

SUMMARY
Tumor necrosis factor (TNF) induces necroptosis, a RIPK3/MLKL-dependent form of inflammatory cell death. In response to infection by Gram-negative bacteria, multiple receptors on macrophages including TLR4, TNF and type I IFN receptors are concurrently activated but it is unclear how they crosstalk to regulate necroptosis. We report that TLR4 activates CASPASE-8 to cleave and remove the deubiquitinase CYLD in a TRIF- and RIPK1-dependent manner to disable necroptosis in macrophages. Inhibiting CASPASE-8 leads to CYLD-dependent necroptosis caused by the TNF produced in response to TLR4 ligation. While LPS-induced necroptosis was abrogated in Tnf−/− macrophages, a soluble TNF antagonist was not able to do so in Tnf+/+ macrophages, indicating that necroptosis occurs in a cell-autonomous manner. Surprisingly, TNF-mediated auto-necroptosis of macrophages requires type I IFN, which primes the expression of key necroptosis-signaling molecules including TNFR2 and MLKL. Thus, the TNF necroptosis pathway is regulated by both negative and positive crosstalk.
Graphical Abstract
INTRODUCTION
Multiple forms of programmed cell death occur following microbial infection, serving to eliminate infected cells and to mount an appropriate host response (Campisi et al., 2014; Vanden Berghe et al., 2014). Apoptosis, which is predominantly dependent on effector caspases such as CASPASE-3 and -7, is thought to generate a tolerogenic response if it occurs in the absence of an inflammatory signal. Pyroptosis, which is dependent on CASPASE-1 and -11, occurs following activation of the inflammasome by microbial products. Pyroptosis serves to eradicate infected cells and the release of cellular contents and damage-associated molecular patterns (DAMPs) following plasma membrane permeabilization amplifies the inflammatory response (Bergsbaken et al., 2009; Chen and Nunez, 2010). In contrast to apoptosis and pyroptosis, which are dependent on various caspases, necroptosis or programmed necrosis has recently emerged as a form of cell death that occurs in the absence of caspase activity. Similar to pyroptosis, necroptosis is also characterized by plasma membrane permeabilization with the release of DAMPs and thus also induces a pro-inflammatory response. Necroptosis may allow the host to circumvent the blockade of caspase-dependent death pathways that may be imposed by a pathogen that encodes caspase inhibitors to block apoptosis or pyroptosis and to retain the ability to mount an inflammatory response to signal danger (Chan et al., 2003; Mocarski et al., 2011; Upton et al., 2010). In this regard, inhibition of host caspases by pathogens and subsequent induction of necroptosis functions effectively as a pathogen-sensing event.
One of the best-characterized inducers of necroptotic death is the cytokine TNF, which paradoxically, can also induce a cell survival response within the same cell. Which response is generated is dependent on the ubiquitination status of the signaling molecule RIPK1 following ligation of TNF receptor 1 (TNFR1); non-degradative Lys63-linked ubiquitination of RIPK1 leads to cell survival whereas inhibiting ubiquitination of RIPK1 leads to necroptosis (Justus and Ting, 2015). In some cellular models, blocking ubiquitination (often using SMAC mimetics) causes RIPK1 to first initiate a caspase-signaling cascade leading to apoptosis (O’Donnell et al., 2007; Wang et al., 2008), but if caspases are also blocked (often using zVAD-fmk), then RIPK1 initiates necroptosis (He et al., 2009; O’Donnell et al., 2011). In other cellular models, blocking caspases is sufficient to trigger necroptosis in the presence of TNF (O’Donnell et al., 2011). In the latter models, the fact that a caspase inhibitor switches the TNF response from survival to necroptosis indicates that a caspase normally generates a pro-survival signal. When that survival signal is blocked, necroptosis is switched on. The molecular mechanism underlying this survival versus necroptosis switch has been clarified over the last few years. Following TNFR1 ligation, CASPASE-8, in a complex with FADD and c-FLIP, delivers a pro-survival signal (Dillon et al., 2012) by cleaving and removing the tumor suppressor CYLD (O’Donnell et al., 2011). CYLD is a deubiquitinating enzyme that is essential for TNF-induced necroptosis (Hitomi et al., 2008; O’Donnell et al., 2011; Vanlangenakker et al., 2010). It disassembles Lys63-linked ubiquitination from RIPK1, a requisite step for necroptosis. Removal of CYLD by CASPASE-8 sustains the ubiquitination of RIPK1, leading to a survival response. Thus, the CASPASE-8:CYLD interaction is an early switch that determines survival versus necroptotic death in the TNFR1 pathway.
With the discovery of RIPK3 as an essential molecule in TNF-induced necroptosis (Cho et al., 2009; He et al., 2009; Zhang et al., 2009), the physiological and patho-physiological roles of necroptosis are starting to become clearer. Excessive RIPK3-dependent necroptosis, often revealed by the genetic deletion of CASPASE-8, leads to embryonic lethality (Kaiser et al., 2011; Oberst et al., 2011), mucosal inflammation (Gunther et al., 2011; Welz et al., 2011) and an impaired T cell response (Ch’en et al., 2011). Furthermore, RIPK3-dependent necroptosis has been reported to be beneficial as well as detrimental for the host during bacterial and viral infections (Cho et al., 2009; Duprez et al., 2011; Polykratis et al., 2014; Robinson et al., 2012; Upton et al., 2010). These in vivo studies indicated that RIPK3-dependent necroptosis is activated subsequent to an interaction between the pathogen and the host’s pathogen-sensing innate cells. Following recognition of LPS derived from Gram-negative bacteria by TLR4 on macrophages, a signature response is the production of TNF. This cytokine is a potent inducer of necroptosis and yet surprisingly, little is known about what effect auto-produced TNF may have on necroptosis in macrophages. In addition to TNF, TLR4 stimulation also leads to the production of type I interferon (IFN) α and β. Type I IFN has also been reported to trigger necroptosis (McComb et al., 2014a; Robinson et al., 2012; Thapa et al., 2013) but whether type I IFN participates in TNF-mediated necroptosis is unclear.
We report here that TLR4, via the TRIF signaling adaptor, rapidly activates CASPASE-8 to cleave and remove CYLD. This CASPASE-8-mediated cleavage event has a self-preservation function because when it is blocked, macrophages undergo auto-necroptosis caused by the TNF that is produced. Furthermore, the ability of TNF to induce necroptosis in macrophages requires IFNα/β priming because in its absence, macrophages have defective expression of key necroptosis-signaling molecules.
