

Abstract
Objective:
To report a novel autoimmune encephalitis in which the antibodies target neurexin-3α, a cell adhesion molecule involved in the development and function of synapses.
Methods:
Five patients with encephalitis and antibodies with a similar pattern of brain reactivity were selected. Antigen precipitation and determination of antibody effects on cultured rat embryonic neurons were performed with reported techniques.
Results:
Immunoprecipitation and cell-based assays identified neurexin-3α as the autoantigen of patients' antibodies. All 5 patients (median age 44 years, range 23–50; 4 female) presented with prodromal fever, headache, or gastrointestinal symptoms, followed by confusion, seizures, and decreased level of consciousness. Two developed mild orofacial dyskinesias, 3 needed respiratory support, and 4 had findings suggesting propensity to autoimmunity. CSF was abnormal in all patients (4 pleocytosis, 1 elevated immunoglobulin G [IgG] index), and brain MRI was abnormal in 1 (increased fluid-attenuated inversion recovery/T2 in temporal lobes). All received steroids, 1 IV immunoglobulin, and 1 cyclophosphamide; 3 partially recovered, 1 died of sepsis while recovering, and 1 had a rapid progression to death. At autopsy, edema but no inflammatory cells were identified. Cultures of neurons exposed during days in vitro (div) 7–17 to patients' IgG showed a decrease of neurexin-3α clusters as well as the total number of synapses. No reduction of synapses occurred in mature neurons (div 18) exposed for 48 hours to patients' IgG. Neuronal survival, dendritic morphology, and spine density were unaffected.
Conclusion:
Neurexin-3α autoantibodies associate with a severe but potentially treatable encephalitis in which the antibodies cause a decrease of neurexin-3α and alter synapse development.
Encephalitis is a severe inflammatory disorder of the brain with many possible causes and a complex differential diagnosis. Studies from different countries and a recent meta-analysis showed that in about 40% of patients with encephalitis the cause is never identified.1,2 Without reliable biomarkers, a response to empiric immunotherapy is frequently used to support that the disorder is immune-mediated, but a lack of response does not rule out an immune-mediated pathogenesis. For example, approximately 40% of patients with anti–NMDA receptor (NMDAR) encephalitis fail first-line immunotherapy (steroids, plasma exchange, or IV immunoglobulin [IVIg]) and require second-line therapies (rituximab or cyclophosphamide).3,4 However, second-line therapies are rarely used in encephalitis of unclear cause unless evidence of autoimmunity is provided. In this setting, the demonstration of autoantibodies to neuronal cell surface proteins is important for 3 reasons. First, they define the disorder as autoimmune regardless of a response to immunotherapy; second, they support the use of second-line immunotherapies or maintenance of intensive care if needed5; and finally, there is evidence that most of the antibodies are pathogenic.6 Some antibodies alter the surface dynamics of the cognate receptors causing their internalization (e.g., NMDAR7,8 or AMPA receptor9), while others block receptor function without altering its surface density (e.g., GABAb receptor10). We report the clinical and immunologic features of a novel form of autoimmune encephalitis in which the antibodies target neurexin-3α, a presynaptic cell adhesion molecule with critical roles in synapse development and function.11 In addition, we show that patients' antibodies alter the formation of synapses.
METHODS
Patients.
Five patients with encephalitis of unclear cause and antibodies against a previously unknown neuronal cell surface autoantigen are the focus of this study. The 5 cases were identified over the last 10 years in the Laboratory of Neuroimmunology at the Institute of Biomedical Research August Pi i Sunyer (IDIBAPS), Hospital Clínic, University of Barcelona, and the Department of Neurology, University of Pennsylvania. The selection of the 5 cases was based on the distinctive pattern of serum and CSF reactivity with neuropil of rat brain, leading to the investigations reported here. Clinical data were provided by the treating physicians. Control samples (total 200) included serum or CSF of 179 patients with different types of neurologic disorders (well-characterized autoimmune encephalitis, suspected autoimmune encephalitis, neurodegenerative diseases, and multiple sclerosis) and 21 healthy blood donors (supplemental data on the Neurology® Web site at Neurology.org).
