

Summary
Insulin-resistant syndromes such as type II diabetes mellitus (T2DM) involve disrupted temporal coordination of hepatic metabolism such that synthesis and secretion of lipid and glucose are inappropriately engaged concurrently. Here we test the hypothesis that a combination of direct and indirect actions of insulin on liver can lead to the metabolic phenotype exhibited in T2DM without a defect in proximal hepatic insulin signaling. First, we show that the insulin-dependent inhibition of Foxo1 and activation of mTorc1 by Akt is both necessary and sufficient for the induction of lipogenesis and the lipogenic gene program. In marked contrast, insulin, acting in vivo independent of hepatocyte insulin signaling can suppress glucose production by reducing serum free fatty acids. These studies support the hypothesis that under conditions of obesity and diabetes, intact hepatic insulin signaling can maintain lipogenesis while excess circulating FFAs become a dominant positive regulator of HGP.
eTOC Paragraph
During diabetes, both gluconeogenesis and lipogenesis occur in the liver. Titchenell et al address the issue of selective insulin resistance to show that direct liver insulin signaling is required for lipogenesis, while liver insulin signaling only indirectly suppresses glucose production by reducing serum free fatty acids coming from adipocytes.
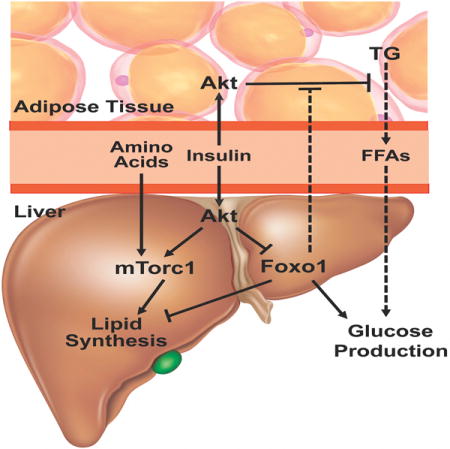
Introduction
The dynamic regulation of hepatic glucose and lipid metabolism is essential for metabolic homeostasis and the adaptation to nutrient availability and deprivation. During periods of fasting, under the influence of low insulin-glucagon ratios, the liver produces glucose while coordinately suppressing the synthesis of new fatty acids. Upon nutrient intake, insulin and increased substrate levels oppose this process, suppressing hepatic glucose production (HGP) and promoting the synthesis of fatty acids for storage and subsequent utilization. A key aspect of insulin-resistant states such as the metabolic syndrome and T2DM is a loss of the normal inverse temporal relationship between glucose and fatty acid synthesis, such that the liver undergoes both anabolic processes concurrently. This metabolic state has been described as “selective hepatic insulin resistance”, which suggests a defect in the insulin signaling pathway that allows preservation of the lipid promoting effects of the hormone despite loss of control of HGP (Brown and Goldstein, 2008). However, an alternative explanation for the hepatic “insulin resistant” state is that insulin signaling is intact allowing de novo lipogenesis (DNL) while another process independent of insulin signaling drives enhanced glucose output. In the studies described below, we explore this possibility by characterizing systematically the direct and indirect pathways by which insulin controls hepatic lipid and glucose metabolism.
Insulin signaling in the liver is mediated by the hepatic insulin receptor (IR), which signals through the downstream kinase, protein kinase B/Akt to coordinate hepatic metabolism (Lu et al., 2012; Taniguchi et al., 2006; Wan et al., 2013; Wan et al., 2011). Akt signals through multiple downstream pathways, including those centered on mechanistic target of rapamycin (mTorc1) and the Foxo family of transcription factors. Recent studies suggest activation of mTorc1 by Akt is required but not sufficient for insulin-induced activation of DNL and the lipogenic gene program (Laplante and Sabatini, 2010; Li et al., 2010; Porstmann et al., 2008; Wan et al., 2011; Yecies et al., 2011). In contrast, Foxo1’s role in hepatic lipid metabolism is less clear, as some studies suggest Foxo1 contributes directly to insulin’s regulation of lipogenic gene expression and DNL in liver while others favor a permissive role for Foxo1 in these processes (Banks et al., 2011; Deng et al., 2012; Haeusler et al., 2014; Lu et al., 2012; Matsumoto et al., 2006; Wan et al., 2011; Zhang et al., 2006). The most well-characterized hepatic function of Foxo1 involves its regulation of carbohydrate metabolism, as liver-specific Foxo1 deletion is sufficient to normalize hyperglycemia and whole-body insulin sensitivity in mice lacking the hepatic insulin receptor, insulin receptor substrates, or the two Akt isoforms expressed in liver, Akt1 and Akt2 (Dong et al., 2008; Lu et al., 2012; Matsumoto et al., 2007; O-Sullivan et al., 2015; Titchenell et al., 2015). Despite Foxo1’s documented role in gluconeogenesis, mice with deletion of both hepatic Akt (or IR) and Foxo1 respond appropriately to insulin in vivo to suppress HGP and gluconeogenic gene expression providing evidence for the cell non-autonomous regulation of hepatic glucose metabolism.
Utilizing mouse loss of function experiments, we demonstrate here that hepatic Foxo1 deletion and mTorc1 activation are both required and sufficient to induce DNL and the lipogenic gene program in the absence of hepatic insulin signaling in vivo. In contrast, insulin’s regulation of glucose output can be achieved in the absence of liver insulin signaling by inhibition of adipocyte lipolysis. These data suggest a model by which the inappropriate manifestation of these regulatory circuits generates the simultaneous biosynthesis of glucose and lipids during “insulin-resistant” states.
Results
Foxo1 deletion fails to normalize postprandial DNL in L-AktDKO and L-IRKO mice
In addition to its effects on glucose homeostasis, insulin is a central regulator of hepatic lipid metabolism (Horton et al., 2002). Deletion of Foxo1 in livers lacking Akt (or IR) normalizes glucose tolerance and reestablishes insulin’s ability to suppress HGP, but it is not known whether loss of Foxo1 also restores insulin’s regulation of hepatic de novo lipogenesis (DNL). We generated mice lacking the essential signaling intermediates of insulin action in liver by injecting AAV-TBG-Cre into Akt1loxp/loxp,Akt2loxp/loxp or Akt1loxp/loxp,Akt2loxp/loxp,Foxo1loxp/loxp mice. Floxed mice injected with AAV-TBG-GFP served as controls and are denoted “LWT” below. Both mice deficient in hepatic Akt (L-AktDKO) and Akt and Foxo1 (L-AktFoxo1TKO) lacked Akt protein and a supraphysiological dose of insulin in vivo failed to induce phosphorylation of the Akt and the mTorc1 substrates PRAS40 and S6, respectively (Figure 1A).
Figure 1. L-AktFoxo1TKO mice have defective lipogenesis.
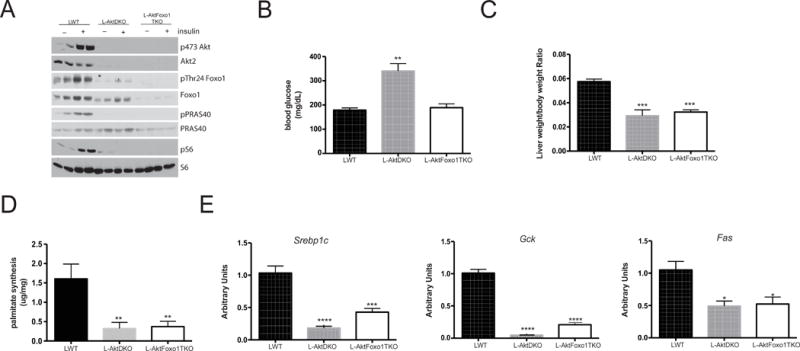
A) Western blot from liver lysates following I.P. insulin injection (2mU/kg) and probed for the indicated proteins B) blood glucose following 3 h of HCD feeding after overnight fast C) liver weight following 6 h refeeding D) de novo lipogenesis following 6 h refeeding E) gene expression analysis following 6 h refeeding. n=4–9 mice/group. **** p<0.0001 vs LWT, *** p<0.001 vs LWT, ** p<0.01 vs LWT, *p<0.05 vs LWT. These data are presented as mean ± s.e.m.