RESULTS
TLR4 activates CASPASE-8 to eliminate the pro-necroptotic molecule CYLD
We previously demonstrated that the deubiquitinating enzyme CYLD is the key substrate cleaved by CASPASE-8 to block TNF-mediated necroptosis (O’Donnell et al., 2011). Upon TNFR1 ligation, CYLD is cleaved by CASPASE-8 at aspartate 215, resulting in the generation of a N-terminal 25 kDa fragment and degradation of the C-terminal fragment containing the catalytic domain (O’Donnell et al., 2011). Since TNF is produced in response to LPS during bacterial infection, we surmised that TNF would activate CASPASE-8 in an autocrine manner to cleave CYLD. Loss of CYLD would then prevent necroptosis and sustain cell survival. Therefore, we sought to confirm that CASPASE-8-mediated cleavage of CYLD occurs after TLR4 ligation. When wild type BMDMs were stimulated with LPS, levels of the 107 kD full-length CYLD (p107) decreased in a time- and dose-dependent manner, coinciding with the appearance of the 25 kD CYLD fragment (p25) generated by CASPASE-8-mediated cleavage (Figure 1A and 1B). This cleavage fragment was evident as early as 30 minutes, implying that CASPASE-8 activity is rapidly induced. This is due to TLR4 as it was not observed in Tlr4−/− BMDMs (Supplemental Figure 1A). The ability of other TLR ligands to induce CYLD cleavage was also tested and only TLR3 and TLR4 ligands could do so after a 4-hour stimulation (Supplemental Figure 1B). Infection of BMDMs with two Gram-negative bacteria, Escherichia coli strain LF82 and Citrobacter rodentium, similarly resulted in the proteolytic processing of CYLD (Figure 1C). Cleavage of CYLD was inhibited by the pan-caspase inhibitor z-VAD-fmk (Supplemental Figure 1C), indicating that this is dependent on the proteolytic activity of caspases. Similar to murine BMDMs, stimulation of the human monocytic cell line THP-1 with LPS also resulted in caspase-dependent cleavage of CYLD (Supplemental Figure 1D). To confirm that the FADD/CASPASE-8 complex cleaves CYLD in macrophages, we compared BMDMs from Ripk3−/−Casp8−/− or Ripk3−/−Fadd−/− mice to Ripk3−/− mice since the Ripk3 deletion rescued the embryonic lethality of the Casp8−/− and Fadd−/− single knockout mice (Dillon et al., 2012; Kaiser et al., 2011; Oberst et al., 2011). The appearance of the CYLD p25 fragment following LPS stimulation and bacterial infection was detected in Ripk3−/− BMDMs, but not in Casp8−/−Ripk3−/− (Figure 1D) or Fadd−/−Ripk3−/− BMDMs (Supplemental Figure 1E). Thus, the FADD/CASPASE-8 complex rapidly cleaves CYLD after TLR4 stimulation.
FIGURE 1. TLR4 activates CASPASE-8 to eliminate CYLD.

(A–B) BMDMs from wild type mice were treated with 100 ng/ml of LPS for the indicated times (A) or with the indicated concentrations of LPS for 4 hours (B). (C) BMDMs from wild type BMDMs were infected with viable E. coli or C. rodentium for 4 hours. (D) BMDMs from wild type, Ripk3−/−, and Ripk3−/−Casp8−/− mice were stimulated with either LPS (100 ng/ml), E. coli (MOI 1), or C. rodentium (MOI 1) for the indicated times. Lysates were subject to western blot analysis using CYLD (D1A10), RIPK3, CASPASE-8, and GAPDH antibodies. All experiments shown are representative of at least three independent experiments with similar results.
TLR4-TRIF signaling and not TNF mediates CASPASE-8 dependent cleavage of CYLD
As discussed earlier, our expectation was that LPS-induced cleavage of CYLD would be dependent on auto-production of TNF, which we sought to confirm using Tnf knockout BMDMs. Surprisingly, the cleavage of CYLD was not dependent on TNF since it was observed in both Tnf+/+ and Tnf−/− BMDMs following LPS stimulation (Figure 2A). To test the possibility that the CASPASE-8-mediated cleavage of CYLD may be secondary to another cytokine produced, we pre-treated BMDMs with the protein synthesis inhibitor cycloheximide prior to LPS stimulation. Cycloheximide did not block CYLD cleavage and degradation, whereas it efficiently blocked the re-synthesis of IκBα following LPS stimulation (Supplemental Figure S2A). Therefore, cleavage of CYLD by CASPASE-8 following TLR4 stimulation is independent of de novo protein synthesis. This suggested a direct activation of CASPASE-8 by TLR4, prompting us to examine the involvement of MYD88 and TRIF. Cleavage of CYLD induced by TLR4 stimulation was not affected in the Myd88−/− BMDMs (Figure 2B), but was abrogated in the Trif−/− BMDMs (Figure 2C), indicating that CASPASE-8 activation was dependent on TRIF. Consistent with the requirement for TRIF in the activation of CASPASE-8, the two molecules are able to form a complex when co-expressed in HEK293 cells (Supplementary Figure S2B). Thus, TLR4 activates CASPASE-8 in a TRIF-dependent but MYD88-independent manner to eliminate CYLD, and not via autocrine production of cytokines.
FIGURE 2. TLR4-TRIF signaling and not TNF mediates CASPASE-8 dependent cleavage of CYLD.

(A) Wild type and Tnf−/− BMDMs were treated with 100 ng/ml of LPS for 4 hours. (B–C) Wild type, Myd88−/−, and Trif−/− BMDMs were treated with 10 ng/ml of LPS for the indicated times. (D) Wild type BMDMs were pre-treated with 30 μM Necrostatin-1 prior to stimulation with LPS for 4 hours. (E) Immortalized BMDMs generated from RosaCreERT2Ripk1fl/fl mice were treated with ethanol or with tamoxifen (4-OHT) to delete RIPK1. Cells were then treated with 100 ng/ml of LPS for the indicated times. Lysates were blotted as indicated. All experiments shown are representative of at least three independent experiments with similar results.
Since the TNF signaling adaptor RIPK1 has been previously implicated in TLR4 signaling (Cusson-Hermance et al., 2005; Meylan et al., 2004) and RIPK1 and CASPASE-8 can associate (Lin et al., 1999), we asked whether RIPK1 has a role in regulating the activation of CASPASE-8 downstream of TLR4. Treatment with the RIPK1 kinase inhibitor Necrostatin-1 prior to LPS stimulation did not inhibit the cleavage of CYLD in BMDMs (Figure 2D), suggesting that the kinase activity of RIPK1 is not required. However, RIPK1 could still regulate the function of CASPASE-8 through a kinase-independent mechanism. We utilized immortalized BMDMs generated from RosaCreERT2Ripk1fl/fl mice and treated those cells with tamoxifen (4-OHT) to acutely delete Ripk1 (Roderick et al., 2014). LPS-induced cleavage of CYLD was abrogated in the RIPK1-deficient macrophages, demonstrating that this is dependent on RIPK1 (Figure 2E). This dependency on RIPK1 for CYLD cleavage may be related to the reduced level of CASPASE-8 observed in RIPK1-deficient BMDMs suggesting that RIPK1 regulates CASPASE-8 expression in macrophages via a mechanism that is unclear at this point.