Standard protocol approvals, registrations, and patient consents.
The study was approved by the institutional review boards of IDIBAPS and University of Pennsylvania and written consent was obtained.
Determination of antibodies, characterization of the target antigen, and development of a cell-based assay.
The techniques involved in these studies are similar to those reported for other neuronal cell surface antibodies and antigens7,12,13 and are described in the supplemental data.
Analysis of the effects of the antibodies on cultures of neurons and confocal microscopy.
To determine the effects of patients' antibodies at the cellular and synaptic levels, patient or control purified immunoglobulin G (IgG) (final concentration 80 μg/mL media) were added to cultures of dissociated rat hippocampal neurons at 7, 10, and 14 days in vitro (div).14 On div 17, cultures were washed with phosphate-buffered saline, fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and blocked for 60 minutes with 1% bovine serum albumin. Cells were then incubated overnight at 4°C using single or double immunolabeling with the following antibodies: anti–leucine-rich repeat transmembrane neuronal 2 (LRRTM2, sheep polyclonal diluted 1:100, AF5589; R&D Systems, Minneapolis, MN), anti-bassoon (mouse monoclonal, 1:400, SAP7F407; Enzo, Farmingdale, NY), or anti-homer1b (rabbit polyclonal, 1:100, PA5-21487; Pierce, Rockford, IL). To determine the levels of cell surface neurexin-3α, human IgG with neurexin-3α antibodies (diluted 1:50) was used as a reagent on the live neuronal cultures before permeabilization and fixation (as previously reported for studies with NMDAR antibodies7,15). After the incubation with primary antibodies, slides were washed and incubated for 1 hour at room temperature with the corresponding secondary antibodies (all diluted 1:1,000; Molecular Probes, Eugene, OR), Alexa Fluor 594 donkey anti-sheep IgG (A-11016), Alexa Fluor 488 goat anti-mouse IgG (A-11001), Alexa Fluor 594 goat anti-rabbit IgG (A-11037), or Alexa Fluor 488 goat anti-human IgG (A11013). Slides were then mounted with ProlonGold with 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI, P36935; Molecular Probes) and results scanned with a confocal microscope (Zeiss LSM710; Oberkochen, Germany) with EC-Plan NEOFLUAR CS 100×/1.3, 63×/1.4, or 20×/0.8 NA oil objectives.
To determine whether patients' antibodies caused alteration of neurexin-3α levels and number of synapses in mature neurons, a similar set of experiments and markers as above were used in cultures of neurons (div 18) exposed for 48 hours to patient or control IgG.
Statistical analysis.
The quantitation of neuronal cell death and number of dendritic spines was analyzed using one-way analysis of variance (ANOVA) and the quantitation of the number of dendrites and complexity with 2-way ANOVA (data shown in supplemental data). Confocal cluster densities of neurexin-3α, LRRTM2, bassoon, or homer1b were analyzed using one-way ANOVA and post hoc testing with Tukey adjustment for multiple comparisons. A p value of <0.05 was considered significant. The α error was set at 0.05. All tests were done using GraphPad (La Jolla, CA) Prism (version 6).
RESULTS
All 5 patients (median age 44 years, range 23–50 years; 4 female) presented with prodromal symptoms (fever, headache, nausea, or diarrhea) that rapidly progressed (1–7 days, median 3) to confusion, decreased level of consciousness, and seizures (table 1). One patient developed myoclonic jerks and 2 mild orofacial dyskinesias. Three patients required intensive care with respiratory support. Four of the patients had history or laboratory findings suggestive of systemic autoimmunity, such as increased antinuclear antibodies (ANA). One had been diagnosed with lupus 7 years earlier (fatigue, Raynaud phenomenon, proteinuria, decreased complement, double-stranded DNA antibodies, without neurologic symptoms), for which she had been treated with chronic oral steroids. The CSF was abnormal in all patients, showing moderate pleocytosis in 4 and mild increase of IgG index in 1 of 2 cases examined. Brain MRI was normal in 4 of 5 patients, and 1 patient had increased fluid-attenuated inversion recovery/T2 signal in the medial temporal lobes. All 5 patients received steroids, 1 also received IVIg, and 1 cyclophosphamide. Two patients died (patient 2 of encephalitis, patient 4 of systemic complications while recovering from the encephalitis) and the other 3 patients had partial recovery after follow-ups of 2, 24, and 26 months. Despite extensive CSF investigations (and neuropathologic studies in 2 patients), no evidence of CNS infection was identified in any of the patients.