As expected, L-AktDKO mice were hyperglycemic following carbohydrate feeding, an effect that was normalized with concomitant Foxo1 deletion (Figure 1B). In contrast, the reduction in liver weight in L-AktDKO mice was not corrected in L-AktFoxo1TKO mice (Figure 1C). To determine the requirement of hepatic insulin signaling in the regulation of DNL, mice were fasted overnight, refed a high-carbohydrate diet (HCD), injected with D2O and analyzed for the incorporation of D2O into liver palmitate. Mice lacking Akt in liver displayed an approximately 80% reduction in DNL (Figure 1D). Unlike its effects on HGP, Foxo1 deletion from liver in L-AktDKO mice was completely without effect on the reduced DNL (Figure 1D). Similarly, the expression of Srebp1c, the master regulator of lipogenic gene transcription, and its targets Gck and Fas were significantly reduced in L-AktDKO and concomitant ablation of Foxo1 failed to restore normal regulation of lipogenic gene expression (Figure 1E). Serum triglyceride was significantly reduced in both L-AktDKO and L-AktFoxo1TKO mice compared to LWT mice (Table S1. Related to Figure 1). In response to HCD, L-AktDKO and L-AktFoxo1TKO mice were hyperinsulinemic. Despite elevated insulin levels, L-AktDKO mice trended to increase serum ketones suggesting increased fatty acid oxidation, an effect normalized by Foxo1 deletion (Table S1. Related to Figure 1).
Similarly to L-AktDKO mice, deletion of hepatic insulin receptor (L-Insulin Receptor KO) also led to reduced liver weight, hepatic DNL and lipogeneic gene expression, which were not reversed by coincident removal of Foxo1 (Figure S1. Related to Figure 1). These data support the notion that liver insulin signaling through Akt is essential for hepatic lipid synthesis, even in the absence of hepatic Foxo1.
mTorc1 activation and Foxo1 inhibition are required to induce DNL downstream of Akt
Recent evidence has indicated that the mTorc1 complex is necessary for DNL and Srebp1c processing in response to feeding and insulin (Li et al., 2010; Wan et al., 2011; Yecies et al., 2011). Insulin failed to promote activation of mTorc1 in livers from L-AktDKO and L-AktFoxo1TKO as assessed by phosphorylation of ribosomal protein S6 (Figure 1A). To determine if activation of mTorc1 is sufficient to drive lipogenesis in the absence of Akt, we deleted Tsc1 in livers of mice lacking hepatic Akt1 and Akt2 (L-AktTsc1TKO). Akt, Tsc1 and Tsc2 protein levels were reduced in livers from L-AktTsc1TKO mice and feeding failed to induce phosphorylation of the canonical Akt targets, Foxo1 and PRAS40 (Figure 2A). Despite the absence of Akt signaling, livers from L-AktTsc1TKO mice exhibited constitutive phosphorylation of ribosomal protein S6 and 4EBP1, indicating activation of mTORC1 independent of nutritional intake (Figure 2A). L-AktTsc1TKO mice displayed an increase of blood glucose (Figure 2B) in response to a meal and a significant reduction in liver weight (Figure 2C). In spite of constitutive activation of mTORC1, HCD feeding failed to induce DNL and increase expression of Srebp1c and its targets Gck and Fas in L-AktTsc1TKO mice (Figure 2D & 2E). Consistent with reduced DNL, L-AktTsc1TKO had a significant reduction in serum triglyceride levels (Table S2). L-AktTsc1TKO mice had elevated serum ketones indicating an increase in fatty acid oxidation during feeding (Table S2). These data add to previous studies indicating that mTorc1 activation is not sufficient to mediate lipid synthesis in the absence of Akt signaling (Wan et al., 2011; Yecies et al., 2011)
Figure 2. Both mTorc1 activation and Foxo1 inhibition are required for DNL in the absence of Akt.
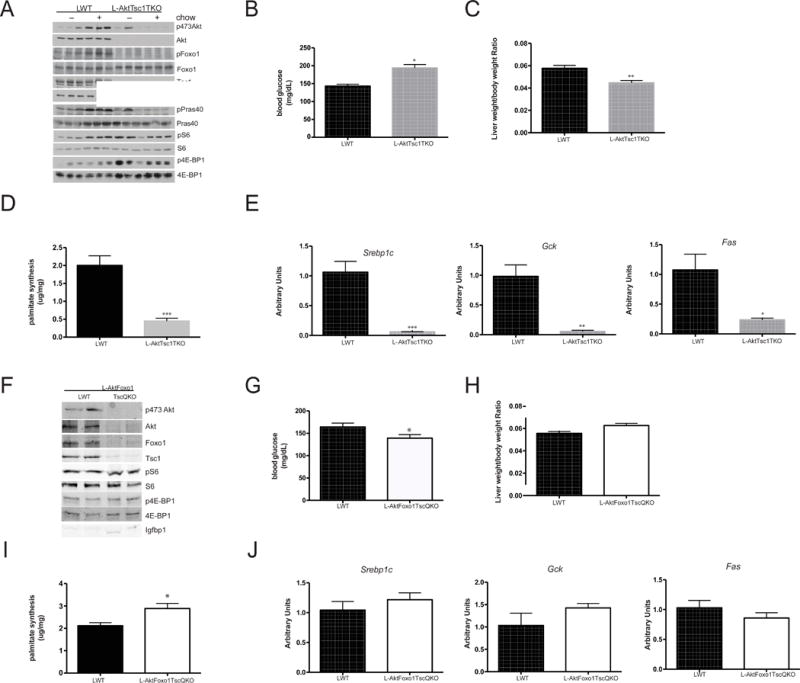
A) Western blot for the indicated proteins from liver lysates following fasting and 4 h refeeding B) blood glucose following 3 h of HCD feeding after overnight fast C) liver weight following 6 h refeeding D) de novo lipogenesis following 6 h refeeding E) gene expression analysis following 6 h refeeding. n=3 mice/group. **** p<0.0001 vs LWT, *** p<0.001 vs LWT, ** p<0.01 vs LWT, *p<0.05 vs LWT F) western blot from liver lysates following 6 h HCD feeding for the indicated proteins G) blood glucose following 3 h of feeding after overnight fast H) liver weight following 6 h refeeding I) de novo lipogenesis following 6 h refeeding J) gene expression analysis following 6 h refeeding. n=5–9 mice/group. **** p<0.0001 vs LWT, *** p<0.001 vs LWT, ** p<0.01 vs LWT, *p<0.05 vs LWT. These data are presented as mean ± s.e.m.