TLR4 induces auto-necroptosis in macrophages via production of TNF
It was reported that blocking caspase activity in LPS-stimulated murine macrophages resulted in necroptosis (He et al., 2011; Kaiser et al., 2013). Similarly, human THP-1 cells undergo necroptosis when stimulated with LPS in the presence of zVAD-fmk (Supplementary Figure 2C). Although auto-produced TNF is not required for CASPASE-8 to cleave CYLD following TLR4 stimulation (Figure 2A), whether auto-produced TNF is required for necroptosis once CASPASE-8 is blocked remains questionable. Previous studies have suggested that TLR4 induces necroptosis independently of TNF after examining the response of Tnf−/− or Tnfr1−/−Tnfr2−/− double knockout macrophages to high doses of LPS (He et al., 2011; Kaiser et al., 2013). In light of the possibility that the effect of a genetic deficiency in a Toll-like receptor signaling molecule can be overlooked due to the use of high amount of the ligand (Bonham et al., 2014; Horng et al., 2002), we conducted our experiments using a lower range of LPS. BMDMs from Tnf+/+ and Tnf−/− mice were stimulated with varying concentrations of LPS up to 1 ng/ml for 16 hours. Under these conditions, LPS alone did not induce cell death in wild type BMDMs whereas in the presence of zVAD-fmk, 1 ng/ml of LPS was sufficient to achieve near maximal death of BMDMs (Figure 3A), a concentration that is 20–500 fold lower than that used previously (He et al., 2011; Kaiser et al., 2013). Using this lower range of LPS, necroptosis was abrogated in the Tnf−/− BMDMs (Figure 3A). When we increased the concentration of LPS by up to 100-fold more, the requirement for TNF was diminished even though there was still a difference in viability between Tnf+/+ and Tnf−/− macrophages (Supplemental Figure 3A). Thus at lower doses of LPS, there is a strong dependency on TNF to induce necroptosis whereas at higher doses, there is diminished dependency. Similar to LPS, E. coli and C. rodentium infection at a low multiplicity of infection (MOI) resulted in necroptosis that is dependent on TNF (Figure 3B). The LPS or bacterial-induced necroptosis in wild type BMDMs was also inhibited by Necrostatin-1 (Nec-1), indicating a RIPK1 kinase dependency (Supplemental Figure 3B and 3C). These results demonstrate that Gram-negative bacteria, as well as stimulation with limiting doses of LPS, induce necroptosis that is dependent on autocrine production of TNF.
FIGURE 3. TLR4 induces auto-necroptosis in macrophages via production of TNF.

(A–B) BMDMs from Tnf+/+ or Tnf−/− mice were pre-treated with 25 μM zVAD-fmk or DMSO prior to stimulation with the indicated concentrations of LPS (A) or infected with E. coli or C. rodentium at the indicated multiplicity of infection (MOI) (B) for 16 hours. Cell death was measured by propidium iodide staining. Data is presented as the mean of three independent experiments performed in triplicate, ± standard deviation. Statistics were performed using T-test. * p<0.05, ** p<0.01, *** p<0.001. (C) Wild type BMDMs were treated with control-FC or TNFR1-FC, in addition to either LPS or TNF, for 24 hours. Viability was determined by CellTiter-Glo assays. Data is presented as the mean of three independent experiments performed in triplicate, ± standard deviation.
To further confirm the role of autocrine TNF, we examined the role of MYD88 since this TLR signaling adaptor protein is required for TNF production in response to LPS (Kawai et al., 1999). Myd88+/+ BMDMs succumbed to necroptosis when treated with LPS or infected with bacteria whereas Myd88−/− BMDMs survived (Supplemental Figure S3D). This result is consistent with the fact that LPS-induced TNF expression is MYD88-dependent (Kawai et al., 1999) and which remains true under necroptotic conditions (Supplemental Figure S3E). Since the other TLR4 signaling adaptor TRIF has been implicated in TLR3 and TLR4-induced necroptosis (He et al., 2011; Kaiser et al., 2013; McComb et al., 2014a) we also examined whether TRIF is required for necroptosis in our low-dose LPS model. Trif−/− BMDMs were more resistant to necroptosis in response to LPS or bacterial infection (Supplemental Figure S3F). The observation that TLR4-induced necroptosis requires TNF (Figure 3A), as well as TRIF (Supplemental Figure S3F), was somewhat puzzling since TNF is not known to signal through TRIF. In addition to its well-known role in the induction of type IFN, TRIF also has a less appreciated role in TNF production as suggested by a previous report (Gais et al., 2010). We therefore considered the possibility that TNF expression may be compromised in the TRIF-deficient BMDMs, which could provide one explanation for why they are more resistant to necroptosis. Consistent with this possibility, TNF expression after LPS stimulation was defective in the TRIF-deficient BMDMs, regardless of the absence or presence of zVAD-fmk (Supplemental Figure S3G).
In addition to using the Tnf and Myd88 knockouts to demonstrate a requirement for TNF in inducing necroptosis following TLR4 ligation, we also utilized a soluble TNF antagonist to inhibit necroptosis. We confirmed the effectiveness of the murine TNFR1-Fc fusion as an antagonist, as it was able to fully block necroptosis of BMDMs induced by the addition of exogenous TNF (Figure 3C). Surprisingly, it had only a modest effect on necroptosis induced by LPS (Figure 3C). Since the genetic deletion of Tnf blocked necroptosis induced by LPS but the soluble mTNFR1-Fc did not, this strongly suggests that death is not dependent on secreted TNF. This observation suggests that death mediated by TNF occurs in a cell-autonomous manner as a consequence of a ligand-receptor interaction in an intracellular compartment that is not accessible to the soluble mTNFR1-Fc antagonist. We have termed this process auto-necroptosis.
Eliminating CYLD protects macrophages against TLR4-induced auto-necroptosis
We previously showed that removal of CYLD by CASPASE-8 generates a survival response in MEFs but when this cleavage event is inhibited, CYLD-dependent necroptosis ensues (O’Donnell et al., 2011). Removal of CYLD following TLR4 stimulation (Figure 1) may serve a similar pro-survival and anti-necroptotic function in macrophages. Therefore, we examined the functional consequence of the loss of CYLD using BMDMs from control or Cyld conditional knockout mice. These conditional knockout mice were generated by crossing Cyldflox/flox mice, which contains LoxP sites flanking exons 8 and 9, with the LysM-Cre strain to selectively delete CYLD in macrophages (Cyld CKO) (Supplemental Figure S4A). Wild type macrophages succumbed to necroptosis in response to LPS and zVAD-fmk whereas CKO cells survived (Figure 4A). A similar result was observed with E. coli or C. rodentium infection (Figure 4B). To rule out the possibility the CYLD-deficient BMDMs were resistant to necroptosis due to a lack of TNF, we examined TNF expression in the CYLD-deficient BMDMs. Under necroptotic conditions, TNF production was slightly elevated in the CYLD-deficient macrophages, arguing against defective TNF production as a reason for their resistance to necroptosis (Supplemental Figure S4B).
FIGURE 4. Elimination of CYLD protects macrophages from TLR4-induced auto-necroptosis.