Table 1.
Main clinical features of 5 patients with neurexin-3α antibodies
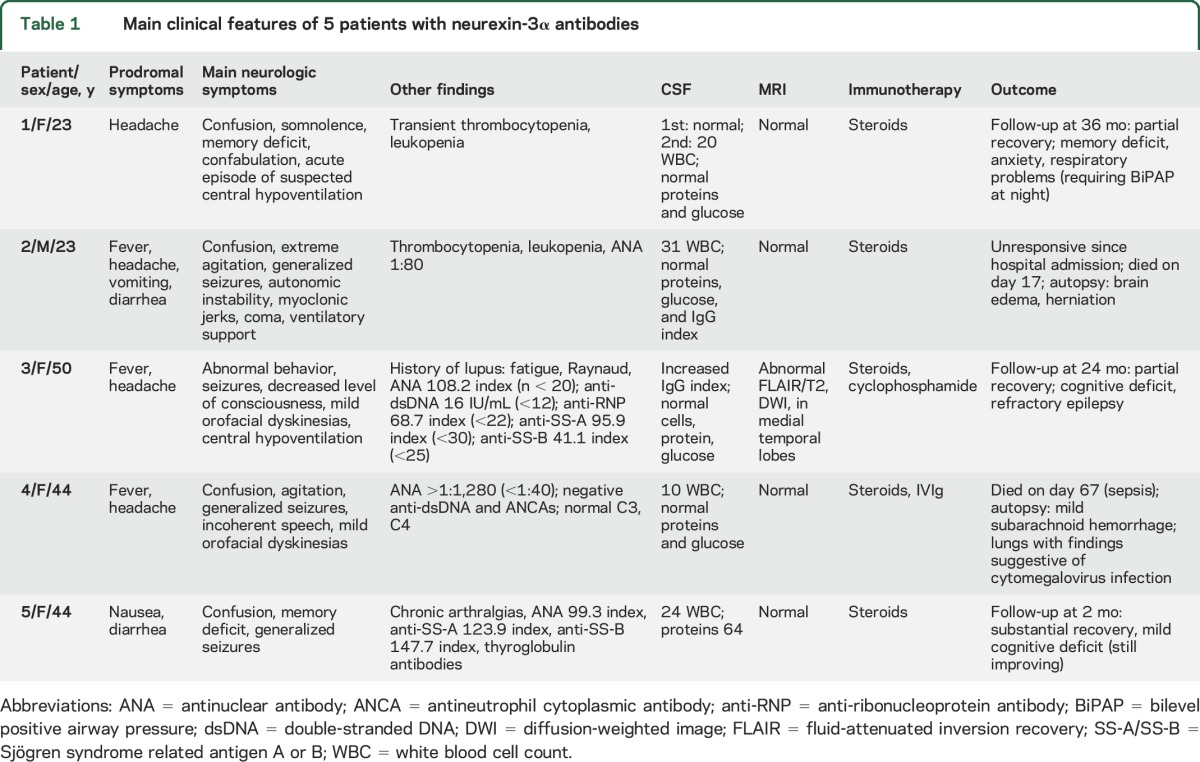
The autopsy of patient 2 showed substantial brain edema without evidence of inflammatory infiltrates and no remarkable systemic findings (blood and CSF studies for 16 different viruses studied at the California Encephalitis Project were all negative). The autopsy of patient 4 showed cytopathic changes in the lung compatible with cytomegalovirus infection. In the brain, there was focal and mild subarachnoid hemorrhage, sparse petechiae in the middle cerebellar peduncles, and moderate meningeal hemorrhage; no remarkable microscopic findings or evidence of CNS cytomegalovirus infection were identified.