As shown above, deletion of Foxo1 alone did not normalize DNL in the absence of Akt; nonetheless, Foxo1 has been implicated in the regulation of hepatic lipid metabolism and the induction of Srebp1c (Deng et al., 2012; Matsumoto et al., 2006; Wan et al., 2011; Zhang et al., 2006). We hypothesized that ablation of both Foxo1 and Tsc1 might stimulate DNL, even in the absence of insulin signaling. In order to test this, we deleted both Foxo1 and Tsc1 in mice lacking liver Akt isoforms (L-AktFoxo1Tsc1QKO) (Figure 2F). Despite the absence of pAkt, there was phosphorylation of the mTorc1 targets S6 and 4EBP1(Figure 2F). Upon refeeding, L-AktFoxo1Tsc1QKO had lower blood glucose levels (Figure 2G) and insulin levels than control mice, suggesting increased insulin sensitivity (Table S2). Despite lower insulin levels, L-AktFoxo1Tsc1QKO mice displayed increased rates of DNL (Figure 2I) and a trend to elevated serum triglyceride levels (Table S2. Related to Figure 2). Unlike the L-AktTsc1TKO mice, L-AktFoxo1Tsc1QKO mice had normal serum ketone levels suggesting fatty acid oxidation was reduced following Foxo1 deletion (Table S2. Related to Figure 2). These changes in lipid homeostasis correlated with restored expression of Srebp1c and Srebp1c-targets Gck and Fas (Figure 2J) despite significant increases in Insig2a expression (Figure S2. Related to Figure 2). These data demonstrate that the coincident inhibition of Foxo1 and activation of mTorc1 by insulin is both necessary and sufficient for activation of DNL and regulation of lipid metabolism by insulin in liver in vivo.
Non-autonomous control of HGP by insulin occurs independently of vagal efferent communication to liver
As shown above, although hepatic insulin signaling through Akt is required for the postprandial regulation of DNL and the lipogenic gene program, insulin can suppress HGP in mice lacking hepatic Akt and Foxo1, suggesting the presence of a cell non-autonomous pathway to regulate carbohydrate metabolism (Lu et al., 2012). The idea that regulation of HGP can be exerted by an intermediary extra-hepatic tissue is supported by the insulin-dependent suppression of HGP in mice lacking the liver insulin receptor (O-Sullivan et al., 2015; Titchenell et al., 2015). We considered central nervous system, alpha cell of the pancreas, and adipocytes as tissues that might mediate the cell non-autonomous actions of insulin in vivo.
In rodents, insulin acts on the brain to regulate HGP via a circuit dependent on the vagus nerve (Pocai et al., 2005a; Pocai et al., 2005b). To test the requirement for this pathway in the non-autonomous regulation of HGP by insulin in L-AktFoxo1TKO mice, we performed hyperinsulinemic-euglycemic clamps on mice with hepatic branch vagotomy (Figure S3A. Related to Experimental Procedure). Insulin concentrations were similar during the clamp portion in both vagotomized and sham-operated L-AktFoxo1TKO mice (Figure S3B. Related to Experimental Procedure). Hepatic vagotomy (HV) did not alter the glucose infusion rate or whole-body glucose disposal (Rd) in L-AktFoxo1 mice (Figure S3C & S3D. Related to Experimental Procedure). Furthermore, insulin suppressed HGP to a similar extent in both sham operated and vagotomized L-AktFoxo1TKO mice (Figure S3E. Related to Experimental Procedure). Hyperinsulinemic-euglycemic clamps performed in vagotomized LWT and L-AktFoxo1TKO at four-fold higher infusion rates of insulin did not reveal any difference in the glucose infusion among the four groups (Figure S3F. Related to Experimental Procedure). These data indicate that insulin suppresses HGP and regulates whole-body insulin sensitivity in L-AktFoxo1TKO independent of the vagal efferent output of the CNS to the liver.
Insulin suppresses HGP independent of liver glucagon signaling and gluconeogenic gene regulation
Glucagon has been implicated in the pathogenesis of diabetes; blockade of glucagon signaling reverses the hyperglycemia of experimental diabetes (Unger and Cherrington, 2012). Infusion of insulin into mice with glucose clamped at physiological levels reduced plasma glucagon levels in control mice (LWT) and trended to doing so in L-AktFoxo1TKO mice (Figure 3A). To test the contribution of liver glucagon action in mediating the extra-hepatic effects of insulin, we blocked hepatic glucagon action by expressing in the liver an inhibitory mutant of the regulatory subunit of PKA (PKA-DN); previously studies have shown that this virus completely blocks glucagon-induced glucose production (Miller et al., 2013). Glucagon did not promote phosphorylation of PKA substrates, including Creb, in hepatocytes isolated from mice injected with AAV expressing PKA-DN (Figure 3B). As expected, expression of PKA-DN in liver lowered fasting blood glucose in LWT mice (Figure 3C) and improved glucose tolerance in both LWT and L-AktFoxo1TKO mice (Figure 3D). Expression of PKA-DN for 2 weeks did not affect circulating triglyceride, free fatty acids or ketone levels (Table S3. Related to Figure 3). Ablation of glucagon signaling in liver led to a significant decrease in the mRNA encoding G6pc and this was not further reduced by infusion of insulin (Figure 3E). A similar trend was observed for Pck1 (Figure 3F). Surprisingly, insulin suppressed HGP in L-AktFoxo1TKO mice expressing PKA-DN in a manner indistinguishable from that in L-AktFoxo1TKO mice (Figure 3G & 3H). These data show that insulin suppresses HGP in vivo by a mechanism independent of glucagon signaling and changes in gluconeogenic gene expression in livers deficient in insulin action.
Figure 3. Inhibition of liver glucagon signaling reduces gluconeogenic gene expression but does not inhibit insulin suppression of HGP.
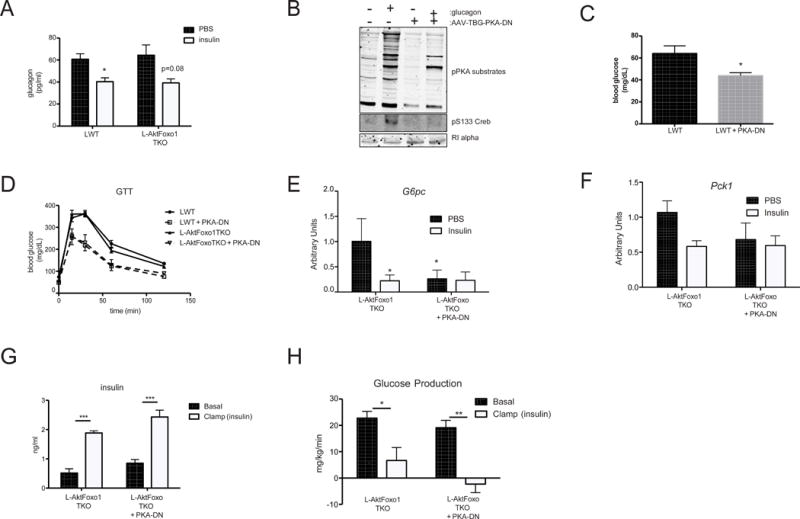
A) plasma glucagon measurement at end of euglycemic clamp with or without 10 mU/kg/min insulin infusion in LWT and L-AktFoxo1TKO mice B) primary hepatocytes isolated from mice transduced with a mutant form of the regulatory subunit of PKA (PKA-DN) and subjected to glucagon treatment for 15 min were probed by Western blot for the indicated proteins C) overnight fasting blood glucose from mice LWT injected with AAV-PKA-DN D) intra-peritoneal glucose tolerance test 2 g/kg E) G6pc mRNA at end of euglycemic clamp in LWT and L-AktFoxo1TKO mice F) Pck1 mRNA at end of euglycemic clamp in LWT and L-AktFoxo1TKO mice G) insulin levels during basal and insulin portions of the clamp H) hepatic glucose production during basal and insulin portions of the clamp. n=3–8 for GTT *** p<0.001 vs LWT, **p<0.01 vs LWT. n=3–4 for hyperinsulinemic-euglycemic clamp studies. *** p<0.001 vs L-AktFoxo1TKO, **p<0.01 vs L-AktFoxo1TKO. *p<0.05 vs L-AktFoxo1TKO. These data are presented as mean ± s.e.m.
