase (SOD) or anti-CD36 blocking antibody was added 30 minutes before the stimulation with HNPs. n=10 experiments. E, Representative cells with ORO staining in different groups described in D. F, Cholesterol efflux. ‡P<0.05 vs V, *P<0.05 vs LDL alone, †P<0.05 vs the LDL+HNP group.
Incubation of macrophages with oxLDL resulted in increased Oil Red O staining, indicative of foam cell formation (Figure 2D and 2E). A combined stimulation of oxLDL and HNPs did not result in any further increase in Oil Red O staining. A similar increase in intracellular Oil Red O staining as seen with oxLDL incubation was observed after macrophages were incubated together with LDL and HNPs but not with HNP vehicle control buffer (Figure 2D and 2E). The HNP-induced foam cell formation (Figure 2D and 2E) and cholesterol efflux (Figure 2F) were largely attenuated by the treatment with superoxide dismutase and anti-CD36 blocking antibody, respectively.
HNPs Activate Platelets
Incubation of platelets with HNPs induced a dose-dependent increase in CD62P expression in human PRP (Figure 3A). Similar results were seen in murine PRP (Figure 3B). The HNP-induced platelet activation was associated with an increase in aggregation in human platelets (Figure 3C), but not in murine platelets (data not shown). The HNP-induced human platelet aggregation resulted in thrombus formation detected in culture supernatants (Figure 3D).
Figure 3.

Human neutrophil peptides (HNPs) induce platelet activation. A and B, Surface expression of CD62P in human and murine platelet-rich plasma (PRP) stimulated with either HNPs (1, 10 μg/mL), vehicle control (V), or human α-thrombin (Thr) (5 U/mL). C, Dose-dependent increase in platelet aggregation in human PRP after stimulation with HNPs at 10 or 100 μg/mL. D, Thrombus formation after the HNP stimulation shown in C. E, HNP-induced human platelet aggregation and synergy with ADP. Human PRP was primed with HNPs (10 μg/mL) or vehicle control followed 2 minutes later by incubation with ADP (1.25 μmol/L). Platelet aggregation is illustrated by representative actual traces (top) and by average traces (bottom, n=7 experiments). F, Thrombus formation after the HNP stimulation shown in E. G, HNP-induced murine platelet aggregation and synergy with ADP. Murine PRP was primed with HNPs (10 μg/mL) or vehicle control followed 2 minutes later by incubation with ADP (5 μmol/L). Platelet aggregation is illustrated by representative actual traces (top) and by average traces (bottom, n=7 experiments). H, Thrombus formation after the HNP stimulation shown in G. *P<0.05 vs V group under identical conditions.
We then examined whether HNPs sensitize the weak agonist ADP for platelet aggregation. When human and murine platelets were primed with HNPs or vehicle control followed 2 minutes later by incubation with ADP (1.25 μmol/L for human, 5 μmol/L for murine cells), a significantly prolonged aggregation was observed in both human (Figure 3E) and murine (Figure 3G) platelets as compared with a transient aggregation after ADP stimulation alone. The increased platelet aggregation by HNP priming was associated with greater thrombus formation as compared with ADP alone (Figure 3F and 3H).
HNPs Mediate Cellular Inflammatory Responses Through LRP Signaling
To understand the signaling mechanisms by which HNPs induced cell adhesion, we repeated the leukocyte adhesion experiments in wild-type and the LDLR and LRP double-gene knockout mice by using an intravital microscopy setup. We observed a dramatic increase in leukocyte rolling in carotid artery 4 hours after HNP challenge in wild-type control mice (Figure 4A and 4B). In contrast, there was a significant decrease in leukocyte rolling in the LDLR−/−/LRP−/− mice (Figure 4C) despite the observation that the plasma level of HNPs and MPO activity were identical in both the wild-type and the gene knockout mice (Figure 4D and 4E). These results suggest a role of LRP signaling in mediating the HNP-induced leukocyte adhesion in vivo.
Figure 4.