(A) BMDMs from Cyldflox/flox (CYLD WT) and LysM-Cre x Cyldflox/flox mice (CYLD conditional knockout, or CKO) were pre-treated with DMSO or 25 μM zVAD-fmk and stimulated with LPS for 16 hours. (B) BMDMs from CYLD WT or CYLD CKO were pre-treated with 25 μM zVAD-fmk prior to infection with E. coli or C. rodentium for 16 hours at the indicated MOI. (A–B) Cell death was analyzed as in Figure 3A. * p<0.05, ** p<0.01, *** p<0.001. (C) Cyld+/+ or Cyld−/− BMDMs pre-treated with 25 μM zVAD-fmk were stimulated with 1 ng/ml of LPS for the indicated time. Triton-soluble lysates were subject to non-reducing SDS-PAGE, and blotted with anti-MLKL and β-actin. The experiment shown is representative of three independent experiments.
MLKL was recently identified as a key effector molecule of necroptosis. It is phosphorylated by RIPK3 to form an oligomeric structure that translocates to the plasma membrane to disrupt membrane integrity and this can be used as a biochemical signature for necroptosis (Cai et al., 2013; Chen et al., 2013; Dondelinger et al., 2014; Wang et al., 2014). TLR4 engagement in the presence of zVAD-fmk did not lead to MLKL oligomerization in CYLD-deficient cells (Figure 4C), demonstrating that removal of CYLD prevents TLR4 from inducing necroptosis. Since TLR4-induced production of TNF could potentially also activate apoptosis, we tested to see if CYLD was involved in apoptosis. We stimulated BMDMs with LPS after pre-treatment with cycloheximide (CHX) in order to sensitize the cells to apoptosis. Wild type and CYLD-deficient BMDMs were equally sensitive to apoptosis induced by LPS in the presence of cycloheximide (Supplemental Figure S4C) indicating that CYLD is not required for this model of apoptosis. As a control in this experiment, LPS-induced necroptosis was abrogated in CYLD-deficient macrophages (Supplemental Figure S4C).
While removal of CYLD by CASPASE-8 will render macrophages resistant to necroptosis, the possibility remains that CYLD may not be the only substrate cleaved by CASPASE-8 to block necroptosis. To test whether there may be additional CASPASE-8 substrates, we reconstituted Cyld−/− BMDMs with wild type Cyld or the cleavage-resistant CyldD215A mutant (O’Donnell et al., 2011) and examined whether the D215A mutation negated the need for caspase inhibition. In contrast to our previous observation in MEFs (O’Donnell et al., 2011), macrophages reconstituted with the cleavage-resistant CYLD-D215A did not undergo necroptosis unless zVAD-fmk was present (Supplementary Figure S4D). This suggests that CYLD may not be the only substrate cleaved by CASPASE-8 following TLR4 ligation. RIPK1 is another necroptosis-signaling molecule that has been implicated as a substrate for CASPASE-8 (Lin et al., 1999; Martinon et al., 2000; McComb et al., 2014b). We therefore used an antibody raised against the N-terminal region of RIPK1 to detect its potential cleavage. LPS stimulation induced the appearance of several smaller fragments of RIPK1 that were abrogated in the presence of zVAD-fmk (Supplementary Figure S4E). This LPS-inducible appearance of these RIPK1 fragments is consistent with the possibility that multiple substrates including CYLD and RIPK1 are cleaved by CASPASE-8 when TLR4 is stimulated. Their removal in a TRIF-dependent manner likely generates a self-preservation signal by preventing the induction of auto-necroptosis in macrophages from the ongoing TNF production during an infection.
TNF-dependent auto-necroptosis in macrophages also requires priming by type I IFN
In addition to TNF production, macrophages also produce type I interferon (IFN) following sensing of Gram-negative bacteria by TLR4 (Doyle et al., 2002; Kawai et al., 2001). Therefore during this interaction between bacteria and macrophages, multiple signaling pathways are engaged including the TLR4, TNFR and IFNAR pathways. How these different pathways cross-regulate each other to control necroptosis is unclear. There is increasing evidence that IFNs can regulate necroptosis (Doyle et al., 2002; Kawai et al., 2001; Robinson et al., 2012; Thapa et al., 2013) but there are conflicting reports on the role of type I IFN in TLR-induced necroptosis (He et al., 2011; Kaiser et al., 2013; McComb et al., 2014a). For these reasons, we wanted to examine whether type I IFN has any role in our low-dose LPS model where we know that autocrine TNF is inducing the necroptosis response. Ifnar1+/+ BMDMs succumbed to necroptosis whereas Ifnar1−/− BMDMs survived (Figure 5A). Similar to Ifnar1−/− BMDMs, Stat2−/− BMDMs were also resistant to low-dose LPS-induced necroptosis confirming a requirement for type I IFN signaling in this model (Supplementary Figure S5A). Infection with E. coli or C. rodentium in the presence of zVAD-fmk similarly induced necroptosis in Ifnar1+/+ but not in Ifnar1−/− BMDMs (Figure 5B). The results from Figures 3A and 5A showed a surprising finding: necroptosis of macrophages following TLR4 ligation requires downstream signaling responses generated by both TNF and type I IFN. This raised the question of the relationship between these two cytokines and suggested that induction of necroptosis by TNF itself maybe dependent on IFNα/β. To test this directly, Ifnar1+/+ and Ifnar1−/− BMDMs were stimulated with murine TNF in the presence of zVAD-fmk. Ifnar1−/− BMDMs were resistant to necroptosis induced by TNF (Figure 5C). The requirement for IFNα/β reveals an unappreciated pathway-extrinsic regulation of the TNF necroptosis pathway in macrophages by another cytokine. IFNα/β may be functioning to directly induce necroptosis following its expression in response to TLR4 stimulation as suggested by others (McComb et al., 2014b) or it may have an indirect role by priming the cells to respond to TNF. To discriminate between the two mechanisms, we used an IFNAR1-neutralizing monoclonal antibody previously shown to be effective in blocking type I IFN responses (Sheehan et al., 2006). Surprisingly, anti-IFNAR1 did not inhibit necroptosis induced by LPS (Figure 5D) whereas it inhibited STAT1 phosphorylation (Figure 5E), which is known to be dependent on IFNα/β, confirming its neutralizing function. The inability of the neutralizing anti-IFNAR1 to block necroptosis argues against acutely produced IFNα/β as a direct trigger of necroptosis. Rather, the combined results from the Ifnar1−/− BMDMs and the neutralizing anti-IFNAR1 experiments are more consistent with a licensing role for type I IFN in priming macrophages for necroptosis.
FIGURE 5. TLR4-induced auto-necroptosis in macrophages also requires type I IFN.

