

Abstract
The ability of adult cardiomyocytes to regenerate is limited, and irreversible loss by cell death plays a crucial role in heart diseases. Autophagy is an evolutionary conserved cellular catabolic process through which long-lived proteins and damaged organelles are targeted for lysosomal degradation. Autophagy is important in cardiac homeostasis and can serve as a protective mechanism by providing an energy source, especially in the face of sustained starvation. Cellular metabolism is closely associated with cell survival, and recent evidence suggests that metabolic and autophagic signaling pathways exhibit a high degree of crosstalk and are functionally interdependent. In this review, we discuss recent progress in our understanding of regulation of autophagy and its crosstalk with metabolic signaling, with a focus on the nutrient-sensing mTOR complex1 (mTORC1) pathway.
Keywords: autophagy, mTORC1, amino acids, glucose, hypoxia, metabolism
1. Introduction
The heart is a high-energy demanding organ as it is required to support the beat-to-beat contraction/relaxation cycle. Myocardial energy reserves are limited, just enough to fuel 10 heart beats. This is further decreased in the failing heart and thus, to meet high energy demand, the heart needs to constantly generate ATP by using free fatty acids (FFAs), glucose, lactate, ketone bodies and amino acids. Although the heart derives energy primarily from the oxidation of FFAs, the heart alters its energy substrate use to adapt to changes in nutrient availability. For example, glucose utilization is increased in response to feeding or hypoxia and the use of ketone bodies and amino acids is increased under starvation, providing metabolic flexibility to ensure cardiac energy homeostasis[1-3]. In response to ischemia, cellular uptake of metabolic substrates such as fatty acids, glucose and oxygen is diminished and sustained ischemia causes energy depletion and eventual cell death [4, 5]. Since adult cardiomyocytes have limited ability to regenerate, cardiomyocyte death is a major cause of heart disease. High calorie diet induces metabolic syndrome in which hyperglycemia and hyperlipidemia mediates cardiotoxicity and heart dysfunction [1-3].
Macroautophagy (hereafter referred to as autophagy) is an intracellular recycling system whereby cytoplasmic components and damaged organelles undergo lysosomal degradation. Autophagy is activated in response to stresses including low nutrient availability, to provide an energy source [6-10]. Autophagy, which means “self-eating” in Greek, was first described by Christian De Duve [11], when he observed the sequestration of cytoplasmic components and organelles into newly emerging double-membrane vesicles called autophagosomes. Autophagy consists of several sequential steps - membrane nucleation, elongation, autophagosome formation, fusion with lysosomes, autophagolysosome formation[6-10, 12]. Autophagy is a highly conserved process from yeast to humans, and is governed by a series of autophagy-related (Atg) proteins [12, 13]. This self-digestion process was initially considered as a cell death mechanism (type II programmed cell death) and indeed excessive autophagy contributes to cardiovascular diseases including ischemia/reperfusion injury, although the functional role of autophagy in I/R injury is still under debate [14-20]. It has been shown that autophagy and autophagic flux are increased by I/R, mainly due to oxidative stress, and that excessive activation of autophagy induced by I/R exerts detrimental effects [14, 21-23]. On the contrary, it has been demonstrated that autophagy induced by I/R plays a protective role in cardiomyocytes [15, 22]. Furthermore, Ma et al., reported that autophagic flux is impaired during reperfusion in part by oxidative stress and this contributes to cardiomyocyte death in I/R injury [16] and it has been shown that enhanced autophagic flux mediates HDAC inhibitor-induced protective effects against I/R [17]. Thus further studies will be required to determine the regulation of autophagy by I/R and its functional role during reperfusion.
Nonetheless it has been established that autophagy plays an important role in cellular homeostasis under basal conditions as well as serves as a protective mechanism against ischemia and starvation. For instance, induction of autophagy plays a critical role in neonatal survival [24]. In the heart, deletion of Atg5, a protein required for autophagosome elongation and maturation, leads to cardiac hypertrophy, left ventricular dilation and contractile dysfunction indicating that autophagy in the heart under baseline conditions is a homeostatic mechanism [25, 26]. Autophagy is rapidly induced in response to nutrient starvation or cellular stress, digesting cellular contents to produce amino acids and fatty acids to synthesize proteins or to produce ATP for cell survival [14, 25, 27-33]. It has been shown that inhibition of autophagy increases myocardial infarction induced by chronic ischemia while induction of autophagy is protective [14, 31, 33-37]. Removal of damaged mitochondria by autophagy also provides cardioprotection by preventing mitochondria death pathways [38-42]. Autophagy is a highly regulated cellular process and it is important to develop a comprehensive understanding of the autophagic signaling complexity involved in maintaining the fine balance between adaptive and maladaptive autophagy. Induction of autophagy in response to nutrient starvation is established to be regulated by several protein kinases including AMPK, mTOR and ULK1. This review summarizes recent progress in our understanding of nutrient-sensing mechanisms that regulate mTOR complex 1 (mTORC1) and the initiation of autophagy.
2. Biology of the mTOR pathway
2.1. mTORC1 negatively regulates autophagy
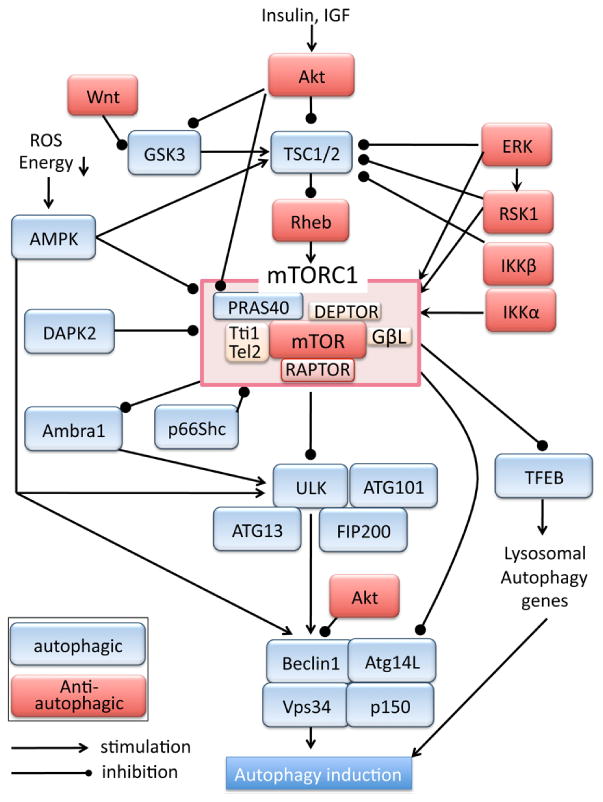
Mechanistic (mammalian) target of rapamycin (mTOR), a serine/threonine kinase, plays a major role in regulating cellular growth and metabolism. mTOR forms two distinct signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [10, 43-45]. mTORC1 constitutes of mTOR, Raptor (regulatory-associated protein of mTOR), GβL/mLST8, Tti1/Tel2, DEPTOR (DEP domain-containing mTOR-interacting protein) and PRAS40 (proline-rich Akt substrate 40 kDa)(figure 1) [46-50]. Raptor is the defining component of mTORC1, acting as an essential scaffold for mTORC1-mediated phosphorylation of downstream target molecules such as 4E-BP1 and p70S6K [48, 51, 52]. PRAS40 is an inhibitory binding protein of mTORC1 [46, 47, 53, 54]. Under nutrient-rich growth conditions, mTORC1 supports cellular growth and suppresses autophagy. On the contrary, in response to nutrient starvation, mTORC1 is inhibited and autophagy is induced to provide energy source. Genetic or pharmacological inhibition of mTORC1 activity has been shown to increase autophagy and provide cardioprotection against stress [14, 31, 33-37, 54]. mTORC1 inhibits the autophagy-initiating molecular complex composed of ULK (Atg1), Atg13, Atg101 and FIP200 through phosphorylation of ULK (figure 1) [55-59]. In addition, ULK stability and activity is also inhibited by phosphorylation and inhibition of AMBRA1 (autophagy/beclin 1 regulator 1) mediated by mTORC1 [60]. ULK1 positively regulates activity of Vps34, a class III phosphatidylinositol 3-kinase (PI3K), which forms molecular complexes with several components of the autophagy machinery including Beclin1 and Atg14L and plays a critical role in vesicle nucleation in autophagy[61]. It has been shown that ULK1 phosphorylates Beclin1 to activate Vps34 activity, thus mTORC1-mediated inhibition of ULK1 results in inhibition of Vps34 activity and autophagy[62, 63]. Further, mTORC1 phosphorylates Atg14L and inhibits the lipid kinase activity of Vps34 [64]. mTORC1-induced inhibition of the Vps34 complex therefore serves as a brake on initiation of autophagy. In addition to directly acting on key components of the autophagic pathway, mTORC1 also transcriptionally inhibits autophagy by phosphorylating and inhibiting TFEB (transcription factor EB), a key regulator of lysosomal and autophagy genes [65, 66].