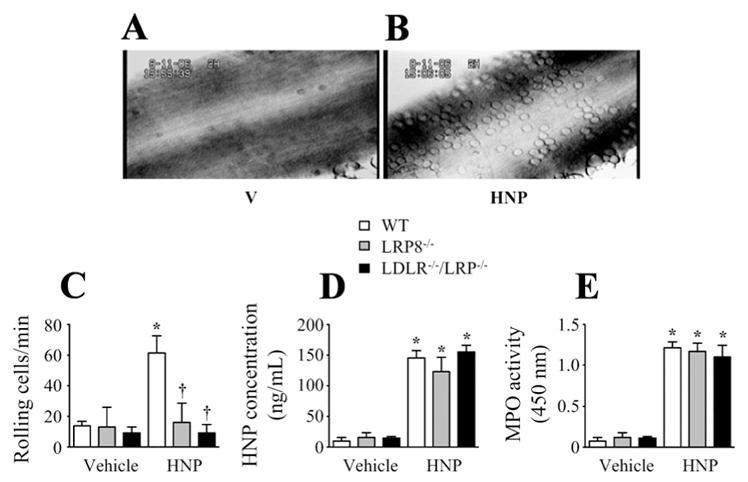
Human neutrophil peptides (HNPs) induce leukocyte adhesion mediated by low-density lipoprotein receptor (LDLR)–related protein 8 (LRP8) in vivo. A and B, Leukocyte rolling in carotid artery was monitored by intravital microscopy in wild-type (WT) mice 4 hours after receiving intravenous injection of vehicle (V) or HNPs (10 mg/kg). C, Mean values of leukocyte rolling in WT, LRP8−/−, or LDLR−/−/LRP−/− mice 4 hours after injection with HNPs. D and E, Mean plasma levels of HNPs and myeloperoxidase (MPO) activity measured in WT, LRP8−/−, or LDLR−/−/LRP−/− mice 4 hours after injection with HNPs. *P<0.05 vs V under identical conditions, †P<0.05 vs WT under identical conditions.
To identify the specific phenotype of LRP in mediating the HNP-induced inflammation, we went further, to examine the role of LRP8 in leukocyte rolling and platelet activation, because LRP8 is the only LRP family member expressed on platelets.40 We first observed a significant decrease in leukocyte rolling in the LRP8−/− mice as compared with the wild-type mice (Figure 4C) given that the plasma level of HNPs and MPO activity were identical in both the wild-type and the gene knockout mice (Figure 4D and 4E).
We then conducted a competitive binding assay in which recombinant human LRP8 was incubated with HNPs at a 1:10 molar ratio 30 minutes before stimulation of human platelets. HNPs alone induced an increase in CD62P expression that was dramatically reduced in the presence of recombinant human LRP8 (Figure 5A). We observed a decrease in CD62P expression in platelets isolated from LRP8−/− mice after HNP stimulation as compared with the wild-type mice, whereas stimulation with thrombin (5 U/mL) resulted in a similar increase in CD62P expression in both groups (Figure 5B). This phenomenon suggests a specific role for LRP8 in mediating the HNP-induced platelet activation.
Figure 5.

Low-density lipoprotein receptor–related protein 8 (LRP8) plays a role in the human neutrophil peptide (HNP)–induced platelet activation and leukocyte rolling. A, Surface expression of CD62P in human platelet-rich plasma (PRP) after stimulation with vehicle control (V) or HNPs (10 μg/mL) in the presence or absence of recombinant human LRP8 (rhLRP8) (n=7 experiments). B, Surface expression of CD62P in murine PRP isolated from wild-type (WT) or LRP8−/− mice after incubation with HNPs (1 or 10 μg/mL) or thrombin (5 U/mL) (n=7 experiments). C, Representative images of platelet aggregation under fluorescence intravital microscope in WT and LRP8−/− mice 4 hours after intravenous injection of HNPs. Low-dose FeCl3 was topically dropped on carotid artery to stimulate platelet aggregation. D, Mean values of fluorescent density of platelets deposited on the vessel wall 5 minutes after FeCl3. E, Time required reaching maximal vessel occlusion after FeCl3 stimulation in mice treated with HNPs. F, Platelet LRP8 is not required for increasing leukocyte rolling. Platelets were depleted by intraperitoneal injection of rat anti-mouse CD41 monoclonal antibody (2 μg per mouse) in WT and LRP8−/− mice 24 hours before intravenous injection of HNPs. Leukocyte rolling in carotid artery was monitored by intravital microscopy 4 hours after receiving HNPs. G, Expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in response to HNP stimulation (10 μg/mL) for 4 hours by primary endothelial cells isolated from WT and LRP8−/− mice (n=5). H, Expression of CD11b in response to HNP stimulation (10 μg/mL) for 1 hour by primary monocytes isolated from bone marrow of WT and LRP8−/− mice (n=5). *P<0.05 vs WT under identical conditions, †P<0.05 vs unstained, ‡P<0.05 vs stained.