Ifnar1+/+ and Ifnar1−/− BMDMs were pre-treated with 25 μM zVAD-fmk prior to stimulation with LPS (A), viable E. coli or C. rodentium (B), or murine TNF (C) for 24 hours. (D) Wild type BMDMs were treated with isotype control or 1 ug/ml of IFNAR1 neutralizing antibody prior to stimulation with the indicated concentrations of LPS for 24 hours. Cell death in (A–D) was measured by propidium iodide staining or by CellTiter-Glo as in Figure 3. * p<0.05, ** p<0.01, *** p<0.001. (E) Wild type BMDMs were treated as in (D) prior to stimulation with LPS for 24 hours. Lysates were blotted with antibodies to phosphorylated STAT1 and total STAT1. Experiment shown is representative of at least three experiments performed independently.
Type I IFN licenses TNF-mediated necroptosis by inducing TNFR2 and MLKL expression
Why TNF requires IFNAR signaling to induce necroptosis was unclear. The defect in the TNF necroptosis pathway in the Ifnar1−/− macrophages could be at the level of the TNF receptors or further downstream. Our group and others have shown that in order for TNFR1 to induce necroptosis, ubiquitination of RIPK1 has to be disrupted to switch RIPK1 from a pro-survival to a pro-death signaling molecule (Arslan and Scheidereit, 2011; O’Donnell et al., 2012; O’Donnell and Ting, 2011). Physiologically, this is achieved by the simultaneous ligation of TNFR1 and TNFR2 whereby TNFR2 ligation leads to the rapid degradation of the TRAF2/cIAP1/2 E3 ubiquitin ligase complex (Chan and Lenardo, 2000; Fotin-Mleczek et al., 2002; Li et al., 2002; Ruspi et al., 2013). The loss of TRAF2/cIAP1/2 reduced RIPK1 ubiquitination, facilitating the conversion of RIPK1 to a death-signaling molecule upon TNFR1 ligation. Consistent with this model, TNFR2 was shown to be necessary for necroptosis in T cells (Chan et al., 2003). Therefore, we hypothesized that there may be a defect with TNFR2 expression in the Ifnar1−/− BMDMs. We first confirmed that TNFR2 has a role in TNF-induced necroptosis in macrophages. We treated wild type BMDMs with either murine TNF, which binds both murine TNFR1 and TNFR2, or with human TNF, which binds only to murine TNFR1 (Lewis et al., 1991). Murine TNF induced necroptosis whereas human TNF did not (Figure 6A), suggesting that ligation of both TNFR1 and TNFR2 is needed for necroptosis. Furthermore, murine TNF was able to induce necroptosis in Tnfr2+/+ but not Tnfr2−/− BMDMs (Figure 6B), confirming the requirement for TNFR2.
FIGURE 6. Type I IFN licenses TNF-mediated necroptosis by inducing TNFR2 and MLKL expression.

(A) Wild type BMDMs were stimulated with indicated amounts of murine or human TNF for 24 hours in the presence of zVAD-fmk. (B) Tnfr2+/+ and Tnfr2−/− BMDMs were stimulated with murine TNF for 24 hours in the presence of zVAD-fmk. (A, B) Viability was determined by CellTiter-Glo assays as in Figure 3C. (C) Ifnar1+/+ and Ifnar1−/− BMDMs stimulated with 1 ng/ml of LPS for 24 hours were stained with a PE-conjugated anti-TNFR2. Data is representative of three independent experiments. (D) Necroptosis in Tnfr2+/+ and Tnfr2−/− BMDMs were analyzed as in Figure 3A & B. (E) Ifnar1+/+ or Ifnar1−/− BMDMs were stimulated with the indicated concentrations of LPS for 4 hours. Triton-soluble lysates were blotted with antibodies for CYLD (D1A10), RIPK1, RIPK3, MLKL, and GAPDH. (F) Ifnar1+/+ or Ifnar1−/− BMDMs were pre-treated with 25 μM zVAD-fmk and stimulated with 10 ng/ml of LPS for the indicated times. MLKL oligomerization was analyzed as in Figure 4C. Experiments were repeated twice with similar results. (G) Ifnar1−/− BMDMs were transduced with retroviruses encoding a control protein, TNFR2, or MLKL, as indicated. Lysates from these cells were blotted to confirm MLKL expression. Ifnar1−/− BMDMs transduced with TNFR2 or MLKL were stained with PE-conjugated anti-TNFR2 to confirm TNFR2 expression that is equivalent to that of Ifnar1+/+ BMDMs stimulated with 10 ng/ml of LPS for 24 hours. These cells were stimulated with LPS in the presence of 25uM of zVAD-fmk. Viability was determined by CellTiter-Glo as in Figure 3C.
While TNFR1 expression is quite ubiquitous, TNFR2 expression appears to be more restricted and is induced on macrophages and other hematopoietic cells upon cellular activation (Bethea et al., 1997; de Kossodo et al., 1994; Tannenbaum et al., 1993). We thus hypothesized that TNFR2 expression induced by LPS stimulation is dependent on IFNα/β. Using flow cytometry, we found that LPS induced the expression of TNFR2 in Ifnar1+/+ but not in Ifnar1−/− BMDMs (Figure 6C). Similar to the Ifnar1−/− BMDMs, TNFR2 expression was also compromised in the Stat2−/− BMDMs (Supplementary Figure S5B). However, the level of Tnfr2 mRNA was not reduced in the Stat2−/− BMDMs (Supplementary Figure S5C), suggesting that a STAT2-dependent gene may regulate TNFR2 expression in a post-transcriptional manner. To test the hypothesis that defective TNFR2 expression on Ifnar1−/− BMDMs could affect necroptosis, we compared the sensitivity of Tnfr2+/+ and Tnfr2−/− BMDMs to necroptosis induced by LPS or bacteria infection. Consistent with our hypothesis, Tnfr2−/− BMDMs were more resistant to necroptosis than Tnfr2+/+ BMDMs (Figure 6D), similar to that seen in Tnf−/− BMDMs (Supplemental Figure S5D).
Since IFNα/β induces a large array of genes collectively known as IFN-inducible genes (ISG), it is possible that in addition to TNFR2, IFNAR1 deficiency may also affect the expression of other necroptosis molecules. Of particular interest is MLKL, the RIPK3 kinase substrate that is essential for TNF-induced necroptosis (Sun et al., 2012; Zhao et al., 2012), which was recently shown to be an ISG (Dillon et al., 2014; Thapa et al., 2013). We examined the protein expression level of MLKL in Ifnar1+/+ and Ifnar1−/− BMDMs after stimulation with varying doses of LPS. In contrast to other necroptosis signaling molecules including CYLD, RIPK1 and RIPK3, MLKL protein expression was reduced in Ifnar1−/− BMDMs (Figure 6E). Since MLKL-deficient macrophages were shown to be resistant to necroptosis (Murphy et al., 2013; Wu et al., 2013), the diminished MLKL expression in the Ifnar1−/− BMDMs also likely contributes to their resistance to necroptosis. Accordingly, MLKL oligomerization was also severely impaired in Ifnar1−/− BMDMs (Figure 6F). In Stat2−/− BMDMs, the basal level of MLKL was also reduced (Supplementary Figure S6A), albeit to a lesser degree than in the Ifnar1−/− BMDMs. MLKL oligomerization was also impaired in the Stat2−/− BMDMs (Supplementary Figure S6B). Consistent with Mlkl being an ISG, its mRNA level was significantly reduced in IFNAR1 and STAT2-deficient BMDMs (Supplementary Figure S6C).