All 5 patients had antibodies in serum and CSF that reacted with the neuropil of rat brain, producing a similar type of reactivity that was not present in control samples (figure 1, A and C). The pattern of immunostaining in the hippocampus (best seen in figure e-1A) was identical in the 5 patients and different from other neuronal cell surface antibodies reported to date. Studies with live hippocampal neuronal cultures showed that the epitopes were on the neuronal cell surface (figure 1B).
Figure 1. Patients' antibodies react with brain tissue and neuronal cultures.

Representative immunoreactivity of patient CSF (A, B) and control CSF (C, D) with sagittal sections of rat brain and cultures of dissociated rat hippocampal neurons. Patients' CSF antibodies (A) show a diffuse immunolabeling of neuropil that is not detected with control CSF (C). Patients' CSF also reacts with the cell surface of live rat hippocampal neurons (B), whereas the control CSF is negative (D). Scale bar for A and C = 2 mm, scale bar for B and D = 20 μm.
To further confirm that the patients' antibodies recognized the same antigen, we performed immunocompetition assays between the sera of 2 of the patients. This showed that preincubation of brain tissue with serum from one patient abrogated the reactivity of biotinylated IgG of the other patient, confirming that the antibodies of both patients recognized the same epitopes (figure e-1).
In order to identify the target antigen, 2 independent neuronal immunoprecipitations were carried out with sera from 2 different patients. Mass spectrometry of the precipitates showed several sequences of neurexin-3α in both cases (table e-1).
To further asses that neurexin-3α was the target antigen of patients' antibodies, patients' serum and CSF were tested in a cell-based assay with HEK293 cells expressing human neurexin-3α or its ligand LRRTM2. Samples from the 5 patients but none of the 200 controls showed specific reactivity with neurexin-3α (a representative case is shown in figure 2, A–F). Moreover, the pattern of brain reactivity of patients' antibodies was abrogated when a representative CSF sample was absorbed with HEK cells expressing neurexin-3α (figure e-2). None of the 5 patients had antibodies against LRRTM2 (figure 2, G–L). To determine if LRRTM2 influenced the antibody recognition of neurexin-3α, patients' sera and CSF were incubated with HEK293 cells coexpressing neurexin-3α and LRRTM2. Cotransfection of both proteins did not modify the intensity of staining of the positive samples (data not shown).
Figure 2. Patients' antibodies react with neurexin-3α but not leucine-rich repeat transmembrane neuronal 2 (LRRTM2).

(A) The serum of a representative patient reacts with HEK293 cells expressing neurexin-3α; in contrast, no reactivity is observed with the serum of a healthy control (D). (B, E) Correspond to the same cells immunolabeled with a commercial neurexin-3α antibody; (C, F) the merged reactivities. In (G–L), a similar experiment is shown with HEK293 cells expressing LRRTM2; no reactivity is observed with the patient's (G) or control (J) serum. Scale bar = 10 μm.
Having demonstrated that patients' antibodies target neurexin-3α and considering the critical role of this protein in synapse development,11 we next determined the effects of the antibodies on the levels of neurexin-3α and formation of synapses, as well as on cell survival, number and complexity of dendrites, and spine density. For these studies, patient's IgG was added at regular intervals to dissociated cell cultures of rat embryonic neurons, div 7 to 14, a period during which there is robust development of neuronal processes and establishment of functional synaptic networks (the effects were examined on day 17).14 Compared to neurons treated with control IgG, those treated with patient IgG had a decrease of cell surface clusters of neurexin-3α, as well as of clusters of neurexin-3α colocalizing with LRRTM2; however, the density of clusters of LRRTM2 was not altered (figure 3). Additionally, neurons treated with patient IgG, but not control IgG, showed a decrease in the total number of synapses, defined by the colocalization of a presynaptic marker (bassoon) with a postsynaptic marker (homer1b), as well as a decrease in the total clusters of bassoon and homer1b (figure 4). No effects were noted on neuronal survival (data not shown), number and complexity of dendrites, or spine density (figure e-3). Similar studies using mature neurons (div 18) exposed for 48 hours to patients' IgG showed a significant decrease of neurexin-3α without affecting LRRTM2 or the number of synapses (figure e-4). Overall, these findings indicate that patients' antibodies cause a specific reduction of neurexin-3α, which in turn decreases the total number of synapses in neurons undergoing development.