Under in vivo conditions in which mice received intravenous injection of HNPs, the platelet intensity/area representing platelet aggregation was significantly greater in the wild-type mice but remained low in the LRP8−/− mice (Figure 5C and 5D). The time to reach maximal occlusion of carotid artery prolonged significantly in the LRP8−/− mice compared with the wild-type mice (Figure 5E).
To verify whether endothelial, monocytic, or platelet LRP8 is required for increased monocyte adhesion in vivo, we examined leukocyte rolling under intravital microscope in the presence and absence of platelets after HNP stimulation in LRP8−/− mice. Our results show that the absence of platelets slightly reduced leukocyte rolling in the wild-type mice, whereas platelet LRP8 is not required for leukocyte adhesion (Figure 5F).
To examine the exact role of endothelial and monocytic LRP8 in contributing to leukocyte rolling, we went further, to measure cell surface markers of activation in vitro endothelial cells and monocytes isolated from wild-type and LRP8−/− mice. We demonstrated that the expression of vascular cell adhesion molecule-1 and ICAM-1 by endothelial cells decreased (Figure 5G), whereas CD11b expression was not significantly altered (Figure 5H) in the absence of LRP8 after HNP stimulation.
Discussion
Several recent advances have suggested that PMNs play a functional role in atherosclerotic progression. By using a small-molecule antagonist to increase peripheral PMNs, Zernecke et al demonstrated a close correlation between murine PMN counts and atherosclerotic lesion size.3 Furthermore, depletion of PMNs significantly reduced plaque formation, confirming a role for PMNs in their formation.3,41 Rotzius et al have shown substantial numbers of PMNs in aortic plaques, especially in the shoulder regions.14
One of the criticisms against the involvement of PMNs in atherosclerosis has been their lack of direct detection within lesions in human atherosclerosis. It has been suggested that the short life span and high turnover of emigrated PMNs coupled with technical limitations in accurately identifying them could account for this discrepancy. Hence, even low numbers of plaque PMNs may represent significant recruitment of cells that have undergone decay, leaving behind only “footprints” revealing their presence. Indeed, it has been shown that the abundant content of HNPs released from activated PMNs are located in luminal site of human atherosclerotic coronary arteries23 and around intimal and medial smooth muscle cells within human atherosclerotic carotid and coronary arteries.20,23 HNPs were found in lesions in which intimal thickening was minimal, suggesting that HNP deposition occurs early in the disease process.20,23 Furthermore, a significant correlation was revealed between HNP skin deposition and the severity of coronary artery diseases, as evaluated by the number of blood vessels associated with focal lesions and stenosis.25
We demonstrated that HNPs may act not only as a biomarker like MPO to predict coronary diseases, as previously reported,42 but also function as an effector to induce or deteriorate inflammatory cardiac events by inducing endothelium-monocyte interaction through increasing the conventional receptor-ligand interaction between ICAM-1 and CD11b, oxidative stress and foam cell formation. In a previous study, we also showed that the stimulation of HUVECs with HNPs dose-dependently increases COX-2 expression.28 The latter is required for integrin-dependent endothelial cell activation and migration.43
On adhesion to the endothelial wall, the monocytes being recruited to the lesion-prone sites play a critical role in the development of atherosclerosis. This process is governed by inflammatory molecules, such as monocyte chemotactic protein-1, endothelin-1, and interleukin-8.44 We demonstrate that HNPs can activate endothelial cells to increase the production of monocyte chemotactic protein-1, a powerful inducer of monocyte chemotaxis. The presence of monocyte chemotactic protein-1 is likely responsible for potentiating monocyte chemotaxis through the endothelium in our in vitro assays. However, there may be other mechanisms contributing to the observed monocyte chemotaxis. We have previously shown in primary human endothelial cells that HNPs increase the production of endothelin-1, another known monocyte chemoattractant,28 and induce the expression of leukocyte function-associated antigen-1 in lymphocytes.33 Besides the chemoattractive properties of HNPs through secondary production of chemokines, HNPs have also demonstrated direct chemotactic activity toward monocytes and dendritic cells at nanomolar concentrations.45