Figure 1. mTORC1 pathway and autophagy.
2.2. Upstream kinases of the mTORC1 pathway
2.2.1. Activation of mTORC1
2.2.1.1 Akt dependent
Growth factors such as IGF and insulin bind their receptors leading to activation of PI3K/Akt signaling. The mTORC1 pathway is one of the most established downstream targets of Akt. Akt phosphorylates and inhibits TSC (tuberous sclerosis)1/2 complex, a GTPase-activating protein (GAP) for the small G-protein Rheb (Ras homolog enriched in brain) [67-69]. Inhibition of TSC1/2 leads to an increase in GTP-bound active Rheb which is a direct activator of mTORC1 (figure 1). In cardiac-specific Rheb transgenic (TG) mice, mTORC1 activity is increased and autophagy induction is suppressed under ischemic conditions, and the hearts are susceptible to ischemic injury [35]. A study using Rheb-deficient mice also suggest that the Rheb-mTORC1 pathway is indispensable for cardiac hypertrophic growth after early postnatal period [70]. Akt also phosphorylates PRAS40, an inhibitory binding protein of mTORC1, and dissociates PRAS40 from Raptor, leading to activation of mTORC1 [46, 47, 54]. Akt also phosphorylates and inhibits glycogen synthesis kinase (GSK) 3α/β, an inhibitor of mTORC1 activation, relieving the inhibitory effects of GSK3s on mTORC1 [71, 72]. Wnt has also been demonstrated to inhibit GSK3-dependent phosphorylation of TSC2, independent of the canonical β-catenin dependent regulation, and thereby stimulate the mTORC1 pathway [72]. In addition, Akt directly inhibits activity of pro-autophagic Vps34 complex through phosphorylation of Beclin1 [73]. Thus Akt plays a major role in activation of mTORC1 to inhibit autophagy. All three known Akt family members, Akt1, Akt2 and Akt3, are expressed in the heart, although Akt1 and Akt2 are the predominant isoforms [74, 75]. It has been suggested that Akt2 regulates cardiac metabolism and Akt1 regulates cardiac growth, but both confer cardioprotection [74-78]. mTORC1 activity is upregulated and autophagy is suppressed in the hearts of high-fat diet-induced obesity mice [35, 79]. Interestingly, Akt2, but not Akt1 nor Akt3 is upregulated by high-fat diet and plays a critical role in activation of mTORC1 as well as in regulation of autophagy flux [80], while caloric restriction compromises mTORC1 activity and increases autophagy in the heart [81, 82].
2.2.1.2. Akt independent
Independent of PI3K/Akt pathway, ERK (Extracellular signal-regulated kinase) activation is reported to inhibit TSC1/2 as well as activate Raptor [83, 84] resulting in mTORC1 activation. Downstream of ERK, p90 ribosomal S6 kinase 1 (RSK1) also inhibits TSC1/2 and activates Raptor to promote mTORC1 activity [86, 87]. ERK and RSK1 activation has been suggested to contribute to phenylephrine induced mTORC1 activation and protein synthesis in adult rat ventricular cardiomyocytes [85]. IKKβ, an upstream kinase of NF-κB signaling pathway, is also found to phosphorylate and inhibit TSC1 activating mTORC1 pathway in non-cardiomyocytes [86]. Importantly, IKKβ dependent mTORC1 activation is also reported in cardiomyocytes [87]. IKKα is also reported to be involved in activation of mTORC1 [88]. These results suggest the close interaction between mTORC1 and NF-κB signaling pathways [87].
2.2.2. Inhibition of mTORC1
2.2.2.1. AMPK dependent
AMP-activated protein kinase (AMPK) is a sensor for metabolic suppression. It is activated by reduction in cellular ATP levels (increase in AMP/ATP ratio) caused by glucose deprivation, or decrease in mitochondrial oxidative phosphorylation during metabolic suppression. A previous study demonstrated that induction of autophagy by in vivo ischemia is attenuated in AMPK dominant-negative TG mouse hearts [14]. AMPK negatively regulates the mTORC1 pathway at multiple steps [29, 58, 59, 89-91]. It phosphorylates and enhances TSC1/2 activity, and also phosphorylates Raptor inducing its binding to 14-3-3, both resulting in inhibition of mTORC1 activation [90, 91]. In addition, AMPK directly phosphorylates and inhibits ULK1 and Beclin1 to induce autophagy [59, 60, 95, 98]. Previous studies have demonstrated a central role for AMPK in the regulation of cardiac metabolism and autophagy. AMPK is activated in the hearts of caloric restriction mice and AMPK inhibition reverses mTORC1 inactivation and diminishes autophagy induction [92]. In high-fat diet induced obesity mice, cardiac AMPK activity is decreased, resulting in activation of mTORC1 and inhibition of autophagy [79], suggesting the central role of AMPK in the regulation of cardiac metabolism and autophagy.
2.2.2.2. AMPK independent
Glycogen synthesis kinase (GSK) 3α/β was originally identified as a negative regulator of glycogen synthesis, but it is now recognized that GSK3 regulates many other cellular functions including apoptosis [93]. GSK3 is a constitutively active kinase and its activity is inhibited by Akt mediated phosphorylation, as mentioned in the previous section. GSK3 inhibits the mTOR pathway by phosphorylating TSC2 [72]. A study in the heart demonstrated that inhibition of GSK3β stimulated mTOR signaling and inhibited autophagy, resulting in increased cardiac damage after prolonged ischemia [94]. Chronic inhibition of GSK3α and resultant overactivation of mTORC1 induces suppression of autophagy, and this contributes to age-related pathologies including cardiac hypertrophy and contractile dysfunction [95]. Death-associated protein kinase 2 (DAPK2) is a calcium/calmodulin (CaM)-regulated serine/threonine kinase and is abundantly expressed in heart, lung, and skeletal muscle [96]. DAPK2 inhibits mTORC1 through phosphorylation of Raptor, and it has been shown to enhance autophagy induced by amino acid deprivation or increase in intracellular calcium by thapsigargin [97]. p66Shc is one of the SHC1 gene encoding proteins and is known as an adaptor molecule. p66Shc has been shown to increase mitochondrial oxidative stress in different cells including cardiac myocytes [104-108] and p66Shc upregulation is also suggested to be associated with type 2 diabetes and obesity [98-101]. Recent studies demonstrate that p66Shc inhibits mTORC1 activity induced by serum or insulin, and thereby limits glucose uptake and metabolism [113] and that p66Shc positively regulates autophagy in human lung adenocarcinoma [102]. Although these studies have linked p66Shc to the regulation of mTORC1, energy metabolism and autophagy, the mechanism by which p66Shc inhibits mTORC1 has not been fully determined nor has it been examined whether mitochondrial distribution and resultant oxidative effect of p66Shc is involved in mTORC1 inhibition. It would be of interest to test the role of p66Shc in metabolism and autophagy in the heart.
3. Nutrient sensing regulation of mTORC1
3.1. Amino-acid dependent regulation of TORC1
3.1.1 mTORC1 activation at the lysosome
In 1977, Mortimore and Schworer demonstrated for the first time that amino acid depletion directly induces formation of autophagosomes in the perfused liver [103]. mTORC1 is a key component in amino acid deprivation-induced autophagy. Withdrawal of amino acids from culture media was shown to rapidly inactivate mTORC1 signaling in mammalian cell lines [104]. The amino acids, leucine, arginine and glutamine, demonstrate particular potency in mTORC1 activation [116-119]. It has not been fully determined whether and how amino acids regulate mTORC1 activity in the heart but many insights into amino acid-dependent regulation of mTORC1 have been derived from studies in non-cardiac cells. Interestingly, the amino acid-dependent regulation of mTORC1 is independent of PI3K/Akt and TSC pathway [104-106], suggesting the existing of alternative amino acid sensing mechanism.
Identification of the Rag subfamily of Ras-related small G-proteins (Rag GTPase) has led to improve understanding of amino acid-dependent regulation of mTORC1 (figure 2) [107, 108]. The Rag family proteins are comprised of four members (RagA, B, C and D) and form heterodimers, RagA/B and RagC/D. Rag proteins play a crucial role in the heart, as loss of RagA/B in cardiomyocytes results in hypertrophic cardiomyopathy [109]. While the Rag complexes do not directly activate mTORC1 kinase activity, they mediate mTORC1 translocation to the lysosome in response to amino acid stimulation [110]. Binding of the Rag complexes to the lysosomal membrane is aided by Ragulator (LAMPTOR1-3 complex) which resides on the lysosome. Ragulator functions as a guanine nucleotide exchange factor (GEF) to activate Rag GTPases leading to enhanced binding of the Rag complexes to mTORC1[111]. Thus the Ragulator-Rag complex serves as a docking site for mTORC1 at lysosomes in response to amino acids (figure 2).
Figure 2. Amino acid-dependent regulation of mTORC1.