Free fatty acids mediate insulin’s suppression of HGP in L-AktFoxo1TKO mice
Consistent with the known anti-lipolytic effects of insulin, serum free fatty acids (FFA) decreased approximately 50–75% in LWT and L-AktFoxo1TKO during a hyperinsulinemic-euglycemic clamp, correlating well with the rate of HGP (Rebrin et al., 1995) (Figure 4A & 4B). Recent studies have shown that infusion of acetate during hyperinsulinemic-euglycemic clamps blocks the effects of insulin on HGP (Perry et al., 2015). We studied the effects of the physiological substrate, FFA, on HGP by infusing intralipid and heparin at a rate to maintain FFA levels at pre-clamp levels during administration of insulin (Figure 4A) (Rebrin et al., 1996). Glycerol was infused in control groups to maintain constant glycerol levels during the hyperinsulinemic-euglycemic clamp protocol (Figure 4C). Intralipid/heparin infusion (lipid) did not affect the rate of glucose infusion or glucose disposal in LWT mice and failed to prevent insulin-mediated suppression of HGP (Figure 4D–G) (Kim et al., 2004a; Kim et al., 2004b; Kim et al., 2001). In contrast, preventing the insulin-dependent decrease in FFA during clamp reduced the glucose infusion rate by 20% and completely blocked suppression of HGP in L-AktFoxo1TKO mice without affecting glucose disposal (Figure 4D–4G). Lipid infusion did not change glucokinase activity in either LWT or L-AktFoxo1TKO mice (Figure 4H). These data show that reduction in HGP from livers lacking the insulin signaling pathway is dependent upon the reduction of serum FFAs by insulin.
Figure 4. Intralipid/heparin infusion prevents insulin suppression of HGP in L-AktFoxo1TKO mice.
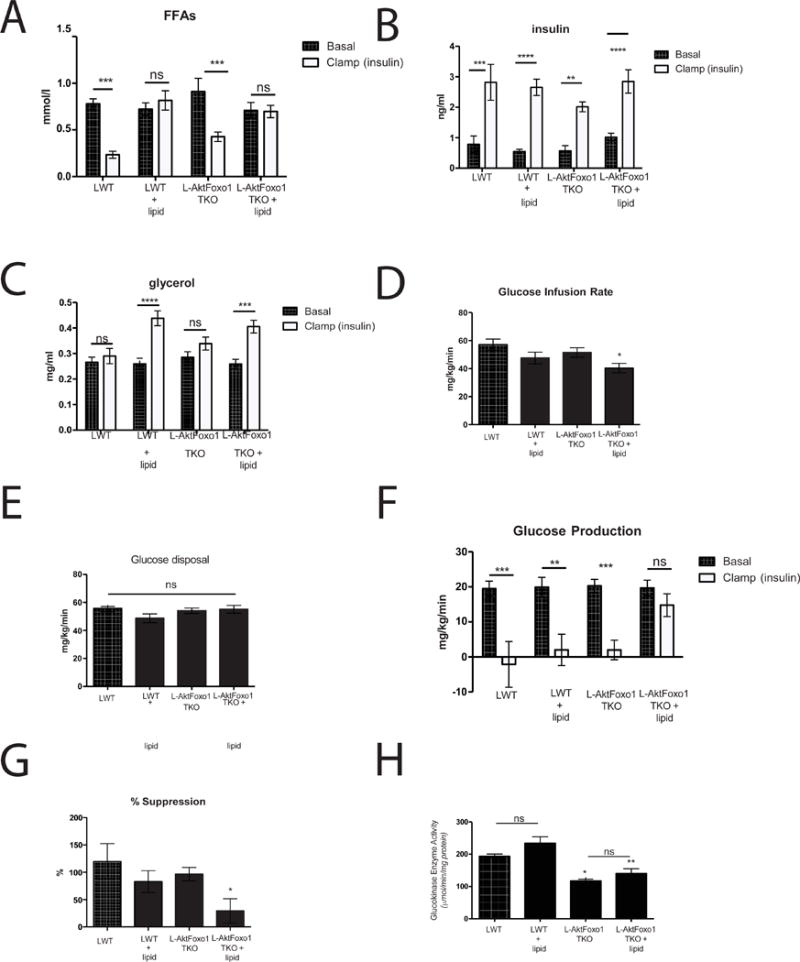
Hyperinsulinemic-euglycemic clamps were performed on unrestrained 5 h fasted LWT and L-AktFoxo1TKO mice using a 2.5 mU/min/kg infusion of insulin and intralipid/heparin infusion. A) free fatty acid levels during basal and insulin portions of the clamp B) insulin levels C) glycerol levels D) steady state glucose infusion rate E) whole-body glucose disposal (Rd) F) hepatic glucose production during basal and insulin portions of the clamp G) percent suppression of hepatic glucose production during the clamp portion compared to basal period. n=5–6 mice/group. H) glucokinase activity at end of clamp. **** p<0.0001 vs basal, *** p<0.001 vs basal, * p<0.05 vs LWT. These data are presented as mean ± s.e.m.
High-dose insulin is sufficient to reduce HGP in L-AktDKO mice
Constitutively active Foxo1 blocks the ability of the liver to respond to its cell non-autonomous regulation by insulin (Lu et al., 2012). In principal, this defect could be caused by two alternative mechanisms: either livers with active Foxo1 fail to respond to a decreases in circulating FFA, or insulin is unable to suppress adipocyte lipolysis in mice with unrestrained activity of Foxo1 in liver. Infusion of a 2.5 mU/kg/min did not increase serum insulin in the already hyperinsulinemic L-AktDKO mice and there was no decrease in glycerol, FFA, or glucose production, consistent with the second hypothesis (Supplemental Figure 4A–4D. Related to Figure 5) To determine if defects in insulin action on the adipocyte were responsible for the increased HGP, we elevated serum insulin levels by performing a hyperinsulinemic-euglycemic using a high infusion rate (20 mU/kg/min) of insulin (Figure 5A). Under these conditions, insulin overcame the adipocyte insulin resistance and reduced circulating FFA, thus increasing the glucose infusion rate and glucose disposal and suppressing HGP in L-AktDKO mice (Figure 5B–F). To determine definitively whether the decrease in FFA by insulin was responsible for the reduction in HGP in L-AktDKO, we blocked the reduction in FFAs by intralipid/heparin infusion (Figure 5G). Maintenance of circulating FFA largely prevented the suppression of HGP by insulin, showing clearly that adipocyte insulin resistance is responsible for the defect in hepatic insulin action on HGP in L-AktDKO mice (Figure 5H).
Figure 5. A high-dose insulin infusion suppresses lipolysis and HGP in L-AktDKO mice.