Once trapped in the arterial wall, monocytes can undergo phenotypic differentiation into macrophages and contribute to atherosclerosis-associated inflammation through formation of foam cells. The oxidation of LDL by reactive oxygen species is a well-known mechanism of the formation of foam cells, a characteristic feature of atherosclerosis at all stages of disease progression. We and others have previously demonstrated the production of reactive oxygen species and reactive nitrogen species, including superoxide anion, H2O2, and nitrotyrosine,27,28 on HNP stimulation in endothelial and epithelial cells. In the present study, we observed a significant production of nitrotyrosine, reflecting peroxynitrite formation when macrophages are stimulated with HNPs. Nitrotyrosine is a known molecule contributing to the oxidation of LDL and thus is likely to contribute to increased foam cell formation in the presence of HNPs. This concept is further supported by the observation that the use of the reactive oxygen species scavenger superoxide dismutase significantly decreases the foam cell formation. In addition, macrophage scavenger receptors CD36 and CD68 are the major receptors for macrophages to internalize oxLDL to become lipid engorged foam cells. Our results suggest that the HNP-induced increase in the surface expression of CD36 has contributed to the further enhancement in foam cell formation.
Platelets play a central role in physiological hemostasis but also contribute to atherothrombosis in pathological conditions. We have identified the ability of HNPs to dose-dependently enhance CD62P expression on human and murine platelets. CD62 is not only a marker of platelet α-degranulation but also is responsible for mediating various inflammatory responses through interactions with P-selectin glycoprotein ligand-1 on monocytes and endothelial cells.46 Furthermore, CD62P is elevated in patients with acute coronary syndrome47 and has been found to promote atherosclerosis in apolipoprotein E−/− mouse models.48 The functional consequence of HNP-mediated platelet activation is demonstrated by the ability of HNPs to independently induce human platelet aggregation and sensitize both murine and human aggregation in response to ADP. The relatively modest response in human platelets, compared with murine platelets, in response to HNP stimulation may be donor specific, and a larger cohort of carefully screened subjects may be required to define maximal response.
We examined the role of LRP in initiating the HNP-induced cell-cell interaction for several reasons: (1) LRP has been shown to regulate integrin CD11b/CD18-mediated adhesion between monocytes and the endothelium49; (2) inhibition of LRP decreases leukocyte pulmonary infiltration and the acid-induced lung injury in a mouse model overexpressing HNPs50; (3) HNPs have been reported to bind to LRP, resulting in HNP internalization by smooth muscle cells31; and (4) the HNP-induced inhibition of aortic contractility was abolished by the use of anti-LRP antibodies and the LRP antagonist receptor-associated protein.31 Furthermore, HNPs and LDL formed stable complexes in solution and on the endothelial cell surface,30 the interaction between HNPs and LDL resulted in LDL retention and slowed LDL degradation in endothelium,24,30 and anti-LDLR antibodies that block LDL binding inhibited the binding of HNPs to HUVECs.30 We thus chose the LRP and LDLR double-gene knockout mice to keep the potential binding background as clean as possible. Our results suggest that LRP is required for HNP signaling by demonstrating a reduced leukocyte flux after HNP stimulation in LRP−/−/LDLR−/− mice and decreased foam cell formation in the macrophages isolated from LRP−/−/LDLR−/− mice. This observation suggests that the LRP family plays an important role in the HNP-induced inflammatory events.