Figure 3. Patient antibodies cause a reduction in neurexin-3α.

Hippocampal neurons treated during 10 days with patient immunoglobulin G (IgG), control IgG, or not treated were examined on days in vitro (div) 17 for the density of cell surface neurexin-3α, the colocalization of neurexin-3α with its ligand leucine-rich repeat transmembrane neuronal 2 (LRRTM2), and the density of LRRTM2. (A) Compared with control IgG or not treated neurons, patient IgG caused a significant reduction in the clusters of cell surface neurexin-3α (left) as well as the colocalization of neurexin-3α with its ligand LRRTM2 (middle), but had no effects on LRRTM (right). (B) Representative pictures of each condition. Note the significant reduction of the clusters of neurexin-3α as well as the colocalization of neurexin-3α with LRRTM2 (merge) in neurons treated with patient IgG. Scale bar = 5 μm. All graphs represent mean ± SD of 3 independent experiments (for each experiment, 10 dendrites per condition); *p < 0.05, **p < 0.01, ****p < 0.0001.
Figure 4. Patient antibodies interfere with the formation of synapses.

Cultures of hippocampal neurons were treated for 10 days with patient immunoglobulin G (IgG) or control IgG or not treated, and the effect on the number of synapses (defined as clusters of the presynaptic marker bassoon colocalizing with the postsynaptic marker homer1b) was determined. The study shows that patient IgG antibodies, but not control IgG, caused a significant reduction of the number of synapses (A) as well as bassoon (B) and homer1b (C). All graphs represent mean ± SD of 5 independent experiments (for each experiment, 10 dendrites per condition); **p < 0.01, ***p < 0.001, ****p < 0.0001.
DISCUSSION
We report a novel type of autoimmune encephalitis characterized by antibodies against extracellular epitopes of neurexin-3α. The disorder is important for 3 reasons: first, symptoms are severe (with a rapid course to death in one patient) but potentially treatable; second, the initial clinical picture in some patients may suggest anti-NMDAR encephalitis; and third, the antibodies cause a decrease of neurexin-3α and formation of synapses.
The combination of young age, rapid decline of the level of consciousness, seizures, orofacial dyskinesias, or central hypoventilation resembles the clinical picture of anti-NMDAR encephalitis.3 This diagnosis was clinically considered in patients 1, 3, and 4, but NMDAR antibody testing with cell-based assay was negative and the pattern of brain immunostaining, although robust and similar in all patients, was clearly different from that associated with NMDAR antibodies. None of the patients developed prolonged psychiatric symptoms or prominent abnormal movements other than mild orofacial dyskinesias or had teratoma, which are frequent features in adults with anti-NMDAR encephalitis. In line with other types of autoimmune encephalitis,13,16 4 of the 5 patients had ANA or other non-neuronal autoantibodies suggesting propensity to autoimmunity. Despite this, only 1 patient received second-line immunotherapy. This is probably due to the rapid course of the disease in patient 2, who died before aggressive immunotherapy could be tried, the limited knowledge on autoimmune encephalitis at the time patients 2 and 4 were studied (2006 and 2008, respectively), and the rapid response to steroids of patient 5.
The absence of brain inflammatory infiltrates in the autopsy of patients 2 and 4, despite both having CSF pleocytosis at symptom onset, is puzzling. In 4 patients, the antibody IgG isotype was investigated; 3 were only IgG1, and 1 IgG1 along with a minor component of IgG4 (data not shown), suggesting the potential for complement activation (not investigated) and recruitment of inflammatory infiltrates. We postulate the autopsy findings could be related to the fulminant disease of patient 2, whose death was likely caused by refractory seizures and brain edema, raising the possibility that mild inflammatory infiltrates could have been missed. Patient 4 died of sepsis and systemic complications after neurologic improvement; the autopsy showed mild subarachnoid hemorrhage. In this case, it is possible that the brain inflammation had resolved by the time of the patient's death (day 67). Nevertheless, the absence of brain inflammation is in contrast to what is found in other autoimmune encephalitis cases and needs further study.17,18
Neurexins are a family of synaptic cell adhesion molecules involved in synapse formation and maturation. They are encoded by 3 genes—NRX1, NRX2, and NRX3—each of them providing 2 alternative splice products, a long α and a short β form.11 Each neurexin can bind to a small number of postsynaptic ligands, including neuroligins, cerebellins, and LRRTMs11,19; neurexin-3α binds specifically to LRRTM2.20,21 The serum and CSF of our 5 patients contained antibodies against neurexin-3α but not LRRTM2.