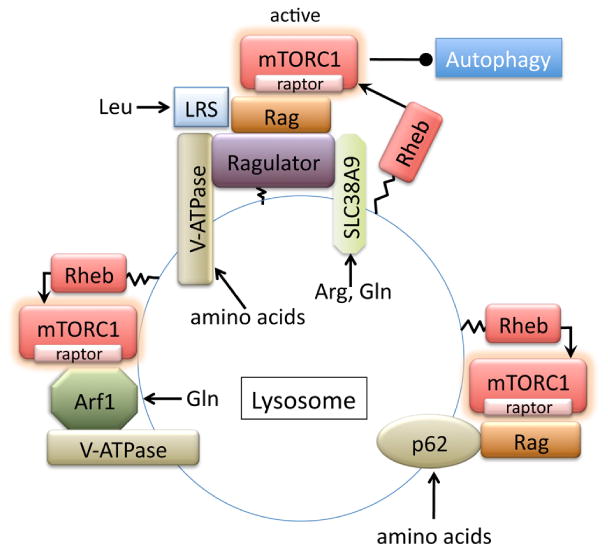
p62, also called sequestosome 1(SQSTM1), is an adaptor protein involved in the regulation of diverse cellular functions through its multi-domain structure. p62 has been reported to regulate mTORC1 activity in response to amino acids, but not to insulin [127]. p62 binds the Rag GTPases and Raptor and this binding is independent of Ragulator, providing an alternative docking site at the lysosome (figure 2) [127]. Rheb, a direct activator of mTORC1, is reported to localize on multiple endomembrane compartments including the lysosome [108, 112]. Thus the recruitment of mTORC1 to the lysosome brings it into proximity with Rheb, resulting in mTORC1 activation [108, 112]. On the contrary, upon amino acid removal, mTORC1 is released from the lysosome, causing it to become cytoplasmic and inactive[129, 130]. This dissociation and inactivation of mTORC1 is positively regulated by the TSC complex translocation to the lysosome induced by amino acid removal. [129, 130].
3.1.2 Amino acid sensing mechanisms
A limited RNAi screen for lysosomal proteins required for mTORC1 activation by amino acids identified vacuolar H+-ATPase (v-ATPase) as a potential amino acid sensing protein [113]. In this paper, it is shown that v-ATPase interacts with and activates Ragulator in response to accumulation of amino acids in the lysosomal lumen, and is needed for the activation of the Rag GTPases and subsequent mTORC1 recruitment to the lysosome. ATP hydrolysis and associated rotation of the v-ATPase, but not the lysosomal proton gradient, appear to be essential for activation of the Ragulator mediated by the v-ATPase. Two recent independent studies have further identified SLC38A9 (number 9 of the solute carrier family 38) as a novel physical and functional component of the lysosomal machinery that controls mTORC1 activity in response to amino acid [114, 115]. SLC38A9 transports amino acids across the lysosomal membrane and binds the Ragulator-Rag GTPases complex in an amino acid-sensitive manner to stimulate mTORC1 activity [114, 115]. These studies also demonstrated the differential regulation of mTORC1 by specific amino acids. Wang et al., showed that SLC38A9 is an arginine transporter and responsible for arginine- but not leucine-induced mTORC1 activation[114]. Rebsamen et al., demonstrated that SLC38A9 has an ability to transport glutamine as well as arginine [115]. Leucyl-tRNA synthetase (LRS), which catalyzes the attachment of leucine to its tRNA, has been shown to directly bind to and regulate RagD, stimulating mTORC1 activity [116]. This non-canonical role of LRS might provide a novel mechanism for leucine-selective mTORC1 regulation.
Although deletion of RagA and RagB in cardiomyocytes results in hypertrophic cardiomyopathy, mTORC1 activity was not substantially impaired in the heart [109], implying the existence of Rag GTPase-independent mechanism for mTORC1 activation. Indeed, a mechanism for amino acid-dependent but Rag-Ragulator-independent mTORC1 activation at the lysosome has recently been discovered [117]. In RagA/B-deficient cells, leucine failed to activate mTORC1, but the ability of glutamine to activate mTORC1 was preserved, suggesting that RagA/B is required for mTORC1 activation by leucine but not glutamine. The study further demonstrated that glutamine-induced mTORC1 recruitment to the lysosome and subsequent activation required v-ATPase and adenosine diphosphate ribosylation factor-1 (Arf1), a key regulator of intracellular vesicle trafficking [117]. These studies show that mTORC1 is differentially regulated by specificamino acids, but there might be an interplay between amino acids. Glutaminolysis, the process by which glutamine is metabolized to glutamate and subsequently to α-ketoglutarate (αKG), is shown to be sufficient to activate mTORC1 signaling through αKG-dependent activation of RagGTPase [118, 119]. The conversion of glutamate to αKG is activated by glutamate dehydrogenase (GDH). Leucine is an allosteric activator of GDH [120], providing a mechanistic link between leucine and glutamine in glutaminolysis-dependent regulation of mTORC1 activation.
3.2. Glucose-dependent regulation of mTORC1
3.2.1. AMPK dependent and independent mechanism
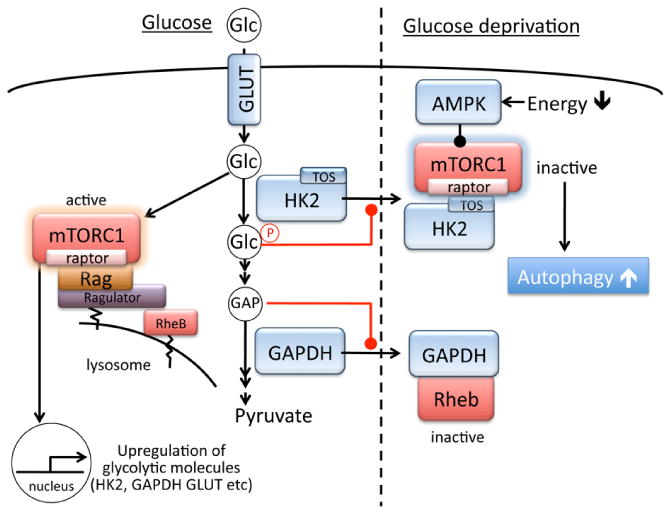
Glucose is an essential energy source and glucose deprivation induces autophagy in many different cell types, which is accompanied with decreased activity of mTORC1. AMPK, activated by reduction of cellular ATP levels, is established to inhibit mTORC1 and induce autophagy in the absence of glucose. Accumulating evidence, however, has revealed that AMPK-independent pathways also regulate mTORC1 activity in the absence of glucose. mTORC1 activity is decreased in response to glucose deprivation in AMPK-α1 and -α2 double knockout MEFs [24, 121]. Similarly mTORC1 is inhibited in TSC1 or TSC2 knockout MEFs subjected to glucose deprivation [119, 138, 139]. These results suggest that mTORC1 inhibition induced by glucose deprivation can take place in an AMPK and TSC1/2 independent manner. It has recently become clear that cells directly sense intracellular glucose levels to regulate the mTORC1 pathway (figure 3). As mentioned above, lysosomes are recognized as an mTORC1 activation site, and it has been shown that glucose deprivation causes mTORC1 to be diffusely distributed in the cytosol in HEK-293T cells [24]. Conversely when mTORC1 is tethered at the lysosome through constitutive activation of RagA expression, glucose deprivation fails to decrease mTORC1 activity even though AMPK is activated [24]. These recent findings suggest that RagGTPases, in addition to their established role in amino acid sensing, participates in the direct sensing of glucose availability to recruit mTORC1 to the lysosome to be activated.
Figure 3. Glucose-dependent regulation of mTORC1.
3.2.2. Glucose-sensing mechanisms
mTOR activation induced by insulin has been shown to require glucose in the heart, suggesting that glucose metabolism has a regulatory role in mTORC1 activation [122, 123]. Glucose-dependent mTORC1 activation is independent of the hexosamine biosynthetic pathway, AMPK, and the pentose phosphate pathway suggesting the contribution of glycolysis to this response. An increase in work load is associated with glucose-6-phosphate (G-6P) accumulation and mTORC1 activation in the heart [122, 123]. The first step of glycolysis is mediated by hexokinases (HKs), which phosphorylate glucose to produce G-6P [124, 125]. Hexokinase-2 (HK2) is the predominant isoform in insulin-sensitive tissues such as skeletal muscle, adipose tissues and heart. HK2 is also upregulated in many types of tumors, associated with the Warburg effect, enhanced aerobic glycolysis[124-126]. In addition to the established role of HK2 in glucose metabolism, HK2 also confers cellular protection. Overexpression of HK2 provides cellular protection against oxidative stress in cardiomyocytes[127-129] and also prevents maladaptive hypertrophy o the heart in vivo [127]. Conversely, heterozygotic HK2 knockout hearts are more susceptible to ischemia/reperfusion injury as well as pressure overload[130, 131]. Studies in the 1960s identified that a significant fraction of total cellular HK2 in the heart is associated with mitochondria via its N-terminal region[132-135]. Mitochondria-associated HKs (mitoHKs) can exert protective effects on mitochondria to prevent mitochondrial death pathways[124, 128, 136-139]. We previously demonstrated that mitoHK2 binding is enhanced by Akt-mediated phosphorylation of HK2 at Thr473, contributing Akt-mediated mitochondria protection [128, 138, 140].