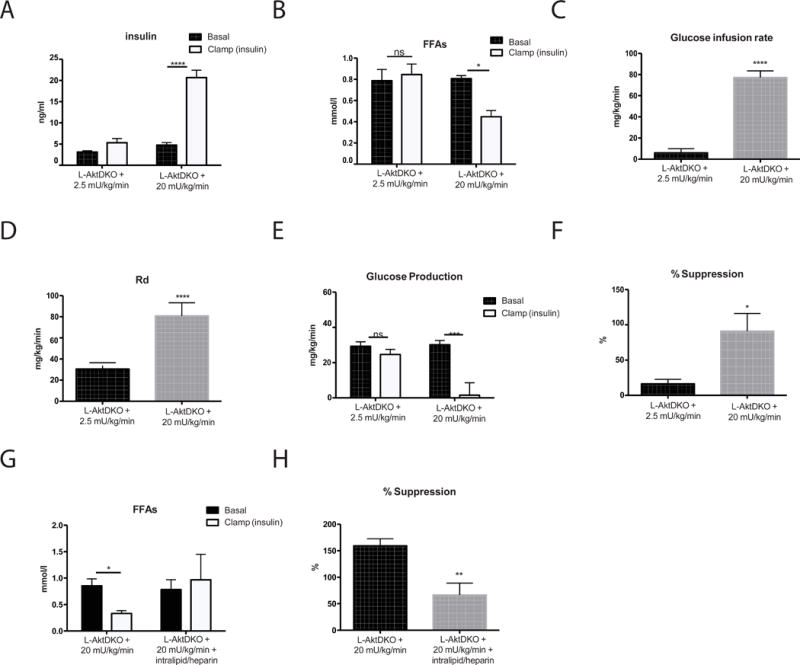
Hyperinsulinemic-euglycemic clamps were performed on unrestrained 5 h fasted L-AktDKO mice using a 2.5 mU/min/kg or 20 mU/min/kg infusion of insulin. A) insulin levels during basal and insulin portions of the clamp B) free fatty acids levels C) steady state glucose infusion rate D) whole body glucose disposal (Rd) E) hepatic glucose production during basal and insulin portions of the clamp F) percent suppression of hepatic glucose production during the clamp portion compared to basal period. n=4 mice/group. **** p<0.001 vs basal, ***p<0.001 vs basal/2.5mU * p<0.05 vs basal/2.5mU. Hyperinsulinemic-euglycemic clamps were performed on unrestrained 5 h fasted L-AktDKO mice using a 20 mU/min/kg infusion of insulin and intralipid/heparin infusion G) free fatty acids levels H) percent suppression of hepatic glucose production during the clamp portion compared to basal period. n=5–6 mice/group. **p<0.01 vs 20mU * p<0.05 vs 20mU. These data are presented as mean ± s.e.m.
Deletion of Foxo1 in liver prevents insulin resistance during diet-induced obesity by maintaining insulin sensitivity in the adipose tissue
In order to assess the relevance of the above experiments to a non-genetic model of T2DM, we tested the role of increased Foxo1 activity in insulin resistance during diet-induced obesity. WT and liver Foxo1 knockout mice (L-Foxo1KO) mice fed Surwit HFD gained significantly more weight than age-matched, chow fed controls (Figure 6A). Deletion of Foxo1 from liver of obese mice significantly improved glucose and insulin tolerance, and reduced hepatic and peripheral insulin resistance as determined by hyperinsulinemic-euglycemic clamp (Figure 6B–6G). Importantly, deletion of Foxo1 exclusively in liver restored insulin’s ability to inhibit adipocyte lipolysis, an action of the hormone absent in DIO mice (Figure 6H). The improvement in hepatic insulin responsiveness in L-Foxo1KO was not due to increased insulin signaling in liver, as phosphorylation of Akt was similar between LWT and L-Foxo1KO mice (Figure 6I).
Figure 6. Liver Foxo1 knockout improves insulin sensitivity during diet-induced obesity.
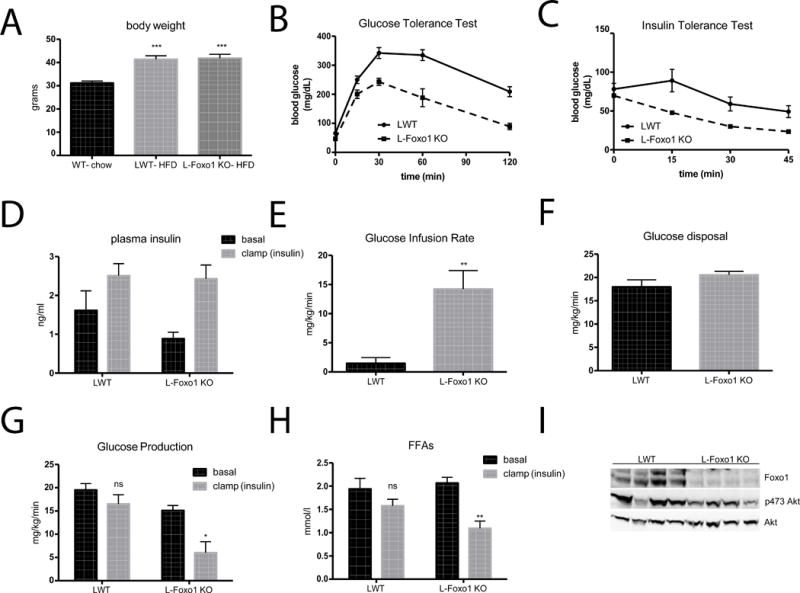
A) Body weight accumulation after 24 weeks of Surwit HFD feeding B) 2g/kg IP glucose tolerance test after overnight fast C) 1 U/kg IP insulin tolerance test D) insulin levels during basal and insulin portions of hyperinsulinemic-euglycemic clamps using 2.5 mU/kg/min insulin infusion E) steady state glucose infusion rate F) whole body glucose disposal (Rd) G) hepatic glucose production during basal and insulin portions of the clamp H) free fatty acids levels during basal and insulin portions of the clamp I) western blot from livers taken at the end of the insulin portion of the clamp. **** p<0.0001 vs basal, ** p<0.01 vs basal, * p<0.05 vs LWT. n=5–6 (A–C), n=4 (D–I). These data are presented as mean ± s.e.m.
Overexpression of glucokinase in liver mimics hepatic deletion of Foxo1 in L-AktDKO mice
The simplest explanation for how constitutive Foxo1 inhibits insulin’s anti-lipolytic effects in adipose tissue is that persistent HGP raises glucose and insulin, the latter promoting down-regulation of insulin signaling in adipocytes. As described above, this effect of Foxo1 deletion on HGP is unlikely to be due solely to reduced expression of the gluconeogenic genes, G6Pase and PEPCK, as their expression does not correlate with the acute regulation of HGP (Figure 3) or hyperglycemia in humans with diabetes (Haeusler et al., 2015). A candidate protein for mediating Foxo1’s effect on net HGP is glucokinase, which is undetectable in livers form L-AktDKO mice, but whose mRNA and protein are partially restored after concomitant deletion of Foxo1 (Figures 1E, 7A) (Haeusler et al., 2014; Lu et al., 2012; Titchenell et al., 2015). To test the hypothesis, we asked whether restoring hepatic glucokinase expression to the liver in L-AktDKO mice was sufficient to normalize glucose homeostasis and insulin responsiveness. Injection of an adeno-associated virus driving glucokinase expression from a liver specific promoter (AAV-TBG-Gck) led to increases in glucokinase protein in both LWT and L-AktDKO livers (Figure 7B). Elevated glucokinase improved glucose tolerance and fasting insulin levels in LWT and prevented the fasting hyperinsulinemia and glucose intolerance in L-AktDKO mice (Figure 7C& 7D). Livers of L-AktDKO mice had reduced levels of glucose-6-phoshate, which were normalized either by expressing glucokinase or deleting Foxo1 (Figure 7E). These data indicate that loss of hepatic Foxo1 normalizes glucose metabolism in Akt-deficient livers at least in part by restoring hepatic glucose phosphorylation.