Neurexins act as a link between presynaptic and postsynaptic compartments, with the intracellular domain interacting with the presynaptic machinery for neurotransmitter release, and the extracellular domain binding to postsynaptic cell adhesion molecules.11 Abrogating the expression of all 3 α neurexins in mice resulted in postnatal death and a dramatic reduction of Ca2+-triggered neurotransmitter release.22 Specific ablation of neurexin-3 showed different presynaptic and postsynaptic functions in distinct brain regions. For example, in the hippocampus, extracellular sequences of presynaptic neurexin-3 mediate transsynaptic regulation of postsynaptic AMPA receptors, whereas in the olfactory bulb, intracellular sequences of neurexin-3 are selectively required for GABA release.23 Mutations in the neurexin genes have been associated with schizophrenia24–26 and autism.24,27–29
Using rat embryonic neurons in the stage of maturation and development of functional synaptic networks (e.g., div 7–17), we found that patients' antibodies caused a specific reduction of the levels of neurexin-3α as well as a decrease of the total number of synapses (defined by the colocalization of the presynaptic marker bassoon30 with the postsynaptic marker homer1b31). A similar study using mature neurons (div 18) exposed for 48 hours to patients' antibodies showed a decrease of the levels of neurexin-3α, without affecting the total number of synapses. Thus, the antibody-mediated decrease of neurexin-3α affects synapse development. These findings are in line with studies showing that disruption of endogenous neurexin–neuroligin interaction by adding a recombinant neurexin reduces the number of presynaptic terminals and the number of inhibitory and excitatory synapses.32,33
Our study has the limitation of a small number of patients. This often occurs in the initial description of a novel autoimmune encephalitis and prevents an estimation of disease frequency. For example, the first 2 reports on the clinical and immunologic findings of anti-NMDAR encephalitis had 4 and 12 patients12,34; however, after the disorder was known and a diagnostic test developed, identification of cases increased and it is now considered one of the most frequent autoimmune encephalitis.35 In some patients with neurexin-3α antibodies, the clinical presentation suggests an infectious etiology and the course of the disease can be fulminant (e.g., patient 2). It is likely that patients with these presentations are not considered for neuronal antibody testing, thus missing the diagnosis.
Future studies should aim to fully characterize the syndrome reported here and the mechanisms by which the antibodies decrease the levels of neurexin-3α, which in turn alter the formation of synapses, and may affect presynaptic (neurotransmitter release) and postsynaptic functions (e.g., AMPA receptor regulation).23 It is unclear whether the antibodies affect the maintenance of synapses in mature neurons; we did not see an effect after 48 hours of exposure but longer treatments may be necessary. The clinical features, immunologic findings, and diagnostic test provided in the current study are the first steps toward these goals.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Thomas C. Südhof (Stanford University) for providing the plasmid with the human sequence of neurexin-3α.
GLOSSARY
- ANA
antinuclear antibodies
- ANOVA
analysis of variance
- div
days in vitro
- IDIBAPS
Institute of Biomedical Research August Pi i Sunyer
- IgG
immunoglobulin G
- IVIg
IV immunoglobulin
- LRRTM2
leucine-rich repeat transmembrane neuronal 2
- NMDAR
NMDA receptor
Footnotes
Editorial, page 2222
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Design/conceptualization of the study: N.G.-A. and J.D.; analysis/interpretation of the data: all authors; statistical analysis and figure development: N.G.-A., J.P., M.P.-P., J.D.; drafting/revising the manuscript: all authors.