We recently reported that HK2 functions as a molecular switch from glycolysis to autophagy through regulation of mTORC1 (figure 3) [141]. Studying the protective effect of HK2 in cardiomyocytes, we observed that 2-deoxy-D-glucose (2-DG), a glucose analogue that is phosphorylated by HKs but not metabolized further, attenuates decrease in mTORC1 activity, inhibits induction of autophagy, and increases cell death induced by glucose deprivation. This suggests a regulatory role of HK2 in mTORC1 inhibition and protective autophagy in the absence of glucose. HK2 knockdown by siRNA-mediated gene silencing also attenuates mTORC1 suppression and inhibits induction of autophagy while HK2 overexpression potentiates the responses in the absence of glucose [141]. These observations suggest that HK2 acts to suppress mTORC1 and thereby stimulates autophagy in response to its substrate (glucose) withdrawal. We demonstrated that HK2 binds to mTORC1 through Raptor and this binding is largely increased by glucose withdrawal in the heart. We identified that HK2 (but not HK1, which is ubiquitously expressed) contains a TOS (mTOR signaling) motif, which is present in p70S6K and 4E-BP1 (mTORC1 substrates), and through which these substrates bind to Raptor and subsequently undergo phosphorylation by mTOR [52, 142]. A TOS motif-deficient mutant of HK2 fails to bind and inhibit mTORC1. Thus HK2 interacts with mTORC1 via binding to Raptor through its TOS motif, functioning as a decoy substrate. Interestingly, the switch between the glycolytic and autophagic effects of HK2 appears to be regulated by G-6P, a product of HK2 activity [141]. Therefore, under low glucose conditions, decreased levels of G-6P induces HK2 interaction with mTORC1 to facilitate autophagy, while under glucose-rich conditions, HK2 produces G-6P which in turn inhibits HK2 binding to mTORC1 to support cellular metabolism and growth (figure 3). It would be of interest to determine whether HK2 binding to mTORC1 prevents localization of mTORC1 to the lysosome and its subsequent activation.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) catalyzes the conversion of glyceraldehyde 3-phosphate (GAP) to D-glycerate 1,3-bisphosphate, the sixth step of glycolysis. GAPDH has also been implicated in several non-glycolytic cellular functions including roles in the nucleus [143]. The role of GAPDH in regulation of autophagy was first described as a potential mechanism by which GAPDH expression confers cell survival against caspase-independent cell death [144]. A later study in the brain identified GAPDH as a Rheb binding protein [145]. The binding of GAPDH to Rheb is increased by decreasing glucose concentration, leading to dissociation of mTORC1 from Rheb and thus inhibition of mTORC1. This inhibitory binding is preserved in TSC1 KO and AMPK silenced cells, but prevented by binding of GAP to GAPDH (ie., substrate binding)(figure 3). This inhibitory binding of GAPDH to Rheb is also suggested to contribute to GLUT1 upregulation-induced activation of mTORC1 [146]. Interestingly, GAPDH is also implicated in regulation of mitochondria specific autophagy in cardiomyocytes [147].
Taken together these findings suggest that glycolytic flux regulates mTORC1 activity to coordinate cellular metabolic status with autophagy development. Conversely, mTORC1 positively regulates glycolysis. For instance, an unbiased genomic, metabolic and bioinformatic study in TSC1/2 deficient cells reveals that mTORC1 signaling activates the genes encoding nearly every step of glycolysis [148]. This is also supported by other studies using TSC1/2 deficient cells demonstrating that mTORC1 activation is sufficient to upregulate GLUT1, HK2, GAPDH, pyruvate kinase muscle isozyme (Pkm2) and lactate gene expression [149, 150]. A muscle-specific mTOR conditional knockout mouse also showed significantly decreased expressions of GLUT4, HK2 and Pkm2 in the heart [151](figure3). Thus mTORC1 activation enhances glycolysis to support cell growth under nutrient rich conditions, while it is negatively regulated by glycolytic molecules under starvation to ensure cellular energy homeostasis through autophagy, suggesting the intrinsic connection between glycolytic and mTORC1/autophagy pathways.
Alternations in cardiac energy metabolism has been suggested to contribute to cardiac disease. In diabetes, there is a shift in cardiac metabolism away from glucose metabolism towards fatty acid metabolism, which is opposite to the changes observed in heart failure induced by pressure overload [152-155]. The metabolic shift in diabetes, especially in type-1 diabetes (insulin-sensitive diabetes), is associated with significant decrease in HK2 [156-159] and insulin treatment restores HK2 levels supporting the role of Akt/mTORC1 pathway in expression of HK2 [156-158, 160]. Hyperglycemia is also reported to decrease GAPDH expression in endothelial cells [161]. In general, autophagy is decreased in type-1 diabetes thus it would be of interest to determine whether these decreases in HK2 and GAPDH expression are causally related to suppression of autophagy.
3.3. Oxygen dependent regulation of mTORC1
3.3.1. HIF-1
Autophagy regulation mediated by oxygen-sensing signaling pathways has also been reported (figure 4). Hypoxia-inducible factor 1 (HIF-1) is a transcriptional factor, and at low levels of oxygen, degradation of the α-subunit of HIF-1 (HIF-1 α) is inhibited, leading to the activation of a transcriptional program to metabolically adapt to the lack of oxygen [162]. Expression of Bnip3, a mitochondrial pro-apoptotic Bcl-2 protein, is induced by hypoxia through HIF-1, contributing to cardiac damage induced by ischemic stress [163-167]. It has been demonstrated that Bnip3 regulates not only cardiac apoptosis but also mitophagy [168, 169]. Interestingly, Bnip3 negatively regulates mTORC1 pathway. A yeast two-hybrid assay identified Bnip3 as a Rheb-binding protein and the binding decreases GTP-bound Rheb levels, playing an important role in hypoxia-induced mTOR inhibition [170]. It has yet to be determined if Bnip3 inhibits the mTOR pathway to regulate autophagy in the heart. In addition to regulation of mTORC1, Bnip3 and Bnip3L (NIX) have been described to inhibit binding of Bcl-2 to Beclin1, releasing the Bcl-2 dependent inhibition of Beclin1, to drive autophagy [171]. Thus Bnip3 may regulate autophagy at multiple steps.
Figure 4. Oxygen-dependent and redox dependent regulation of mTORC1.
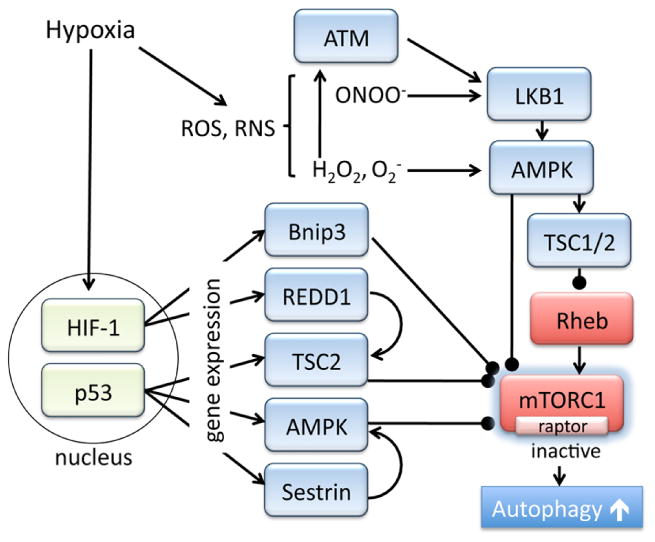
REDD1 (regulated in DNA damage and development 1, also known as RTP801, DDIT4 and Dig2) is a 25 kDa protein which is ubiquitously expressed in various tissues, and is highly induced by hypoxia. The expression of REDD1 is regulated by several transcription factors including HIF-1 and ATF4 (a regulator of ER stress responses), as well as by post-translational regulation (through ubiquitin-proteasome system) [172, 173]. Previous studies demonstrated that REDD1 is rapidly induced by stress and subsequently inhibits mTORC1 activation [174-176]. REDD1 inhibits the interaction of TSC2 with 14-3-3, resulting in greater TSC2 dependent inhibition of mTORC1 [177]. Although the role of REDD1 in the heart has not been fully examined, REDD1 knockdown impairs autophagy in hypertrophied cardiomyocytes [178]. In addition to mTORC1 regulation, a recent study discovered that REDD1 forms a complex with TXNIP, a pro-oxidant protein, and induces ROS, suppresses ATG4B activity and activates autophagy [179].