Figure 7. Glucokinase overexpression is sufficient to normalize glucose tolerance and insulin sensitivity in L-AktDKO mice.
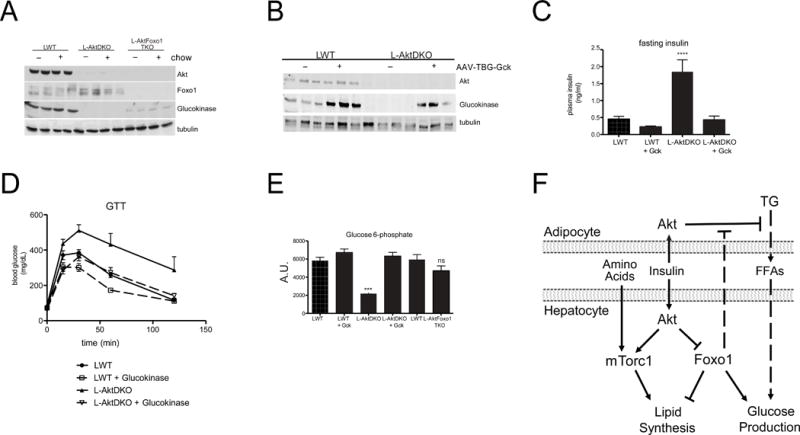
A) Western blot from liver lysates following overnight fasted or 4 hour after refeeding probed for the indicated proteins B) Western blot from liver lysates 4 hour after refeeding probed for the indicated proteins C) fasting insulin levels n=5–6 **** p<0.0001 D) 2g/kg IP glucose tolerance test after overnight fast E) steady-state glucose-6-phosphate levels 4 hour following refeeding (methods) n=3–4 *** p<0.001. These data are presented as mean ± s.e.m. F) Model of the signaling pathways mediating insulin dependent regulation of hepatic glucose production and de novo lipogenesis. Following a meal, insulin signals directly in the liver via Akt to inhibit Foxo1 and activate mTorc1, both of which are required for feeding-induced de novo lipogenesis. However, unlike the absolute requirement for direct insulin action to stimulate postprandial lipogenesis, insulin utilizes both intra-hepatic and extra-hepatic pathways (adipocyte) to regulate hepatic glucose production. In the absence of hepatic insulin signaling, Foxo1 cell non-autonomously inhibits insulin’s suppressive effects on adipocyte lipolysis leading to elevated FFAs and subsequently glucose production, which becomes the dominant regulator of HGP.
Discussion
Substantial data have accumulated over the years to support the notion that insulin acts directly on the mammalian liver to antagonize glucose production and promote lipid synthesis (Brown and Goldstein, 2008). Thus, the observation that obesity in humans and rodents is associated with concurrent increased lipogenesis and HGP has been perceived as a paradox: how can the “insulin resistant” liver respond to the effect of insulin to simulate DNL but be resistant to hormone in the reduction of glucose output? Observations in both humans and preclinical models have shown clearly that the lipogenesis and steatosis associated with insulin resistance depend on intact insulin signaling in liver (Leavens et al., 2009; Semple et al., 2009). The most widely accepted explanation for this phenomenon is that obesity is associated with blockade of insulin signaling at a point downstream of where the pathway bifurcates into two branches that regulate carbohydrate and lipid metabolism, respectively. An alternative hypothesis is insulin signaling remains intact in the “insulin resistant” liver but that a liver cell non-autonomous pathway drives glucose production independent of liver insulin action. As a first step towards testing this model, we have addressed several critical questions: is there a signaling pathway in liver that is both necessary and sufficient for mediating the effects of insulin on DNL?; and what is the pathway by which insulin can suppress HGP in the absence of insulin signaling in liver?
Several years ago, we reported that insulin suppresses HGP in livers lacking the major insulin receptor downstream target, Akt, as long as Foxo1 is deleted concomitantly (Lu et al., 2012). More recently, we provided evidence that under these conditions the effect of insulin is liver cell non-autonomous, as hepatocytes deficient in insulin receptor respond to insulin in vivo, once Foxo1 is deleted (Titchenell et al., 2015). In this study, we asked if insulin regulates hepatic lipogenesis and the lipogenic gene program by a similar cell-non-autonomous mechanism. We demonstrated that hepatic Akt signaling downstream of insulin action is required for the postprandial induction of DNL and the lipogenic gene program even in the absence of Foxo1. Moreover, activation of mTorc1 alone is not sufficient to drive lipid synthesis and induce Srebp1c. Rather, in order for Akt to stimulate DNL, Akt must both suppress Foxo1 and activate mTorc1. This conclusion is supported by the data that deletion of hepatic Tsc1, and Foxo1 together, but not each gene on its own, is sufficient to activate DNL and Srebp1c and lipogenic gene expression in hepatocytes lacking Akt.
The concept that direct and indirect mechanisms may contribute to the regulation of HGP by insulin dates back several years (Cherrington et al., 1998). We considered three candidate pathways as potential mediators of insulin’s cell non-autonomous effects on: the central nervous system, alpha cell of the pancreas, and adipose tissue. In recent years, there has accumulated overwhelming evidence that nutrient and hormone action in the central nervous system regulates hepatic carbohydrate metabolism (Myers and Olson, 2012). In rodents insulin acts in the hypothalamus to open brain K(ATP) channels, leading to a reduction in liver G6pc mRNA and HGP (Pocai et al., 2005a; Pocai et al., 2005b), and subsequent studies localized the site of action of insulin to AgRP neurons (Konner et al., 2007). Rossetti and colleagues showed that the route of communication between the hypothalamus and liver is via a vagal efferent, as section of its hepatic branch blocked the effect of central infusion of insulin on HGP. In our studies, vagotomy did not affect suppression of HGP in WT or L-AktFoxo1TKO mice, indicating the lack of requirement for this peripheral nerve in mediating a reduction of HGP in response to peripheral insulin suggesting that the mechanism is independent of the CNS. These results are consistent with the observation that hepatic vagotomy does not alter HGP during a peripheral infusion of insulin (Lam et al., 2005) and that actions of insulin in brain is probably not shared in all species (Ramnanan et al., 2012).
In the present study we found that insulin acts in the adipose tissue to reduce circulating free fatty acids, thus suppressing HGP in livers devoid of insulin signaling. Consistent with our study, Perry et al. suggested that the effects of insulin on HGP in L-AktFoxo1TKO mice could be antagonized by concurrent infusion of acetate and, consistent with the present experiments, concluded that insulin controls HGP via regulation of adipocyte lipolysis (Perry et al., 2015). One significant difference between the current study and that of Perry et al. was that the latter found that infusion of acetate during hyperinsulinemic-euglycemic clamps blocks the insulin-dependent decrease in HGP in WT mice, whereas we found that an intact insulin-signaling pathway in liver was sufficient to mediate the suppression of HGP even when the decrease in FFA was abrogated. The Shulman group has reported results similar to ours in wild-type mice using the physiological substrate FFAs, i.e. intralipid/heparin infusion failed to prevent insulin-dependent suppression of HGP (Kim et al., 2004a; Kim et al., 2004b; Kim et al., 2001). Similarly, in dogs the liver autonomous insulin-signaling pathway is dominant in the control of HGP (Edgerton et al., 2006). These studies suggest that the effect of the indirect pathway in regulation of HGP comes into play only under conditions of insulin resistance induced by HFD feeding, genetic modification of insulin signaling or pharmacological manipulations such as infusion of somatostatin. The contextual dependence of cell non-autonomous regulation of HGP by insulin may explain some of the contradictory data regarding the importance of indirect insulin signaling (Moore et al., 2012). Overall, using novel genetic mouse models and nutritional stress, our studies illuminate the controversy regarding the relative contribution of the direct and indirect pathways to the regulation of HGP and bring a consistent view to a seemingly contradictory literature.