STUDY FUNDING
Supported in part by NIH RO1NS077851 (J.D.); Fondo de Investigaciones Sanitarias, FEDER, Spain (FIS PI12/00611, FG; FIS 14/00203, J.D.); Instituto de Salud Carlos III, Madrid, Spain (CM14/00081, TA; CD 14/00155, E.M.-H.); Asociación Española de Pediatría research grant from Dodot-Procter & Gamble (T.A.); and Fundació CELLEX (J.D.).
DISCLOSURE
N. Gresa-Arribas, J. Planagumà, M. Petit-Pedrol, I. Kawachi, S. Katada, C. Glaser, M. Simabukuro, T. Armangue, E. Martínez-Hernández, and F. Graus report no disclosures relevant to the manuscript. J. Dalmau has a research grant from Euroimmun and receives royalties from patents for the use of Ma2 and NMDAR as autoantibody tests. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis-associated hospitalizations in the United States, 1998–2010. Neurology 2014;82:443–451. [DOI] [PubMed] [Google Scholar]
- 2.Glaser CA, Honarmand S, Anderson LJ, et al. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin Infect Dis 2006;43:1565–1577. [DOI] [PubMed] [Google Scholar]
- 3.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nosadini M, Mohammad SS, Ramanathan S, Brilot F, Dale RC. Immune therapy in autoimmune encephalitis: a systematic review. Expert Rev Neurother 2015;15:1391–1419. [DOI] [PubMed] [Google Scholar]
- 5.Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann NY Acad Sci 2015;1338:94–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012;135:1606–1621. [DOI] [PubMed] [Google Scholar]
- 9.Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol 2015;77:381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain A, Lancaster E, Dalmau J, Balice-Gordon RJ. Autoantibodies in the CSF of anti-GABA receptor encephalitis patients block activation of GABA receptors in vitro (abstract M116, 140th annual meeting of the American Neurological Association). Ann Neurol 2015;78(suppl 19):S77. [Google Scholar]
- 11.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008;455:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchhalter JR, Dichter MA. Electrophysiological comparison of pyramidal and stellate nonpyramidal neurons in dissociated cell culture of rat hippocampus. Brain Res Bull 1991;26:333–338. [DOI] [PubMed] [Google Scholar]
- 15.Planaguma J, Leypoldt F, Mannara F, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015;138:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011;77:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krueger DD, Tuffy LP, Papadopoulos T, Brose N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr Opin Neurobiol 2012;22:412–422. [DOI] [PubMed] [Google Scholar]
- 20.Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci 2010;30:7495–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Sudhof TC. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 2013;154:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Missler M, Zhang W, Rohlmann A, et al. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 2003;423:939–948. [DOI] [PubMed] [Google Scholar]
- 23.Aoto J, Foldy C, Ilcus SM, Tabuchi K, Sudhof TC. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat Neurosci 2015;18:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gauthier J, Siddiqui TJ, Huashan P, et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet 2011;130:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rujescu D, Ingason A, Cichon S, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet 2009;18:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikeda M, Aleksic B, Kinoshita Y, et al. Genome-wide association study of schizophrenia in a Japanese population. Biol Psychiatry 2011;69:472–478. [DOI] [PubMed] [Google Scholar]
- 27.Feng J, Schroer R, Yan J, et al. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett 2006;409:10–13. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Hu Z, Xun G, et al. Mutation analysis of the NRXN1 gene in a Chinese autism cohort. J Psychiatr Res 2012;46:630–634. [DOI] [PubMed] [Google Scholar]
- 29.Vaags AK, Lionel AC, Sato D, et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet 2012;90:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tom DS, Sanmarti-Vila L, Langnaese K, et al. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol 1998;142:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayashi MK, Tang C, Verpelli C, et al. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 2009;137:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 2000;101:657–669. [DOI] [PubMed] [Google Scholar]
- 33.Levinson JN, Chery N, Huang K, et al. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem 2005;280:17312–17319. [DOI] [PubMed] [Google Scholar]
- 34.Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol 2005;58:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Granerod J, Ambrose HE, Davies NW, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835–844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.