LRP is a large family consisting of several members.49 To identify the specific phenotype of LRP in mediating the HNP-induced inflammation, we examined the effects of HNP8 in leukocyte rolling and platelet activation because only LRP8 is expressed in platelets.40 HNPs alone induce an increase in CD62P expression that was dramatically reduced in the presence of recombinant human LRP8, indicating competitive inhibitory effects between the recombinant human LRP8 and HNPs in platelet activation. Our results also suggest that the HNP-induced platelet activation is independent of the thrombin pathway. The requirement of LRP8 signaling in mediating the HNP-induced inflammatory responses is further supported by using LRP8−/− mice, where reduced leukocyte rolling, platelet activation, and thrombus formation are seen after stimulation with HNPs. The endothelial but not monocyte LRP8 appears to play a major role in mediating the HNP-induced cell-cell interaction, which is supported by our in vitro data following direct stimulation of HNPs on these cell types isolated from wild-type and LRP8−/− mice. Interestingly, our data also suggest that platelet LRP8 is not required for leukocyte adhesion, although platelets have been reported to contribute to leukocyte rolling.51,52
It is noteworthy that LRP8−/− mice do not experience hypercholesterolemia as LDLR knockout mice do53; thus, the HNP-induced platelet activation is likely independent of modification of lipid metabolism, although this issue is not addressed in the present study. Furthermore, LRP8−/− mice have been shown to have decreased platelet activation and prolonged carotid artery occlusion time in response to ADP and thrombin stimulation, compared with littermate controls.54 Our study examining platelet responses to HNPs in LRP8−/− may provide novel ligand-membrane protein interaction in addition to conventional ADP and thrombin pathways.
In summary, HNPs can initiate the interaction between the endothelium and monocytes, induce leukocyte transendothelial migration, and increase foam cell formation and platelet activation and aggregation through LRP8 signaling pathways. The role of HNPs in the pathogenesis of inflammatory cardiovascular diseases warrants further investigation to confirm their potential as a therapeutic target.
Acknowledgments
The authors thank Dr F. Syeda for the initial participation in pilot studies of platelet aggregation assays, M. Mallik for her help in migration assay, and Dr J. Yin for help in the intravital microscopy setting.
Sources of Funding
This work was supported by the Canadian Institutes of Health Research (MOP8558 to A.S.S.; MOP69042 and MOP77818 to H.Z.) and by the Ontario Thoracic Society and McLaughlin Foundation (to H.Z.). Dr Henriques is a recipient of the John D. Schultz Science Student Scholarship, Heart and Stroke Foundation of Ontario and the Alexander Graham Bell Canada Graduate Scholarship, Natural Sciences and Engineering Research Council. Dr Robinson is the Canada Research Chair for Leukocyte Migration. Dr Zhang is a recipient of Ontario Premier’s Research Excellence Award.
Footnotes
Disclosures
None.
References
- 1.Dorweiler B, Torzewski M, Dahm M, Kirkpatrick CJ, Lackner K, Vahl C. Subendothelial infiltration of neutrophil granulocytes and liberation of matrix-destabilizing enzymes in an experimental model of human neointima. Thromb Haemost. 2008;99:373–381. doi: 10.1160/TH07-06-0387. [DOI] [PubMed] [Google Scholar]
- 2.van Leeuwen M, Gijbels MJ, Duijvestijn A, Smook M, van de Gaar MJ, Heeringa P, de Winther MP, Tervaert JW. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:84–89. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- 3.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 4.Baetta R, Corsini A. Role of polymorphonuclear neutrophils in atherosclerosis: current state and future perspectives. Atherosclerosis. 2009;210:1–13. doi: 10.1016/j.atherosclerosis.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 5.Rotzius P, Soehnlein O, Kenne E, Lindbom L, Nystrom K, Thams S, Eriksson E. ApoE−/−/lysozyme MEGFP/EGFP mice as a versatile model to study monocyte and neutrophil trafficking in atherosclerosis. Atherosclerosis. 