Several studies have reported elevated liver Foxo1 activity during the progression of insulin resistance (Aoyama et al., 2006; Qu et al., 2006). In our study, we found that mice with livers deficient for Akt or mice subjected to HFD feeding, constitutively active Foxo1 blocked the effects of insulin on liver not by altering hepatic signaling or metabolic pathways, but by desensitizing adipose tissue to the anti-lipolytic actions of insulin. We suggest that the communication between liver and adipose tissue was provided by chronic hyperglycemia secondary to decreased hepatic glucose utilization, as expression of glucokinase was sufficient to mimic loss of hepatic Foxo1 and improve fasting hyperinsulinemia in L-AktDKO mice. Indeed, deletion of three Foxo isoforms in liver increases glucokinase expression and improves glucose tolerance and insulin sensitivity during high-fat diet (Haeusler et al., 2014; Xiong et al., 2013). Similar beneficial outcomes are observed followed increased hepatic glucokinase expression or activity in both genetic and diet-induced diabetic and insulin resistant models (Desai et al., 2001; Ferre et al., 1996; Pfefferkorn, 2013; Torres et al., 2009). These data define the mechanism by which hepatic Foxo1 improves insulin sensitivity and suggest normalizing hepatic glucose utilization improves glucose homeostasis multiple direct and indirect mechanisms.
In summary, we have elucidated the direct and indirect mechanisms mediating insulin’s coordinated physiological regulation of hepatic lipid synthesis and glucose production in vivo (Figure 7F). Furthermore, these data support the notion that during “insulin-resistant” pathological states in humans, portal fatty acids levels are adequate to drive gluconeogenesis, but that insulin signaling remains intact in liver, allowing uncontrolled DNL via the pathways we have defined in these experiments. These data indicate an essential role of FFA in determining rates of HGP during both genetic and physiological insulin resistance. Whether elevated FFAs alone are sufficient to increase HGP during diabetes independent of changes in other extra-hepatic mechanism such increased glucagon secretion is unclear. Nonetheless, this hypothesis implies that there is no inherent defect in insulin signaling in the livers of individuals with T2DM, but rather a metabolic dysregulation that allows FFA to become a governing regulator of HGP. The precise nature of the hepatic abnormalities leading to the dominant action of FFA on glucose production will require further experimentation.
Experimental Procedures
Mice
Male mice (mus musculus) were used in all experiments. The L-AktDKO and L-AktFoxo1TKO were described previously (Lu et al., 2012). Akt1loxP/loxP, Akt2loxP/loxP, Akt1loxP/loxP, Akt2loxP/loxP, and FoxO1loxP/loxP, Akt1loxP/loxP, Akt2loxP/loxP, and Tsc1loxP/loxP Akt1loxP/loxP, Akt2loxP/loxP, Tsc1loxP/loxP, FoxO1loxP/loxP mice were injected at 6- to 8-weeks of age with 1011 genomic copies per mouse adeno-associated-virus containing a liver specific promoter, thyroxine-binding globulin (TBG), driving either GFP or Cre recombinase to generate LWT, L-AktDKO, L-AktFoxo1TKO, L-AktTsc1TKO, and L-AktTsc1Foxo1QKO. The “LWT” group consisted of GFP injected littermates floxed for the indicated genotypes. Experiments were performed 2 to 3 weeks after virus injection. To block acutely liver glucagon action, an adeno-associated-virus encoding a dominant negative form of the regulatory subunit for PKA (PKA-DN) was injected at the same time as AAV-GFP or AAV-Cre. To express glucokinase in liver, an adeno-associated-virus encoding glucokinase (Gck) was injected at the same time as AAV-GFP or AAV-Cre. All mice experiments were reviewed and approved by the University of Pennsylvania IACUC in accordance with the guidelines of the NIH.
Liver protein extraction and western blotting
Protein lysates were prepared from frozen livers or hepatocytes in a modified RIPA buffer as described previously (Titchenell et al., 2015). The following antibodies were used for immunoblotting: pAkt (#4060), Akt (#2964), pFoxo1 (#9464), Foxo1 (#9452), PRAS40 (#2610) pS6 (#2215), S6 (#2217), p4EBP1 (#2855), 4EBP1 (#9452), Tsc1 (#6935), pPKA substrates (#9624), pS133 Creb (#9198), RI alpha (#5675) were from Cell Signaling Technology. Igfbp1 (sc-6000) and Tsc2 (sc-893) were from Santa Cruz. pPRAS40 (07-888) was from Millipore Corporation.
mRNA isolation and real-time PCR
Total RNA was isolated from frozen livers using the NucleoSpin RNA kit (Clontech). Complementary DNA was synthesized using Moloney murine leukemia virus reverse transcriptase and the relative expression of the genes of interest was quantified by real-time PCR using the SYBR Green Dye-based assay.
Primary hepatocytes isolation
Primary hepatocytes were isolated as previously described (Miller et al., 2011). Cells were lysed in modified RIPA buffer described above and subjected to western blot analysis with indicated antibodies.
Metabolic Measurements
For glucose tolerance test, overnight fasted mice were administered glucose at 2 g/kg body weight via intraperitoneal injection. Insulin concentration was measured using an ultra sensitive ELISA (Linco).
Selective hepatic branch vagotomy
Hepatic branch vagotomy was performed immediately before the placement of an indwelling catheter in the internal jugular vein as previously described (Pocai et al., 2005b). Briefly, the hepatic branch of this vagal trunk was isolated and then transected by microcautery, severing the hepatic vagus, thereby minimizing the possibility of regeneration. Anatomical nerve transaction was verified at sacrifice by microscopic observation of the absence of vagal nerve fibers.
Hyperinsulinemic-euglycemic clamp
Euglycemic clamps were performed on unrestrained and awake 5 hour fasted mice as described previously (Wan et al., 2013). Where indicated, an Intralipid and heparin infusion was performed to raise free fatty acids levels immediately before hyperinsulinemic-euglycemic clamps (Kim et al., 2001). Prior to the initiation of the insulin portion of the clamp, a 100 ul bolus of 10% intralipid/heparin (3 U) or 2.25% glycerol was administered I.V. During the insulin clamp, intralipid was infused at 5 ml/kg/h and heparin at the rate of 6 U/h. A glycerol solution was infused in control mice at a rate of 4.5 mg/h. At the end of the 2 hr clamp, mice were sacrificed by pentobarbital injection, and livers were quickly removed, freeze clamped in liquid nitrogen, and stored at −80°C for future uses.
De novo lipogenesis
The amount of newly made hepatic lipid was determined as previously described (Wan et al., 2011). Briefly, mice were fed HCD (D12450B, Research Diets) for 2 days and fasted overnight. Mice were fed for 3 h then injected I.P. with D2O and allowed access to food for a following 3 h. Plasma and liver samples were taken at 6 h post feeding. Palmitate was analyzed by using GC/MS and the absolute amount of newly made palmitate was assumed equivalent to the de novo lipogenesis rate.