These results thus far support the model that defective TNFR2 and MLKL expression in the absence of IFNα/β priming contributes to the resistance of the Ifnar1−/− BMDMs to necroptosis. To test whether TNFR2 and MLKL expression are the only defects or whether additional defects exist, we complemented Ifnar1−/− BMDMs with Tnfr2 or Mlkl. Retroviral transduction of Ifnar1−/− BMDMs with Tnfr2 restored the expression of TNFR2 but was not sufficient to reverse their resistance to necroptosis (Figure 6G). Likewise, retroviral transduction of Ifnar1−/− BMDMs with Mlkl restored their expression of MLKL but this was also insufficient to reverse their resistance to necroptosis (Figure 6G). Simultaneous reconstitution of Ifnar1−/− BMDMs with both TNFR2 and MLKL also failed to reverse the resistance of these cells to necroptosis (Figure 6G). This suggests that in addition to TNFR2 and MLKL, there are additional defects in the TNF necroptosis pathway in the absence of IFNα/β priming. Nonetheless, the reduction in TNFR2 and MLKL observed in the Ifnar1−/− cells plays a key role as evidenced by the resistance of the TNFR2 and MLKL-deficient macrophages to necroptosis. This underscores the critical licensing role played by type I IFN in TNF-mediated necroptosis.
DISCUSSION
This current study shows that the determination of survival versus death in macrophages is controlled by multiple pathways (Figure 7). We demonstrated that TLR4 activates CASPASE-8 to proteolyze and remove CYLD. This removal is a mechanism for protecting against necroptosis that can be triggered by the TNF produced during an infection by Gram-negative bacteria. We had previously reported the existence of an early NF-kappaB-independent but ubiquitin-dependent cell death checkpoint in the TNFR1 pathway (Justus and Ting, 2015; Legarda-Addison et al., 2009; O’Donnell et al., 2007; O’Donnell and Ting, 2010). Thus, TLR4 via its activation of CASPASE-8 sustains this early ubiquitin-dependent checkpoint in the TNFR1 pathway by removing the CYLD deubiquitinase. However, when CASPASE-8 is inhibited, this early ubiquitin-dependent checkpoint is disrupted and macrophages undergo auto-necroptosis mediated by the TNF that is produced during the infection. This process occurs in a cell-autonomous manner, likely mediated by a ligand-receptor interaction in cis. This auto-necroptosis response further requires licensing by type I IFN, which is necessary for the expression of TNFR2, MLKL and potentially other necroptosis-signaling molecules. In the absence of TNFR2 and MLKL, the ability of TNF to induce necroptosis is severely compromised. Our data thus demonstrates an unappreciated pathway-extrinsic regulation of the TNF necroptosis pathway by the TLR4 and type I IFN receptors in a negative and positive manner, respectively.
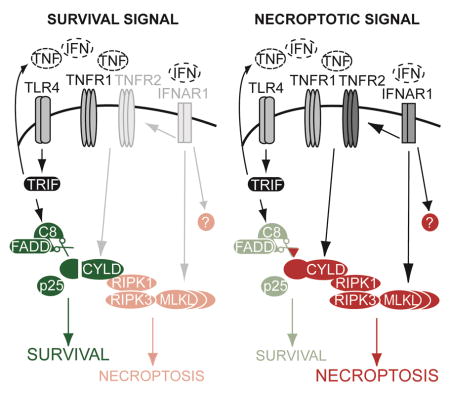
FIGURE 7. Regulation of survival and necroptosis in macrophages.

During an infection, the determination of TNF-induced necroptosis is subject to cross-regulation by other receptor pathways. I: TLR4 activates CASPASE-8 through a TRIF-dependent manner to proteolyze and remove CYLD. CYLD is essential for TNF-induced necroptosis, and so its removal prevents macrophages from undergoing necroptosis that can be induced by the TNF produced during bacterial infection. II: Inhibition of CASPASE-8 stabilizes CYLD and this results in necroptosis mediated by auto-produced TNF signaling via CYLD. Type I IFN plays a licensing role because it is required for the induction of TNFR2, MLKL and other uncharacterized molecules that are essential for TNF-mediated necroptosis.
We previously demonstrated that whether CYLD is cleaved by CASPASE-8 or not is the key determinant of survival versus necroptotic death in MEFs following TNFR1 ligation (O’Donnell et al., 2011). Our current study demonstrates that TLR4, in a TRIF-dependent mechanism, activates CASPASE-8 to remove CYLD and this has an inhibitory effect on the TNF death pathway. We have genetically defined some of requirements for the cleavage of CYLD: it requires TRIF, RIPK1, FADD and CASPASE-8. An interaction between TRIF and CASPASE-8 has been suggested previously (Estornes et al., 2012; Maelfait et al., 2008). Indeed, the two molecules can interact in co-transfection experiments. Alternatively, TRIF can interact with RIPK1 in a RHIM-dependent manner (Kaiser and Offermann, 2005; Meylan et al., 2004) and in turn, RIPK1 can recruit CASPASE-8 (Wang et al., 2008). Therefore it is likely that TRIF, RIPK1, FADD and CASPASE-8 form a proteolytic complex that confers cell survival when macrophages are activated. This remains to be directly confirmed because the analysis of endogenous TRIF-containing complexes in primary macrophages has been hampered by the lack of suitable immunoprecipitating antibodies against TRIF. Our current analysis of how TLR4 regulates CYLD cleavage in macrophages has revealed a couple of aspects that are different from our previous analysis of CYLD cleavage in MEFs. While RIPK1 was not required for CYLD cleavage in response to TNFR1 ligation in MEFs (O’Donnell et al., 2011), it appears to have a role in the TLR4-induced response in macrophages, in part by regulating the expression of CASPASE-8 itself, or RIPK1 could also serve to recruit CASPASE-8 to TRIF. Another differing aspect is that we did not detect any RIPK1 cleavage in response to TNFR1 stimulation in MEFs whereas this was observed in response to TLR4. In conjunction, mutating the cleavage site of CYLD was sufficient to sensitize MEFs to undergo TNF-induced necroptosis and negating the need for caspase inhibition, whereas this was insufficient in macrophages. Thus, multiple substrates may be cleaved by CASPASE-8 in macrophages following TLR4 stimulation, indicating receptor and cell type differences in the regulation of CYLD cleavage.