3.3.2. p53
Another intriguing regulatory mechanism of mTORC1 pathway was obtained from studies of p53, a key tumor suppressor protein. Under physiological conditions, p53 expression is inhibited by MDM2-dependent proteasomal degradation while its expression is increased in response to stress including hypoxia. In addition to the established role of p53 in cell death, it is becoming increasingly recognized that basal or low levels of p53 expression plays an important role in the maintenance of redox state as well as energy homeostasis [180-186]. p53 transcriptionally regulates expression of various molecules which inhibit mTORC1 activity, including AMPK, TSC2 as well as sestrins [187-190]. Sestrin1 and sestrin2 inhibit mTORC1 through activation of AMPK and are involved in the induction of autophagy in tumor cells [188-190]. Intriguingly sestrin2 is expressed in the heart and the expression is increased in response to in vivo ischemia [191]. Furthermore, sestrin2 KO hearts show impaired activation of AMPK and increased cardiac damage induced by ischemia/reperfusion [191]. Thus sestin2 may provide cardioprotection through inhibition of mTORC1 and activation of autophagy.
3.3.3. Oxidative stress dependent mechanism
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are formed by the incomplete reduction of oxygen. It has been demonstrated that the levels of ROS/RNS are increased by starvation or hypoxia, and play a regulatory role in the induction of autophagy [192-194]. For example, ROS, specifically hydrogen peroxide (H2O2), directly activate autophagy during nutrient starvation by oxidization and inhibition of ATG4 and subsequent increase in LC3 lipidation [192]. Activation of AMPK by H2O2, superoxide anion (O2-) or peroxynitrite (ONOO-) has been demonstrated in non-cardiac [195-198] and cardiac cells [199, 200], suggesting a redox dependent regulation of AMPK (figure 4). Mitochondria are an important source of ROS (H2O2 and O2-) which can interact with NO to generate ONOO- and it has been demonstrated that mitochondria-derived ROS are critical in AMPK activation and autophagy under stress conditions [195, 197, 201-203]. ROS and RNS positively regulate AMPK activity through direct and indirect mechanisms. H2O2 results in oxidative modification of AMPKα subunit to increase its catalytic activity [198, 204], and ONOO- induces activation of liver kinase B1 (LKB1), an upstream kinase of AMPK [200, 205]. Recent studies in non-cardiac cells also suggest that ataxia-telangiectasia mutated (ATM) kinase, best known for its role in nuclear DNA damage, functions as a redox sensor in the cytosol to activate AMPK through LKB1, stimulating autophagy [206, 207]. In the heart, ATM has been shown to play a regulatory role in cardiac remodeling [208]. However, the role of ATM in regulating AMPK, mTORC1 and autophagy in the heart has yet to be determined.
3.4. Fatty acid and mTORC1/autophagy
FFAs are the major fuel for the heart. However FFAs-sensing mechanisms in direct regulation of mTORC1 and autophagy in the heart has not been demonstrated. FFAs are transported into cardiomyocytes and esterified to fatty acyl CoA by fatty acyl CoA synthase. Fatty acyl CoA is then transported to mitochondria for beta-oxidation to produce ATP or converted to triglycerides (TGs) and stored in lipid droplets. In pancreatic β-cells, high concentration of palmitic acid activates mTOR, decreases autophagic flux and induces cell death, suggesting the involvement of impaired autophagy in lipotoxicity [209]. In the heart, loss of long-chain acyl-CoA synthase isoform 1 reduces FFA oxidation by >90% and increases glucose usage 8-fold to compensate, which in turn activates mTORC1 and suppresses autophagy [210]. The observations support the reciprocal relationship between FFAs and glucose oxidation (Randle Cycle) [211] as well as the glucose-sensing autophagy inhibition described above. During starvation, FFAs are released from TGs by the process of lipolysis. mTORC1-autophagy pathway is shown to be involved in lipid breakdown by degradation of lipid droplets in hepatocytes [212]. A recent study in liver also showed that TAK1-dependent AMPK activation induced by starvation inactivates mTORC1 resulting in induction of autophagy, as well as activation of PPARα, a key transcription factor in regulation of FFA oxidation, facilitating lipid breakdown [213]. A critical role of autophagy in lipolysis is supported by a study in adipocytes demonstrating that ULK1 and ULK2 enhance lipid breakdown by inducing autophagy[214]. Interestingly, ULK1 and ULK2 have distinct non-autophagic functions in regulation of lipid metabolism; ULK1 stimulates FFA oxidation and inhibits FFA uptake while ULK2 has opposing effects [214]. A recent seminal study in mouse embryonic fibroblasts provides new insight into the role of autophagy in regulation of FFAs trafficking to mitochondria during starvation. Rambold et al., demonstrated that autophagy breaks down cellular membranes to supply FFAs to lipid droplets from where FFAs generated by lipolysis are further transferred into mitochondria to produce ATP. This study also demonstrated that mitochondrial fusion and resultant continuous mitochondrial network are required to distribute and efficiently oxidize transferred FFAs [215]. These recent findings indicate that autophagy plays a crucial role in lipid metabolism and FFAs utilization under nutrient starvation to preserve cellular energy homeostasis and thus it would be of interest to determine if these mechanisms operate in the heart.
Conclusion remark
We have described some recent advances in our understanding of nutrient sensing mechanisms in the regulation of mTORC1 and autophagy. Autophagy is a key cellular catabolic process in which mTORC1 serves as a convergent point in nutrient-sensing pathways. However, the physiological and pathophysiological role and significance of the nutrient-sensing regulation of mTORC1 signaling pathways in the heart need to be further investigated.
Although direct inhibition of mTORC1 facilitates autophagy which could confer cardiac protection, mTORC1 also regulates a myriad of cellular functions in other organs. For example, mTORC1 deficiency leads to skeletal muscle dystrophy in mice [216]. From a whole-body metabolic perspective, mTORC1 positively regulates β cell size and proliferation and insulin secretion in the pancreas, and inhibits ketogenesis and PPARα activity in the liver [217, 218]. Thus global inhibition of mTORC1 may result in widespread systemic disturbances and have a detrimental outcome. It will be important to identify the regulatory molecules, particularly heart-specific molecules, that selectively modulate the mTORC1 pathway to confer cardioprotective autophagy as these would provide potential therapeutic targets that can be explored in the treatment of cardiovascular diseases.
It is intriguing that autophagy is activated in response to nutrient deprivation to provide metabolic defense, and this is tightly regulated by diverse metabolic molecules. Alternation in energy resources and metabolism is an important factor in the progression of cardiac disease. Understanding the complexity of the crosstalk between autophagic and metabolic pathways during the progression of cardiac disease will aid in the development of therapeutic strategies to prevent or treat heart failure.
Highlights.
Autophagy is important for maintaining cardiac homeostasis.
mTORC1 plays a major role in regulating cellular growth and metabolism and inhibits autophagy
mTORC1 serves as a convergent point in nutrient-sensing pathways.
Acknowledgments
This work was supported by National Institutes of Health grant 2R56HL097037 and American heart Association 15GRNT2297009 to S.M.
Abbreviations
- 2-DG
2-deoxy-D-glucose
- α KG
α-ketoglutarate
- AMBRA1
autophagy/beclin 1 regulator 1
- AMPK
AMP-activated protein kinase
- Arf1
adenosine diphosphate ribosylation factor-1
- Atg
autophagy-related
- ATM
ataxia-telangiectasia mutated kinase
- Bcl-2
B-cell lymphoma 2
- Bnip3
BCL2/Adenovirus E1B 19kDa Interacting Protein 3
- Bnip3L (NIX)
BCL2/Adenovirus E1B 19kDa Interacting Protein 3-Like
- DAPK2
Death-associated protein kinase 2
- ERK
Extracellular signal-regulated kinase
- FFAs
free fatty acids
- G-6P
glucose-6-phosphate
- GAP
GTPase-activating protein
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GDH
glutamate dehydrogenase
- GEF
guanine nucleotide exchange factor
- GLUT1
glucose transporter 1
- GSK
glycogen synthesis kinase
- HDAC
histone deacetylases
- HEK
human embryonic kidney
- HIF-1
hypoxia-inducible factor 1
- HK
hexokinase
- IGF
insulin-like growth factor
- IKK
IkappaB kinase
- I/R
ischemia/reperfusion
- LKB1
liver kinase b1
- LRS
Leucyl-tRNA synthetase
- MDM2
mouse double minute 2 homolog
- MEFs
mouse embryonic fibroblasts
- mitoHK
mitochondria associated hexokinase
- mTOR
Mechanistic (mammalian) target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- NF-κB
nuclear factor-κB
- PI3K
phosphatidylinositol 3-kinase
- Pkm2
pyruvate kinase muscle isozyme
- PPARα
peroxisome proliferator-activated receptor-α
- PRAS40
proline-rich Akt substrate 40 kDa
- Rag
Ras-related small G-proteins
- Raptor
regulatory-associated protein of mTOR
- REDD1
regulated in DNA damage and development 1
- Rheb
Ras homolog enriched in brain
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RSK1
p90 ribosomal S6 kinase 1
- siRNA
small interfering RNA
- SLC38A9
number 9 of the solute carrier family 38
- SQSTM1
sequestosome 1
- TAK1
Transforming growth factor-β-activated kinase-1
- TFEB
transcription factor EB
- TG mice
transgenic mice
- TXNIP
thioredoxin-interacting protein
- TSC
tuberous sclerosis complex
- ULK
unc51-like kinase
- v-ATPase
vacuolar H+-ATPase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none.