Metabolites
Supernatants from pulverized liver tissue samples treated with 80:20 methanol:water were dry ice, extracted, and reconstituted in LC/MS grade water and analyzed via reverse-phase ion-pairing chromatography coupled to an Exactive orbitrap mass spectrometer (ThermoFisher Scientific, San Jose, CA) in negative ion mode (Lu et al., 2010) or analyzed on an Thermo Finnigan TSQ Quantum Ultra triple quadrupole mass spectrometer (Thermo Electron Corp., San Jose, CA) operating in positive ion selected reaction monitoring (SRM) mode, coupled with a Shimadzu LC-10AD HPLC system (Shimadzu, Columbia, MD), on a Luna NH2 column (250 mm × 2 mm, 5 μm particle size; Phenomenex, Torrance, CA) at a basic pH with a running time of 40 min as described previously (Bajad et al., 2006). The data analyses were performed using MAVEN software as previously described (Melamud et al., 2010).
Glucokinase activity
Glucokinase activity from flash frozen liver was detected using spectrophotometric assay as previously described (Zelent et al., 2006).
Statistical analysis
All data are presented as mean ± s.e.m. Statistical analysis was performed using one-way analysis of variance when more than two groups were compared, two-analysis of variance when two conditions were involved and an unpaired 2 tailed Students’s t test when only two groups of data were concerned. A P <0.05 was considered statistically significant.
Supplementary Material
Highlights.
Insulin autonomously regulates hepatic lipid synthesis in vivo
Activation of mTORC1 is not sufficient to induce lipogenesis in the absence of Akt
Inhibition of Foxo1 and activation of mTORC1 are sufficient to drive lipogenesis
Foxo1 controls adipocyte lipolysis to non-autonomously regulate glucose production
Acknowledgments
The authors would like to thank D. Accili (Columbia University) for sharing the Foxo1loxP/loxP mice, G. Schwartz (Albert Einstein College of Medicine) for instruction in vagotomy, and Mitch Lazar for careful reading of the manuscript. Deuterium-labeled palmitate levels were measured by the Stable Isotope Tracer Kinetic Service Center in the University of Pennsylvania. The Viral Vector Core and Metabolomics Core of University of Pennsylvania supported by the Diabetes and Endocrinology Research Center (NIH DK19525) provided viruses. This work was supported by the US National Institutes of Health grant R01 DK056886 (M.J.B.), NRSA individual postdoctoral fellowship F32 DK101175 (P.M.T.) and the Samuel Chiaffa Memorial Fund (P.M.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
P.M.T. conceived the hypothesis, designed and performed the experiments and analyzed the data. W.J.Q., contributed to discussion, performed experiments and analyzed data. M.L., W.L., J.R., C.L performed experiments and analyzed the data. Q.C., B.R.M., H.C., J.C. provided technical assistance. M.J.B. conceived the hypothesis and directed the project. P.M.T. and M.J.B. prepared the manuscript.
References
- Aoyama H, Daitoku H, Fukamizu A. Nutrient control of phosphorylation and translocation of Foxo1 in C57BL/6 and db/db mice. International journal of molecular medicine. 2006;18:433–439. [PubMed] [Google Scholar]
- Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. Journal of chromatography A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Banks AS, Kim-Muller JY, Mastracci TL, Kofler NM, Qiang L, Haeusler RA, Jurczak MJ, Laznik D, Heinrich G, Samuel VT, et al. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metab. 2011;14:587–597. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Cherrington AD, Edgerton D, Sindelar DK. The direct and indirect effects of insulin on hepatic glucose production in vivo. Diabetologia. 1998;41:987–996. doi: 10.1007/s001250051021. [DOI] [PubMed] [Google Scholar]
- Deng X, Zhang W, I OS, Williams JB, Dong Q, Park EA, Raghow R, Unterman TG, Elam MB. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem. 2012;287:20132–20143. doi: 10.1074/jbc.M112.347211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai UJ, Slosberg ED, Boettcher BR, Caplan SL, Fanelli B, Stephan Z, Gunther VJ, Kaleko M, Connelly S. Phenotypic correction of diabetic mice by adenovirus-mediated glucokinase expression. Diabetes. 2001;50:2287–2295. doi: 10.2337/diabetes.50.10.2287. [DOI] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton DS, Lautz M, Scott M, Everett CA, Stettler KM, Neal DW, Chu CA, Cherrington AD. Insulin’s direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest. 2006;116:521–527. doi: 10.1172/JCI27073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre T, Pujol A, Riu E, Bosch F, Valera A. Correction of diabetic alterations by glucokinase. Proc Natl Acad Sci U S A. 1996;93:7225–7230. doi: 10.1073/pnas.93.14.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Camastra S, Astiarraga B, Nannipieri M, Anselmino M, Ferrannini E. Decreased expression of hepatic glucokinase in type 2 diabetes. Molecular metabolism. 2015;4:222–226. doi: 10.1016/j.molmet.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun. 2014;5:5190. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O’Brien WR, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004a;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, Mozell RL, Tan G, Stricker-Krongrad A, Hirsch DJ, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004b;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3281–3282. doi: 10.1073/pnas.1000323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Analytical chemistry. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Analytical chemistry. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121:2518–2528. doi: 10.1172/JCI45942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Advances in nutrition. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, Olson DP. Central nervous system control of metabolism. Nature. 2012;491:357–363. doi: 10.1038/nature11705. [DOI] [PubMed] [Google Scholar]
- O-Sullivan I, Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, DePinho RA, Stolz DB, Kahn CR, Schwartz MW, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn JA. Strategies for the design of hepatoselective glucokinase activators to treat type 2 diabetes. Expert opinion on drug discovery. 2013;8:319–330. doi: 10.1517/17460441.2013.748744. [DOI] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature. 2005a;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005b;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnanan CJ, Edgerton DS, Cherrington AD. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell Metab. 2012;15:656–664. doi: 10.1016/j.cmet.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin K, Steil GM, Getty L, Bergman RN. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes. 1995;44:1038–1045. doi: 10.2337/diab.44.9.1038. [DOI] [PubMed] [Google Scholar]
- Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest. 1996;98:741–749. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres TP, Catlin RL, Chan R, Fujimoto Y, Sasaki N, Printz RL, Newgard CB, Shiota M. Restoration of hepatic glucokinase expression corrects hepatic glucose flux and normalizes plasma glucose in zucker diabetic fatty rats. Diabetes. 2009;58:78–86. doi: 10.2337/db08-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Leavens KF, Hunter RW, Koren S, von Wilamowitz-Moellendorff A, Lu M, Satapati S, Chu Q, Sakamoto K, Burgess SC, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013;18:99–105. doi: 10.1016/j.cmet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, Kaestner KH, Sabatini DM, Birnbaum MJ. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011;14:516–527. doi: 10.1016/j.cmet.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Tao R, DePinho RA, Dong XC. Deletion of hepatic FoxO1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PLoS One. 2013;8:e74340. doi: 10.1371/journal.pone.0074340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelent D, Golson ML, Koeberlein B, Quintens R, van Lommel L, Buettger C, Weik-Collins H, Taub R, Grimsby J, Schuit F, et al. A glucose sensor role for glucokinase in anterior pituitary cells. Diabetes. 2006;55:1923–1929. doi: 10.2337/db06-0151. [DOI] [PubMed] [Google Scholar]
- Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.