While TNF is not required for the CYLD cleavage observed upon TLR4 stimulation, it is required for the necroptosis induced upon caspase inhibition. Our result differs from previous studies suggesting that TNF is not required (He et al., 2011; Kaiser et al., 2013; McComb et al., 2014a; Robinson et al., 2012) and this is likely due to the low doses of LPS used in the current study compared to the high doses used in the previous studies. We further confirmed that the LPS results were reflective of that seen during bacterial infection by infecting macrophages with a low MOI of viable C. rodentium and E. coli. The genetic requirement for a molecule in a TLR pathway could be missed when higher doses of ligand are utilized as illustrated by Bonham et al. in their study of the adaptor molecule TIRAP in TLR9 signaling (Bonham et al., 2014). Surprisingly, we were unable to block this TNF-dependent death process using a soluble TNFR1-Fc antagonist indicating that this is not mediated by secreted TNF. Instead, this observation strongly suggests that TNF is engaging its receptors within the cell and activating downstream signaling in a cell-autonomous manner. The biological reason for why macrophages are wired to undergo auto-necroptosis upon caspase inhibition is unclear. We speculate that this may serve a tripwire function in a sentinel cell such as a macrophage where pathogen or host-mediated alterations in caspase activity following an infection could trigger necroptosis as a danger signal.
In contrast to the inhibitory effect TLR4 exerts on the TNF necroptosis pathway, type I IFN receptor signaling exerts a positive effect on this death pathway in a STAT2-dependent manner. Our analysis unequivocally showed that LPS and TNF-induced necroptosis required IFNα/β-mediated responses. This requirement for type I IFN in order for TNF to trigger necroptosis was quite unexpected and prompted us to examine the basis of this crosstalk. We initially directed our attention at TNFR2 and found that its expression after LPS stimulation was impaired in the Ifnar1−/− macrophages and it is required for necroptosis to occur. In addition to TNFR2, expression of MLKL was also reduced in the Ifnar1−/− BMDMs. There are likely additional defects in the Ifnar1−/− cells, as reconstitution with Tnfr2 and Mlkl did not reverse their resistance to death. Since the other molecules essential for TNF necroptosis described to date (CYLD, RIPK1 and RIPK3) are present in the Ifnar1−/− BMDMs, it is likely other molecules essential for necroptosis remain to be characterized.
The requirement for IFNAR1 raises the question whether IFNα/β produced following sensing of microbes directly induces necroptosis, as suggested by a recent report (McComb et al., 2014a), or whether basal level of IFNα/β in BMDMs cultures prime these cells to respond to TNF. Our finding that a neutralizing anti-IFNAR1 did not inhibit necroptosis of BMDMs indicates that the IFNα/β acutely produced in response to LPS does not directly trigger necroptosis. Rather, prior exposure of the BMDMs to IFNα/β is necessary to license and prime the necroptosis pathway via the induction of essential signaling molecules. The requirement of IFNAR1 as an essential regulator for necroptosis is likely restricted to certain cell types as suggested by genetic crosses involving the Casp8−/− mice. The embryonic lethality of Casp8−/− mice was rescued by crossing the mice to Ripk3−/− mice (Kaiser et al., 2011; Oberst et al., 2011) but not by crossing to Stat1−/− mice that are defective in type I IFN signaling (Thapa et al., 2013), indicating that the necroptosis pathway in the cell type affected in the Casp8−/− embryos is not dependent on IFN signaling. A report by Duprez et al demonstrated that RIPK3-dependent necroptosis was responsible for the morbidity and mortality in a mouse model of sterile sepsis induced by TNF (Duprez et al., 2011). Prior to the study by Duprez et al., Huys et al., using a similar model of TNF-induced sepsis, showed that IFNAR1 was essential for driving this disease (Huys et al., 2009), further indicating that the interplay of type I IFN and TNF is relevant in vivo.
In summary, our studies showed that necroptosis of macrophages is regulated in a complex manner by multiple crosstalk between the TLR4, TNF and IFN receptors and highlights the insights gained from studying the TNF death pathway in the context of other pathways that are concurrently activated.
EXPERIMENTAL PROCEDURES
Generation of CYLD conditional knockout mice
Mice expressing loxP-flanked (floxed) Cyld exon 8 and 9 were generated by Ingenious Targeting Laboratories (Ronkonkoma, New York). These mice were backcrossed to C57BL/6 for 10 generations, and were then crossed with LysM-Cre transgenic mice, which express the Cre recombinase from a myeloid-specific promoter, to selectively delete CYLD from monocytes and macrophages (LysM-Cre Cyldflox/flox, or CYLD conditional knockout, or CYLD CKO). For all experiments performed, Cyldflox/flox littermates were used as controls (CYLD WT).
Bone Marrow-Derived Macrophages (BMDMs)
BMDMs were generated by culturing bone marrow progenitors in L929 conditioned medium. Immortalization of BMDM from Ripk1 conditional knockouts was conducted by infection with J2 oncovirus (Roberson and Walker, 1988).
Cell Death Assays
Cell death was analyzed by propidium iodide staining followed by flow cytometry or with the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega).
Western blot analysis
Cellullar lysates obtained using 1% Triton X-100 were resolved by reducing SDS-PAGE. For MLKL oligomerization, lysates generated using either 2% SDS or M2 buffer containing 1% Triton X-100 were resolved by SDS-PAGE under non-reducing conditions (without beta-mercaptoethanol). Blotting was conducted with standard techniques.
Statistical Analysis
Cell death, cell viability, and intracellular staining data are presented as the mean ± standard deviation. Statistics were performed using T-test. * p<0.05, ** p<0.01, *** p<0.001. Quantitative real-time PCR data is presented as the mean, ± standard deviation. Statistics were performed using Two-Way ANOVA. Bonferroni posts-tests were performed to compare the triplicate means at different time points. ** p<0.01, *** p<0.001.
Supplementary Material
HIGHLIGHTS.
TLR4 activates CASPASE-8 to remove CYLD and disable the TNF necroptosis pathway.
Autocrine TNF induces necroptosis in a cell-autonomous manner.
Priming of macrophages by Type I IFN is needed for necroptosis induced by LPS or TNF.
Type I IFN primes TNFR2 and MLKL expression, which are needed to induce necroptosis.
Acknowledgments
We thank Dr. Shizuo Akira, Dr. Douglas Green, Dr. Shao-Cong Sun and Dr. Adolfo Garcia-Sastre for providing mice and femurs/tibia, Dr. Nicolas Barnich for providing AIEC strain LF82, and Dr. Kate Fitzgerald for providing J2 virus. We would also like to thank Dr. Marie Anne O’Donnell for helpful discussions. This work was supported by National Institutes of Health (NIH) grants AI052417, AI104521, DK072201 (A.T.T.), DK080070 (E.M.), AI095245, DK072201 (J.M.B.), AI075118 (M.A.K.) and NIAID contract HHSN272201000054C (T.M.M). This work is also supported by a Senior Research Award #253097 from the Crohn’s and Colitis Foundation of America (A.T.T.). D.L. is a recipient of a Career Development Award from the Crohn’s and Colitis Foundation of America. S.J.J. is supported in part by a Public Health Service Institutional Research Training Award (AI07647) and a Helmsley Trust fellowship. E.M. is supported by a grant from the Broad Medical Foundation (E.M.). J.M.B. is supported by the Burroughs Wellcome Trust Fund, the Leukemia and Lymphoma Society, and the Irma-Hirschl and Monique Weill-Caulier Charitable Trust Funds. R.L.A. is supported by a NIAID T32 Cross-Disciplinary Training Program in Transplant Research (AI78892).