References
- 1.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005 Jul;85(3):1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 2.Wang ZV, Li DL, Hill JA. Heart failure and loss of metabolic control. J Cardiovasc Pharmacol. 2014 Apr;63(4):302–13. doi: 10.1097/FJC.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013 Aug 16;113(5):603–16. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013 Aug 2;113(4):428–38. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 6.Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. The Journal of Clinical Investigation. 2015;125(1):55–64. doi: 10.1172/JCI73943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishida K, Taneike M, Otsu K. The role of autophagic degradation in the heart. J Mol Cell Cardiol. 2015 Jan;78C:73–9. doi: 10.1016/j.yjmcc.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Orogo AM, Gustafsson AB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circ Res. 2015 Jan 30;116(3):489–503. doi: 10.1161/CIRCRESAHA.116.303791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014 Jan 31;114(3):549–64. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Duve C, Wattiaux R. Functions of Lysosomes. Annu Rev Physiol. 1966;28(1):435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 12.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 13.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010 Aug;6(6):764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007 Mar 30;100(6):914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 15.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006 Oct 6;281(40):29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 16.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012 Jun 26;125(25):3170–81. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014 Mar 11;129(10):1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren J, Taegtmeyer H. Too much or not enough of a good thing--The Janus faces of autophagy in cardiac fuel and protein homeostasis. J Mol Cell Cardiol. 2015 Jul;84:223–6. doi: 10.1016/j.yjmcc.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Wang ZV, Hill JA. Protein quality control and metabolism: bidirectional control in the heart. Cell Metab. 2015 Feb 3;21(2):215–26. doi: 10.1016/j.cmet.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, et al. Blockade of Hsp20 Phosphorylation Exacerbates Cardiac Ischemia/Reperfusion Injury by Suppressed Autophagy and Increased Cell Death. Circ Res. 2009 Dec 4;105(12):1223–31. doi: 10.1161/CIRCRESAHA.109.200378. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006 Jun;40(6):846–52. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 22.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007 Jan;14(1):146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 23.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011 Jun;14(11):2179–90. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013 Jan 31;493(7434):679–83. doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007 May;13(5):619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 26.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010 Jul;6(5):600–6. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 27.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 Jan 11;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005 Jan 28;120(2):237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 29.Takagi H, Matsui Y, Hirotani S, Sakoda H, Asano T, Sadoshima J. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy. 2007 Jul-Aug;3(4):405–7. doi: 10.4161/auto.4281. [DOI] [PubMed] [Google Scholar]
- 30.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010 Dec 3;330(6009):1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005 Sep 27;102(39):13807–12. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013 Dec;123(12):5284–97. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanamori H, Takemura G, Goto K, Maruyama R, Ono K, Nagao K, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. 2011 Jun;300(6):H2261–71. doi: 10.1152/ajpheart.01056.2010. [DOI] [PubMed] [Google Scholar]
- 34.Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol. 2009 Dec 15;54(25):2435–46. doi: 10.1016/j.jacc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 35.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012 Mar 6;125(9):1134–46. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res. 2011 Jul 15;91(2):330–9. doi: 10.1093/cvr/cvr073. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, He L, Chen F, He X, Cai Y, Zhang G, et al. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One. 2014;9(11):e112891. doi: 10.1371/journal.pone.0112891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013 Apr 26;340(6131):471–5. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010 Sep 3;285(36):27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorn GW, 2nd, Kitsis RN. The Mitochondrial Dynamism-Mitophagy-Cell Death Interactome: Multiple Roles Performed by Members of a Mitochondrial Molecular Ensemble. Circ Res. 2015 Jan 2;116(1):167–82. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013 Jan 11;288(2):915–26. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015 Jan 16;116(2):264–78. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 43.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010 Oct 22;40(2):310–22. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013 Feb 7;49(3):379–87. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015 Jan;125(1):25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007 Mar 23;25(6):903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007 Jul 6;282(27):20036–44. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 48.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002 Jul 26;110(2):163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 49.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003 Apr;11(4):895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 50.Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010 Jun 25;285(26):20109–16. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hara K, Maruki Y, Long X, Yoshino K-i, Oshiro N, Hidayat S, et al. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell. 2002;110(2):177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 52.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003 May 2;278(18):15461–4. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 53.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007 Jul 13;282(28):20329–39. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci U S A. 2013 Jul 30;110(31):12661–6. doi: 10.1073/pnas.1301455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009 May 1;284(18):12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009 Apr;20(7):1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009 Apr;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011 Mar 22;108(12):4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–16. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 61.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010 Jun;20(6):355–62. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jaber N, Dou Z, Chen J-S, Catanzaro J, Jiang Y-P, Ballou LM, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proceedings of the National Academy of Sciences. 2012 Feb 7;109(6):2003–8. doi: 10.1073/pnas.1112848109. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013 Jul;15(7):741–50. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013 Dec;9(12):1983–95. doi: 10.4161/auto.26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011 Jun 17;332(6036):1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012 Mar 7;31(5):1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002 Sep;4(9):648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 68.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr Biol. 2003;13(15):1259–68. doi: 10.1016/s0960-9822(03)00506-2. 2015/09/12. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Huang BPH, Luciani DS, Wang X, Johnson JD, Proud CG. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2008;45(6):812–20. doi: 10.1016/j.yjmcc.2008.07.016. 2015/09/12. [DOI] [PubMed] [Google Scholar]
- 70.Tamai T, Yamaguchi O, Hikoso S, Takeda T, Taneike M, Oka T, et al. Rheb (Ras homologue enriched in brain)-dependent mammalian target of rapamycin complex 1 (mTORC1) activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J Biol Chem. 2013 Apr 5;288(14):10176–87. doi: 10.1074/jbc.M112.423640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995 Dec 21-28;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 72.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006 Sep 8;126(5):955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 73.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012 Nov 16;338(6109):956–9. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006 Oct 27;281(43):32841–51. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006 May 2;113(17):2097–104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 76.Kunuthur SP, Mocanu MM, Hemmings BA, Hausenloy DJ, Yellon DM. The Akt1 isoform is an essential mediator of ischaemic preconditioning. J Cell Mol Med. 2012 Aug;16(8):1739–49. doi: 10.1111/j.1582-4934.2011.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miyamoto S, Del Re DP, Xiang SY, Zhao X, Florholmen G, Brown JH. Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J Cardiovasc Transl Res. 2010 Aug;3(4):330–43. doi: 10.1007/s12265-010-9192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011 Jul;91(3):1023–70. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liang L, Shou XL, Zhao HK, Ren GQ, Wang JB, Wang XH, et al. Antioxidant catalase rescues against high fat diet-induced cardiac dysfunction via an IKKbeta-AMPK-dependent regulation of autophagy. Biochim Biophys Acta. 2015 Feb;1852(2):343–52. doi: 10.1016/j.bbadis.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Xu X, Ren J. MTOR overactivation and interrupted autophagy flux in obese hearts: a dicey assembly? Autophagy. 2013 Jun 1;9(6):939–41. doi: 10.4161/auto.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y, Han X, Hu N, Huff AF, Gao F, Ren J. Akt2 knockout alleviates prolonged caloric restriction-induced change in cardiac contractile function through regulation of autophagy. J Mol Cell Cardiol. 2014 Jun;71:81–91. doi: 10.1016/j.yjmcc.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, et al. Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J Mol Cell Cardiol. 2011 Jan;50(1):117–27. doi: 10.1016/j.yjmcc.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 83.Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, et al. ERK1/2 Phosphorylate Raptor to Promote Ras-dependent Activation of mTOR Complex 1 (mTORC1) J Biol Chem. 2011 Jan 7;286(1):567–77. doi: 10.1074/jbc.M110.159046. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and Functional Inactivation of TSC2 by Erk. Cell. 2005;121(2):179–93. doi: 10.1016/j.cell.2005.02.031. 2015/09/12. [DOI] [PubMed] [Google Scholar]