2009;202:111–118. doi: 10.1016/j.atherosclerosis.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 7.Avanzas P, Arroyo-Espliguero R, Cosín-Sales J, Aldama G, Pizzi C, Quiles J, Kaski JC. Markers of inflammation and multiple complex stenoses (pancoronary plaque vulnerability) in patients with non-ST segment elevation acute coronary syndromes. Heart. 2004;90:847–852. doi: 10.1136/hrt.2003.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ovbiagele B, Lynn MJ, Saver JL, Chimowitz MI. Leukocyte count and vascular risk in symptomatic intracranial atherosclerosis. Cerebrovasc Dis. 2007;24:283–288. doi: 10.1159/000105681. [DOI] [PubMed] [Google Scholar]
- 9.Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–670. doi: 10.1161/01.ATV.0000156877.94472.a5. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi H, Mori T, Kawano T, Kono S, Sasaki J, Arakawa K. Band neutrophil count and the presence and severity of coronary atherosclerosis. Am Heart J. 1996;132:9–12. doi: 10.1016/s0002-8703(96)90384-1. [DOI] [PubMed] [Google Scholar]
- 11.Sweetnam PM, Thomas HF, Yarnell JW, Baker IA, Elwood PC. Total and differential leukocyte counts as predictors of ischemic heart disease: the Caerphilly and Speedwell studies. Am J Epidemiol. 1997;145:416–421. doi: 10.1093/oxfordjournals.aje.a009123. [DOI] [PubMed] [Google Scholar]
- 12.Kaski JC, Avanzas P, Arroyo-Espliguero R. Neutrophil count and complex lesions in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2005;25:e112. doi: 10.1161/01.ATV.0000168419.71423.b8. author reply e112. [DOI] [PubMed] [Google Scholar]
- 13.Rho YH, Chung CP, Oeser A, Solus J, Asanuma Y, Sokka T, Pincus T, Raggi P, Gebretsadik T, Shintani A, Stein CM. Inflammatory mediators and premature coronary atherosclerosis in rheumatoid arthritis. Arthritis Rheum. 2009;61:1580–1585. doi: 10.1002/art.25009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng CN, Bjorkstrom NK, Malmberg KJ, Lindbom L, Eriksson EE. Distinct Infiltration of Neutrophils in Lesion Shoulders in ApoE−/− Mice. Am J Pathol. 2010;177:493–500. doi: 10.2353/ajpath.2010.090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TD, Gonzalez ML, Kumar P, Chary-Reddy S, Grammas P, Pereira HA. CAP37, a novel inflammatory mediator: its expression in endothelial cells and localization to atherosclerotic lesions. Am J Pathol. 2002;160:841–848. doi: 10.1016/S0002-9440(10)64907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganz T, Lehrer RI. Defensins. Curr Opin Immunol. 1994;6:584–589. doi: 10.1016/0952-7915(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 17.Rehaume LM, Hancock RE. Neutrophil-derived defensins as modulators of innate immune function. Crit Rev Immunol. 2008;28:185–200. doi: 10.1615/critrevimmunol.v28.i3.10. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Garcia M, Oliva H, Climent N, Garcia F, Gatell JM, Gallart T. Human immature monocyte-derived dendritic cells produce and secrete α-defensins 1–3. J Leukoc Biol. 2007;82:1143–1146. doi: 10.1189/jlb.0507295. [DOI] [PubMed] [Google Scholar]
- 19.Kokryakov VN, Harwig SS, Panyutich EA, Shevchenko AA, Aleshina GM, Shamova OV, Korneva HA, Lehrer RI. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993;327:231–236. doi: 10.1016/0014-5793(93)80175-t. [DOI] [PubMed] [Google Scholar]
- 20.Quinn K, Henriques M, Parker T, Slutsky AS, Zhang H. Human neutrophil peptides: a novel potential mediator of inflammatory cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2008;295:H1817–H1824. doi: 10.1152/ajpheart.00472.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ihi T, Nakazato M, Mukae H, Matsukura S. Elevated concentrations of human neutrophil peptides in plasma, blood, and body fluids from patients with infections. Clin Infect Dis. 1997;25:1134–1140. doi: 10.1086/516075. [DOI] [PubMed] [Google Scholar]
- 22.Panyutich AV, Panyutich EA, Krapivin VA, Baturevich EA, Ganz T. Plasma defensin concentrations are elevated in patients with septicemia or bacterial meningitis. J Lab Clin Med. 1993;122:202–207. [PubMed] [Google Scholar]