Footnotes
AUTHOR CONTRIBUTIONS
D.L. and S.J.J. designed and performed the majority of the experiments. D.L. and S.J.J. contributed equally to this work. N.R. performed experiments. R.L.A. performed the real-time quantitative PCR experiments. W. L. and T.M.M. provided Ifnar1−/− mice. J.Z. provided the femurs and tibia from Ripk3−/− and Ripk3−/− Fadd−/− mice for BMDM isolation. E.M. provided AIEC strain LF82. M.Z. and M.A.K. generated the RosaCreERT2Ripk1fl/fl BMDMs. J.M.B. provided technical and conceptual guidance during the experimental studies and in drafting of the manuscript. D.L., S.J.J., and A.T.T. wrote the manuscript. A.T.T. directed the studies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arslan SC, Scheidereit C. The prevalence of TNFalpha-induced necrosis over apoptosis is determined by TAK1-RIP1 interplay. PLoS One. 2011;6:e26069. doi: 10.1371/journal.pone.0026069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nature reviews Microbiology. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethea JR, Ohmori Y, Hamilton TA. A tandem GC box motif is necessary for lipopolysaccharide-induced transcription of the type II TNF receptor gene. The Journal of Immunology. 1997;158:5815–5823. [PubMed] [Google Scholar]
- Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell. 2014;156:705–716. doi: 10.1016/j.cell.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, Ward Y, Wu L-G, Liu Z-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nature Cell Biology. 2013 doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi L, Cummings RJ, Blander JM. Death-defining immune responses after apoptosis. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2014;14:1488–1498. doi: 10.1111/ajt.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. The Journal of experimental medicine. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. European Journal of Immunology. 2000;30:652–660. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chan FKM, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. The Journal of biological chemistry. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He W-T, Song Y, Yang C, Li W, Zheng X, Chen P, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Research. 2013 doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FKM. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- de Kossodo S, Critico B, Grau GE. Modulation of the transcripts for tumor necrosis factor-alpha and its receptors in vivo. European Journal of Immunology. 1994;24:769–772. doi: 10.1002/eji.1830240342. [DOI] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8- cFLIP(L) complex. Cell reports. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong Y-N, et al. RIPK1 Blocks Early Postnatal Lethality Mediated by Caspase-8 and RIPK3. Cell. 2014 doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell reports. 2014 doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Doyle S, Vaidya S, Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Estornes Y, Toscano F, Virard F, Jacquemin G, Pierrot A, Vanbervliet B, Bonnin M, Lalaoui N, Mercier-Gouy P, Pacheco Y, et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012;19:1482–1494. doi: 10.1038/cdd.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA, Wajant H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. Journal of Cell Science. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- Gais P, Tiedje C, Altmayr F, Gaestel M, Weighardt H, Holzmann B. TRIF signaling stimulates translation of TNF-alpha mRNA via prolonged activation of MK2. J Immunol. 2010;184:5842–5848. doi: 10.4049/jimmunol.0902456. [DOI] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proceedings of the National Academy of Sciences. 2011 doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a Molecular Signaling Network that Regulates a Cellular Necrotic Cell Death Pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Huys L, Van Hauwermeiren F, Dejager L, Dejonckheere E, Lienenklaus S, Weiss S, Leclercq G, Libert C. Type I interferon drives tumor necrosis factor-induced lethal shock. J Exp Med. 2009:1873–1882. doi: 10.1084/jem.20090213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justus SJ, Ting AT. Cloaked in ubiquitin, a killer hides in plain sight: the molecular regulation of RIPK1. Immunol Rev. 2015;266:145–160. doi: 10.1111/imr.12304. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like Receptor 3-mediated necrosis via TRIF, RIP3 and MLKL. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. The Journal of Immunology. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- Legarda-Addison D, Hase H, O’Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 2009;16:1279–1288. doi: 10.1038/cdd.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M, Tartaglia LA, Lee A, Bennett GL, Rice GC, Wong GH, Chen EY, Goeddel DV. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–347. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Holler N, Richard C, Tschopp J. Activation of a proapoptotic amplification loop through inhibition of NF-kappaB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett. 2000;468:134–136. doi: 10.1016/s0014-5793(00)01212-6. [DOI] [PubMed] [Google Scholar]
- McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, Gamero AM, Mossman KL, Sad S. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proceedings of the National Academy of Sciences. 2014a doi: 10.1073/pnas.1407068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb S, Shutinoski B, Thurston S, Cessford E, Kumar K, Sad S. Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J Immunol. 2014b;192:5671–5678. doi: 10.4049/jimmunol.1303380. [DOI] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nature Reviews Immunology. 2011 doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity. 2013 doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- O’Donnell MA, Hase H, Legarda D, Ting AT. NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS One. 2012;7:e41238. doi: 10.1371/journal.pone.0041238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17:418–424. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MA, Ting AT. Chronicles of a death foretold: dual sequential cell death checkpoints in TNF signaling. Cell Cycle. 2010;9:1065–1071. doi: 10.4161/cc.9.6.10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MA, Ting AT. RIP1 comes back to life as a cell death regulator in TNFR1 signaling. FEBS J. 2011;278:877–887. doi: 10.1111/j.1742-4658.2011.08016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, Mccormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Lee TH, Chan FKM, Pasparakis M, Kelliher MA. Cutting Edge: RIPK1 Kinase Inactive Mice Are Viable and Protected from TNF-Induced Necroptosis In Vivo. Journal of immunology (Baltimore, Md: 1950) 2014 doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson SM, Walker WS. Immortalization of cloned mouse splenic macrophages with a retrovirus containing the v-raf/mil and v-myc oncogenes. Cell Immunol. 1988;116:341–351. doi: 10.1016/0008-8749(88)90236-5. [DOI] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature Immunology. 2012 doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, Kelliher MA. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci U S A. 2014;111:14436–14441. doi: 10.1073/pnas.1409389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruspi G, Schmidt EM, McCann F, Feldmann M, Williams RO, Stoop AA, Dean JLE. TNFR2 increases the sensitivity of ligand-induced activation of the p38 MAPK and NF-κB pathways and signals TRAF2 protein degradation in macrophages. Cellular Signalling. 2013 doi: 10.1016/j.cellsig.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. 2006;26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Tannenbaum CS, Major JA, Hamilton TA. IFN-gamma and lipopolysaccharide differentially modulate expression of tumor necrosis factor receptor mRNA in murine peritoneal macrophages. The Journal of Immunology. 1993;151:6833–6839. [PubMed] [Google Scholar]
- Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proceedings of the National Academy of Sciences. 2013 doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host and Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature Reviews Molecular Cell Biology. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Fulda S, Vandenabeele P. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death and Differentiation. 2010 doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang L-F, Wang F-S, Wang X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Molecular Cell. 2014 doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Research. 2013 doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (New York, NY) 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu Z-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proceedings of the National Academy of Sciences. 2012 doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.