- 85.Rolfe M, McLeod LE, Pratt PF, Pround CG. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2) Biochem J. 2005;388(3):973–84. doi: 10.1042/BJ20041888. 2005-06-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee D-F, Kuo H-P, Chen C-T, Hsu J-M, Chou C-K, Wei Y, et al. IKKbeta Suppression of TSC1 Links Inflammation and Tumor Angiogenesis via the mTOR Pathway. Cell. 2007;130(3):440–55. doi: 10.1016/j.cell.2007.05.058. 2015/09/12. [DOI] [PubMed] [Google Scholar]
- 87.Dhingra R, Gang H, Wang Y, Biala AK, Aviv Y, Margulets V, et al. Bidirectional Regulation of Nuclear Factor-kB and Mammalian Target of Rapamycin Signaling Functionally Links Bnip3 Gene Repression and Cell Survival of Ventricular Myocytes. Circulation: Heart Failure. 2013 2013 Mar 1;6(2):335–43. doi: 10.1161/CIRCHEARTFAILURE.112.000061. [DOI] [PubMed] [Google Scholar]
- 88.Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent Activation of mTORC1 Complex Involves Phosphorylation of mTOR (Mammalian Target of Rapamycin) by IkB Kinase alpha (IKKalpha) J Biol Chem. 2014 Sep 5;289(36):25227–40. doi: 10.1074/jbc.M114.554881. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011 Jan 28;331(6016):456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008 Apr 25;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003 Nov 26;115(5):577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 92.Zheng Q, Zhao K, Han X, Huff AF, Cui Q, Babcock SA, et al. Inhibition of AMPK accentuates prolonged caloric restriction-induced change in cardiac contractile function through disruption of compensatory autophagy. Biochim Biophys Acta. 2015 Feb;1852(2):332–42. doi: 10.1016/j.bbadis.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 93.Lal H, Ahmad F, Woodgett J, Force T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ Res. 2015 Jan 2;116(1):138–49. doi: 10.1161/CIRCRESAHA.116.303613. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential Roles of GSK-3beta During Myocardial Ischemia and Ischemia/Reperfusion. Circ Res. 2011 Aug 19;109(5):502–11. doi: 10.1161/CIRCRESAHA.111.249532. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest. 2013 Apr 1;123(4):1821–32. doi: 10.1172/JCI64398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kawai T, Nomura F, Hoshino K, Copeland NG, Gilbert DJ, Jenkins NA, et al. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene. 1999 Jun 10;18(23):3471–80. doi: 10.1038/sj.onc.1202701. [DOI] [PubMed] [Google Scholar]
- 97.Ber Y, Shiloh R, Gilad Y, Degani N, Bialik S, Kimchi A. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell Death Differ. 2015;22(3):465–75. doi: 10.1038/cdd.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pagnin E, Fadini G, Toni Rd, Tiengo A, Calo L, Avogaro A. Diabetes Induces p66shc Gene Expression in Human Peripheral Blood Mononuclear Cells: Relationship to Oxidative Stress. The Journal of Clinical Endocrinology & Metabolism. 2005;90(2):1130–6. doi: 10.1210/jc.2004-1283. [DOI] [PubMed] [Google Scholar]
- 99.Xi G, Shen X, Radhakrishnan Y, Maile L, Clemmons D. Hyperglycemia-Induced p66shc Inhibits Insulin-Like Growth Factor I-Dependent Cell Survival via Impairment of Src Kinase-Mediated Phosphoinositide-3 Kinase/AKT Activation in Vascular Smooth Muscle Cells. Endocrinology. 2010;151(8):3611–23. doi: 10.1210/en.2010-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ranieri SC, Fusco S, Panieri E, Labate V, Mele M, Tesori V, et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proceedings of the National Academy of Sciences. 2010 Jul 27;107(30):13420–5. doi: 10.1073/pnas.1008647107. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tomilov AA, Ramsey JJ, Hagopian K, Giorgio M, Kim KM, Lam A, et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell. 2011;10(1):55–65. doi: 10.1111/j.1474-9726.2010.00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zheng Z, Yang J, Zhao D, Gao D, Yan X, Yao Z, et al. Downregulated adaptor protein p66Shc mitigates autophagy process by low nutrient and enhances apoptotic resistance in human lung adenocarcinoma A549 cells. FEBS J. 2013;280(18):4522–30. doi: 10.1111/febs.12416. [DOI] [PubMed] [Google Scholar]
- 103.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 1977 Nov 10;270(5633):174–6. doi: 10.1038/270174a0. [DOI] [PubMed] [Google Scholar]
- 104.Hara K, Yonezawa K, Weng Q-P, Kozlowski MT, Belham C, Avruch J. Amino Acid Sufficiency and mTOR Regulate p70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J Biol Chem. 1998 Jun 5;273(23):14484–94. doi: 10.1074/jbc.273.23.14484. 1998. [DOI] [PubMed] [Google Scholar]
- 105.Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb Binding to Mammalian Target of Rapamycin (mTOR) Is Regulated by Amino Acid Sufficiency. J Biol Chem. 2005 Jun 24;280(25):23433–6. doi: 10.1074/jbc.C500169200. 2005. [DOI] [PubMed] [Google Scholar]
- 106.Wang X, Campbell LE, Miller CM, Proud CG. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J. 1998;334(1):261–7. doi: 10.1042/bj3340261. 1998-08-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10(8):935–45. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008 Jun 13;320(5882):1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim YC, Park HW, Sciarretta S, Mo J-S, Jewell JL, Russell RC, et al. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun. 2014;5(4241) doi: 10.1038/ncomms5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010 Apr 16;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell. 2012;150(6):1196–208. doi: 10.1016/j.cell.2012.07.032. 2015/09/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saito K, Araki Y, Kontani K, Nishina H, Katada T. Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J Biochem. 2005 Mar;137(3):423–30. doi: 10.1093/jb/mvi046. [DOI] [PubMed] [Google Scholar]
- 113.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011 Nov 4;334(6056):678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015 Jan 9;347(6218):188–94. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015 Mar 26;519(7544):477–81. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012 Apr 13;149(2):410–24. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 117.Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015 Jan 9;347(6218):194–8. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis Activates Rag-mTORC1 Signaling. Mol Cell. 2012;47(3):349–58. doi: 10.1016/j.molcel.2012.05.043. 2015/09/12. [DOI] [PubMed] [Google Scholar]
- 119.Lorin S, Tol MJ, Bauvy C, Strijland A, Pous C, Verhoeven AJ, et al. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy. 2013 Jun 1;9(6):850–60. doi: 10.4161/auto.24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fahien LA, Teller JK, Macdonald MJ, Fahien CM. Regulation of glutamate dehydrogenase by Mg2+ and magnification of leucine activation by Mg2+ Mol Pharmacol. 1990 Jun;37(6):943–9. [PubMed] [Google Scholar]
- 121.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010 May 5;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013 Jun;2(3):e004796. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res. 2007 Oct 1;76(1):71–80. doi: 10.1016/j.cardiores.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003 Aug;10(16):1535–51. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 125.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003 Jun;206(Pt 12):2049–57. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 126.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007 Jun;39(3):211–22. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 127.McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. 2013 Dec;2(6):e000355. doi: 10.1161/JAHA.113.000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013 Aug 16;288(33):23798–806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008 Feb;28(3):1007–17. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L, et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ Res. 2011 Jan 7;108(1):60–9. doi: 10.1161/CIRCRESAHA.110.223115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012 Jul;4(7):633–46. doi: 10.1002/emmm.201200240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mayer SE, Mayfield AC, Haas JA. Heart muscle hexokinase: subcellular distribution and inhibition by glucose 6-phosphate. Mol Pharmacol. 1966 Sep;2(5):393–405. [PubMed] [Google Scholar]
- 133.Rose IA, Warms JV. Mitochondrial hexokinase. Release, rebinding, and location. J Biol Chem. 1967 Apr 10;242(7):1635–45. [PubMed] [Google Scholar]
- 134.Sui D, Wilson JE. Structural determinants for the intracellular localization of the isozymes of mammalian hexokinase: intracellular localization of fusion constructs incorporating structural elements from the hexokinase isozymes and the green fluorescent protein. Arch Biochem Biophys. 1997 Sep 1;345(1):111–25. doi: 10.1006/abbi.1997.0241. [DOI] [PubMed] [Google Scholar]
- 135.Xie GC, Wilson JE. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Arch Biochem Biophys. 1988 Dec;267(2):803–10. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- 136.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004 Dec 3;16(5):819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 137.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004 Jan;24(2):730–40. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008 Mar;15(3):521–9. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 139.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006 Aug 7;25(34):4683–96. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 140.Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015 Feb;22(2):248–57. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation induced autophagy through TORC1 inhibition. Mol Cell. 2014 Feb 20;53(4):521–33. doi: 10.1016/j.molcel.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol. 2002 Apr 16;12(8):632–9. doi: 10.1016/s0960-9822(02)00762-5. [DOI] [PubMed] [Google Scholar]