- 23.Barnathan ES, Raghunath PN, Tomaszewski JE, Ganz T, Cines DB, Higazi Aa-R. Immunohistochemical localization of defensin in human coronary vessels. Am J Pathol. 1997;150:1009–1020. [PMC free article] [PubMed] [Google Scholar]
- 24.Higazi AA, Lavi E, Bdeir K, Ulrich AM, Jamieson DG, Rader DJ, Usher DC, Kane W, Ganz T, Cines DB. Defensin stimulates the binding of lipoprotein (a) to human vascular endothelial and smooth muscle cells. Blood. 1997;89:4290–4298. [PubMed] [Google Scholar]
- 25.Nassar H, Lavi E, Akkawi S, Bdeir K, Heyman SN, Raghunath PN, Tomaszewski J, Higazi AA. α-Defensin: link between inflammation and atherosclerosis. Atherosclerosis. 2007;194:452–457. doi: 10.1016/j.atherosclerosis.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 26.Krylov AV, Kisseleva EP, Aleshina GM, Shamova OV, Kokryakov VN. Effects of defensin and lactoferrin on functional activity of endothelial cells in vitro. Bull Exp Biol Med. 2007;144:331–334. doi: 10.1007/s10517-007-0325-2. [DOI] [PubMed] [Google Scholar]
- 27.Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Neutrophil antimicrobial peptide α-defensin causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2006;43:357–363. doi: 10.1016/j.jvs.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Syeda F, Tullis E, Slutsky AS, Zhang H. Human neutrophil peptides upregulate expression of COX-2 and endothelin-1 by inducing oxidative stress. Am J Physiol Heart Circ Physiol. 2008;294:H2769–H2774. doi: 10.1152/ajpheart.00211.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bdeir K, Cane W, Canziani G, Chaiken I, Weisel J, Koschinsky ML, Lawn RM, Bannerman PG, Sachais BS, Kuo A, Hancock MA, Tomaszewski J, Raghunath PN, Ganz T, Higazi AA, Cines DB. Defensin promotes the binding of lipoprotein (a) to vascular matrix. Blood. 1999;94:2007–2019. [PubMed] [Google Scholar]
- 30.Higazi AA, Nassar T, Ganz T, Rader DJ, Udassin R, Bdeir K, Hiss E, Sachais BS, Williams KJ, Leitersdorf E, Cines DB. The α-defensins stimulate proteoglycan-dependent catabolism of low-density lipoprotein by vascular cells: a new class of inflammatory apolipoprotein and a possible contributor to atherogenesis. Blood. 2000;96:1393–1398. [PubMed] [Google Scholar]
- 31.Nassar T, Akkawi S, Bar-Shavit R, Haj-Yehia A, Bdeir K, Al-Mehdi AB, Tarshis M, Higazi AA. Human α-defensin regulates smooth muscle cell contraction: a role for low-density lipoprotein receptor-related protein/α2-macroglobulin receptor. Blood. 2002;100:4026–4032. doi: 10.1182/blood-2002-04-1080. [DOI] [PubMed] [Google Scholar]
- 32.Khine AA, Del Sorbo L, Vaschetto R, Voglis S, Tullis E, Slutsky AS, Downey GP, Zhang H. Human neutrophil peptides induce interleukin-8 production through the P2Y6 signaling pathway. Blood. 2006;107:2936–2942. doi: 10.1182/blood-2005-06-2314. [DOI] [PubMed] [Google Scholar]
- 33.Vaschetto R, Grinstein J, Del Sorbo L, Khine AA, Voglis S, Tullis E, Slutsky AS, Zhang H. Role of human neutrophil peptides in the initial interaction between lung epithelial cells and CD4+ lymphocytes. J Leukoc Biol. 2007;81:1022–1031. doi: 10.1189/jlb.0706435. [DOI] [PubMed] [Google Scholar]
- 34.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- 35.Stein S, Lohmann C, Schafer N, Hofmann J, Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Boren J, McBurney MW, Landmesser U, Luscher TF, Matter CM. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 31:2301–2309. doi: 10.1093/eurheartj/ehq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Cai S, Peterson BR, Kris-Etherton PM, Heuvel JP. Development of a cell-based, high-throughput screening assay for cholesterol efflux using a fluorescent mimic of cholesterol. Assay Drug Dev Technol. 2011;9:136–146. doi: 10.1089/adt.2010.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reheman A, Yang H, Zhu G, Jin W, He F, Spring CM, Bai X, Gross PL, Freedman J, Ni H. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2009;113:1809–1817. doi: 10.1182/blood-2008-04-148361. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Porro G, Orzech N, Mullen B, Liu M, Slutsky AS. Neutrophil defensins mediate acute inflammatory response and lung dysfunction in dose-related fashion. Am J Physiol Lung Cell Mol Physiol. 2001;280:L947–L954. doi: 10.1152/ajplung.2001.280.5.L947. [DOI] [PubMed] [Google Scholar]