- 143.Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem. 2005 May 1;95(1):45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- 144.Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007 Jun 1;129(5):983–97. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 145.Lee MN, Ha SH, Kim J, Koh A, Lee CS, Kim JH, et al. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol Cell Biol. 2009 Jul;29(14):3991–4001. doi: 10.1128/MCB.00165-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Buller CL, Heilig CW, Brosius FC., 3rd GLUT1 enhances mTOR activity independently of TSC2 and AMPK. Am J Physiol Renal Physiol. 2011 Sep;301(3):F588–96. doi: 10.1152/ajprenal.00472.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Phosphorylation by Protein Kinase Cdelta (PKCdelta) Inhibits Mitochondria Elimination by Lysosomal-like Structures following Ischemia and Reoxygenation-induced Injury. J Biol Chem. 2013 Jun 28;288(26):18947–60. doi: 10.1074/jbc.M113.466870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of ametabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010 Jul 30;39(2):171–83. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bhaskar PT, Nogueira V, Patra KC, Jeon SM, Park Y, Robey RB, et al. mTORC1 hyperactivity inhibits serum deprivation-induced apoptosis via increased hexokinase II and GLUT1 expression, sustained Mcl-1 expression, and glycogen synthase kinase 3beta inhibition. Mol Cell Biol. 2009 Sep;29(18):5136–47. doi: 10.1128/MCB.01946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011 Mar 8;108(10):4129–34. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang P, Shan T, Liang X, Deng C, Kuang S. Mammalian target of rapamycin is essential for cardiomyocyte survival and heart development in mice. Biochem Biophys Res Commun. 2014 Sep 12;452(1):53–9. doi: 10.1016/j.bbrc.2014.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kolwicz SC, Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011 May 1;90(2):194–201. doi: 10.1093/cvr/cvr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. 2014 Apr;171(8):2080–90. doi: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taegtmeyer H, Lubrano G. Rethinking cardiac metabolism: metabolic cycles to refuel and rebuild the failing heart. F1000Prime Rep. 2014;6:90. doi: 10.12703/P6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. 2011 May 1;90(2):202–9. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gurel E, Ustunova S, Kapucu A, Yilmazer N, Eerbeek O, Nederlof R, et al. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol Biol Rep. 2013 Jul;40(7):4153–60. doi: 10.1007/s11033-013-2495-5. [DOI] [PubMed] [Google Scholar]
- 157.Katzen HM, Soderman DD, Wiley CE. Multiple forms of hexokinase. Activities associated with subcellular particulate and soluble fractions of normal and streptozotocin diabetic rat tissues. J Biol Chem. 1970 Aug 25;245(16):4081–96. [PubMed] [Google Scholar]
- 158.Xue W, Cai L, Tan Y, Thistlethwaite P, Kang YJ, Li X, et al. Cardiac-specific overexpression of HIF-1{alpha} prevents deterioration of glycolytic pathway and cardiac remodeling in streptozotocin-induced diabetic mice. Am J Pathol. 2010 Jul;177(1):97–105. doi: 10.2353/ajpath.2010.091091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang Y, Feng W, Xue W, Tan Y, Hein DW, Li XK, et al. Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes. 2009 Jun;58(6):1391–402. doi: 10.2337/db08-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Katzen HM. The effect of diabetes and insulin in vivo and in vitro on a low Km form of hexokinase from various rat tissues. Biochem Biophys Res Commun. 1966 Aug 23;24(4):531–6. doi: 10.1016/0006-291x(66)90352-4. [DOI] [PubMed] [Google Scholar]
- 161.Madsen-Bouterse S, Mohammad G, Kowluru RA. Glyceraldehyde-3-phosphate dehydrogenase in retinal microvasculature: implications for the development and progression of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010 Mar;51(3):1765–72. doi: 10.1167/iovs.09-4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001 Apr;8(4):367–76. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- 164.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007 Aug 1;405(3):407–15. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Webster KA, Graham RM, Bishopric NH. BNip3 and signal-specific programmed death in the heart. J Mol Cell Cardiol. 2005 Jan;38(1):35–45. doi: 10.1016/j.yjmcc.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 166.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, et al. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem. 2006 Jan 20;281(3):1442–8. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 167.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002 Aug 9;91(3):226–31. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 168.Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010 Aug;3(4):374–83. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010 Oct;6(7):855–62. doi: 10.4161/auto.6.7.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007 Dec 7;282(49):35803–13. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- 171.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009 May;29(10):2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S, et al. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 2009 Aug;10(8):866–72. doi: 10.1038/embor.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Canal M, Romani-Aumedes J, Martin-Flores N, Perez-Fernandez V, Malagelada C. RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front Cell Neurosci. 2014;8:313. doi: 10.3389/fncel.2014.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004 Dec 1;18(23):2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005 Jul;25(14):5834–45. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Corradetti MN, Inoki K, Guan KL. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem. 2005 Mar 18;280(11):9769–72. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 177.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008 Jan 15;22(2):239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu C, Xue R, Wu D, Wu L, Chen C, Tan W, et al. REDD1 attenuates cardiac hypertrophy via enhancing autophagy. Biochem Biophys Res Commun. 2014 Nov 7;454(1):215–20. doi: 10.1016/j.bbrc.2014.10.079. [DOI] [PubMed] [Google Scholar]
- 179.Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. doi: 10.1038/ncomms8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009 Oct;9(10):691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 181.Green DR, Chipuk JE. p53 and metabolism: Inside the TIGAR. Cell. 2006 Jul 14;126(1):30–2. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 182.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006 Jul 14;126(1):107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 183.Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator) J Biol Chem. 2009 Jan 16;284(3):1748–54. doi: 10.1074/jbc.M807821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009 Mar 3;106(9):3431–6. doi: 10.1073/pnas.0813210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008 May;10(5):611–8. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- 186.Puzio-Kuter AM. The Role of p53 in Metabolic Regulation. Genes Cancer. 2010 Apr;2(4):385–91. doi: 10.1177/1947601911409738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007 Apr 1;67(7):3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 188.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008 Aug 8;134(3):451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, et al. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009 May 15;8(10):1571–6. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- 190.Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013 Dec 3;18(6):792–801. doi: 10.1016/j.cmet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015 Feb;29(2):408–17. doi: 10.1096/fj.14-258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007 Apr 4;26(7):1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014 Jul 18;115(3):348–53. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res Nov 8. 113(11):1253–64. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009 May 15;46(10):1386–91. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, et al. The regulation of AMP-activated protein kinase by H(2)O(2) Biochem Biophys Res Commun. 2001 Sep 14;287(1):92–7. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 197.Han Y, Wang Q, Song P, Zhu Y, Zou MH. Redox regulation of the AMP-activated protein kinase. PLoS One. 2010;5(11):e15420. doi: 10.1371/journal.pone.0015420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010 Oct 22;285(43):33154–64. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Leon H, Atkinson LL, Sawicka J, Strynadka K, Lopaschuk GD, Schulz R. Pyruvate prevents cardiac dysfunction and AMP-activated protein kinase activation by hydrogen peroxide in isolated rat hearts. Can J Physiol Pharmacol. 2004 Jun;82(6):409–16. doi: 10.1139/y04-050. [DOI] [PubMed] [Google Scholar]
- 200.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004 Oct 15;279(42):43940–51. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 201.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013 Jan;25(1):50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 202.Mackenzie RM, Salt IP, Miller WH, Logan A, Ibrahim HA, Degasperi A, et al. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin Sci (Lond) 2013 Mar;124(6):403–11. doi: 10.1042/CS20120239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One. 2011;6(2):e17234. doi: 10.1371/journal.pone.0017234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Klaus A, Zorman S, Berthier A, Polge C, Ramirez S, Michelland S, et al. Glutathione S-transferases interact with AMP-activated protein kinase: evidence for S-glutathionylation and activation in vitro. PLoS One. 2013;8(5):e62497. doi: 10.1371/journal.pone.0062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, et al. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006 Mar 10;281(10):6366–75. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 206.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010 Mar 2;107(9):4153–8. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A. 2013 Aug 6;110(32):E2950–7. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Foster CR, Singh M, Subramanian V, Singh K. Ataxia telangiectasia mutated kinase plays a protective role in beta-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Mol Cell Biochem. 2011 Jul;353(1-2):13–22. doi: 10.1007/s11010-011-0769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE. Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem. 2015 Mar 6;290(10):6071–85. doi: 10.1074/jbc.M114.605345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Grevengoed TJ, Cooper DE, Young PA, Ellis JM, Coleman RA. Loss of long-chain acyl-CoA synthetase isoform 1 impairs cardiac autophagy and mitochondrial structure through mechanistic target of rapamycin complex 1 activation. FASEB J. 2015 Nov;29(11):4641–53. doi: 10.1096/fj.15-272732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963 Apr 13;1(7285):785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 212.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009 Apr 30;458(7242):1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014 Aug;124(8):3566–78. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ro SH, Jung CH, Hahn WS, Xu X, Kim YM, Yun YS, et al. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013 Dec;9(12):2103–14. doi: 10.4161/auto.26563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015 Mar 23;32(6):678–92. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008 Nov;8(5):411–24. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 217.Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss A, Gould A, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A. 2008 Jul 8;105(27):9250–5. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010 Dec 23;468(7327):1100–4. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]