- 39.Kuebler WM, Kuhnle GE, Groh J, Goetz AE. Leukocyte kinetics in pulmonary microcirculation: intravital fluorescence microscopic study. J Appl Physiol. 1994;76:65–71. doi: 10.1152/jappl.1994.76.1.65. [DOI] [PubMed] [Google Scholar]
- 40.Riddell DR, Vinogradov DV, Stannard AK, Chadwick N, Owen JS. Identification and characterization of LRP8 (apoER2) in human blood platelets. J Lipid Res. 1999;40:1925–1930. [PubMed] [Google Scholar]
- 41.Soehnlein O, Weber C. Myeloid cells in atherosclerosis: initiators and decision shapers. Semin Immunopathol. 2009;31:35–47. doi: 10.1007/s00281-009-0141-z. [DOI] [PubMed] [Google Scholar]
- 42.Roman RM, Camargo PV, Borges FK, Rossini AP, Polanczyk CA. Prognostic value of myeloperoxidase in coronary artery disease: comparison of unstable and stable angina patients. Coron Artery Dis. 2010;21:129–136. doi: 10.1097/MCA.0b013e328333f50d. [DOI] [PubMed] [Google Scholar]
- 43.Zaric J, Ruegg C. Integrin-mediated adhesion and soluble ligand binding stabilize COX-2 protein levels in endothelial cells by inducing expression and preventing degradation. J Biol Chem. 2005;280:1077–1085. doi: 10.1074/jbc.M410006200. [DOI] [PubMed] [Google Scholar]
- 44.Daugherty A, Webb NR, Rateri DL, King VL. Thematic review series: the immune system and atherogenesis: cytokine regulation of macrophage functions in atherogenesis. J Lipid Res. 2005;46:1812–1822. doi: 10.1194/jlr.R500009-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Soehnlein O, Weber C, Lindbom L. Neutrophil granule proteins tune monocytic cell function. Trends Immunol. 2009;30:538–546. doi: 10.1016/j.it.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Lindemann S, Kramer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost. 2007;5(suppl 1):203–211. doi: 10.1111/j.1538-7836.2007.02517.x. [DOI] [PubMed] [Google Scholar]
- 47.Bigalke B, Lindemann S, Ehlers R, Seizer P, Daub K, Langer H, Schonberger T, Kremmer E, Siegel-Axel D, May AE, Gawaz M. Expression of platelet collagen receptor glycoprotein VI is associated with acute coronary syndrome. Eur Heart J. 2006;27:2165–2169. doi: 10.1093/eurheartj/ehl192. [DOI] [PubMed] [Google Scholar]
- 48.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 49.Spijkers PP, da Costa Martins P, Westein E, Gahmberg CG, Zwaginga JJ, Lenting PJ. LDL-receptor-related protein regulates β2-integrin-mediated leukocyte adhesion. Blood. 2005;105:170–177. doi: 10.1182/blood-2004-02-0498. [DOI] [PubMed] [Google Scholar]
- 50.Bdeir K, Higazi AA, Kulikovskaya I, Christofidou-Solomidou M, Vinogradov SA, Allen TC, Idell S, Linzmeier R, Ganz T, Cines DB. Neutrophil α-defensins cause lung injury by disrupting the capillary-epithelial barrier. Am J Respir Crit Care Med. 2010;181:935–946. doi: 10.1164/rccm.200907-1128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Lang S, Zhai Z, Li L, Kahr WH, Chen P, Brkic J, Spring CM, Flick MJ, Degen JL, Freedman J, Ni H. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood. 2009;114:425–436. doi: 10.1182/blood-2008-03-145821. [DOI] [PubMed] [Google Scholar]
- 52.Totani L, Evangelista V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol. 2010;30:2357–2361. doi: 10.1161/ATVBAHA.110.207480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evangelho JS, Casali KR, Campos C, De Angelis K, Veiga AB, Rigatto K. Hypercholesterolemia magnitude increases sympathetic modulation and coagulation in LDLr knockout mice. Auton Neurosci. 2011;159:98–103. doi: 10.1016/j.autneu.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Robertson JO, Li W, Silverstein RL, Topol EJ, Smith JD. Deficiency of LRP8 in mice is associated with altered platelet function and prolonged time for in vivo thrombosis. Thromb Res. 2009;123:644–652. doi: 10.1016/j.thromres.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]