

Summary
One-carbon (1C) units for purine and thymidine synthesis can be generated from serine by cytosolic or mitochondrial folate metabolism. The mitochondrial 1C pathway is consistently overexpressed in cancer. Here we show that most but not all proliferating mammalian cell lines use the mitochondrial pathway as the default for making 1C units. CRISPR-mediated mitochondrial pathway knockout activates cytosolic 1C-unit production. This reversal in cytosolic flux is triggered by depletion of a single metabolite, 10-formyl-THF, and enables rapid cell growth in nutrient-replete conditions. Loss of the mitochondrial pathway, however, renders cells dependent on extracellular serine to make 1C units and on extracellular glycine to make glutathione. HCT-116 colon cancer xenografts lacking mitochondrial 1C pathway activity generate the 1C units required for growth by cytosolic serine catabolism. Loss of both pathways precludes xenograft formation. Thus, either mitochondrial or cytosolic 1C metabolism can support tumorigenesis with the mitochondrial pathway required in nutrient poor conditions.
eTOC blurb
Using genetic and metabolomic approaches, Ducker et al. dissect the roles of cytosolic and mitochondrial folate metabolism in cell proliferation, revealing that most cells default to mitochondria for making 1C units, simultaneously generating glycine, NADH and NADPH. Upon loss of the mitochondrial pathway, however, cytosolic metabolism supports tumor growth.
Introduction
Tetrahydrofolate (THF) is the cofactor required for the activation and transfer of one-carbon (1C) units for nucleotide biosynthesis and methionine regeneration. Dietary folate is essential, and folate deficiency is a leading cause of birth defects (World Health Organization, 2008). Pharmacological inhibition of 1C metabolism with folate analogues (antifolates) was the first effective chemotherapy, and antifolates remain mainstays of cancer treatment. Unfortunately, existing agents, which broadly inhibit folate-mediated reactions, result in substantial side effects, including impaired hematopoiesis and damage to the gastrointestinal epithelium. Despite extensive research into antifolates, the biological function of specific folate enzymes in different physiological and pathological contexts is only now being elucidated.
In cancer, certain 1C genes are consistently overexpressed. These include DHFR (dihydrofolate reductase), an enzyme strongly inhibited by current antifolates, and TYMS (thymidylate synthase), the target of the important chemotherapeutic 5-fluorouracil (Huennekens, 1994; Longley et al., 2003). Equally upregulated are two genes of mitochondrial 1C transformations: SHMT2 (mitochondrial serine hydroxymethyl transferase) and MTHFD2 (mitochondrial methylenetetrahydrofolate dehydrogenase) (Jain et al., 2012; Lee et al., 2014; Nilsson et al., 2014). Together these enzymes, both of which lie in a pathway essential for embryonic development (Di Pietro et al., 2002; Momb et al., 2013), transform serine into glycine and a formyl unit attached to THF (Figure 1a). Interestingly, production of serine itself is frequently upregulated in cancer, with the first enzyme of serine synthesis, 3-phosphoglycerate dehydrogenase (PHGDH), often genomically amplified in breast cancer and melanoma (Locasale et al., 2011; Possemato et al., 2011). Expression of both serine biosynthesis and the mitochondrial 1C pathway can be driven by the transcription factor ATF4, which can be activated by mTORC1 and NRF2-KEAP1 signaling (Ben-Sahra et al., 2016; DeNicola et al., 2015). Thus, cancers commonly overexpress the enzymes to make serine and convert it into glycine and mitochondrial 10-formyl-tetrahydrofolate (10-formyl-THF).
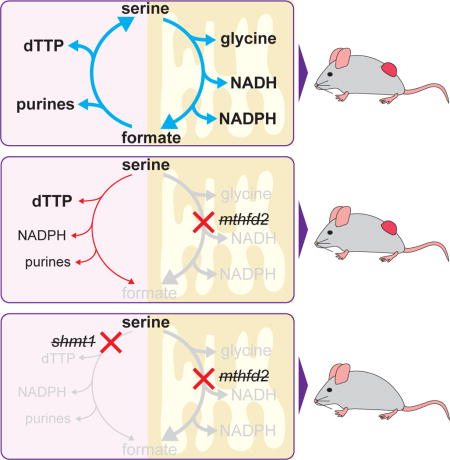
Figure 1. Mitochondrial folate metabolism mutants are deficient in 10-formyl-THF.
a. Folate mediated 1C metabolism occurs in linked cytosolic and mitochondrial pathways. b. Western blot of 1C metabolic enzymes in HEK 293T cells deleted for specific genes using CRISPR/Cas9. c. Doubling times of deletion cell lines cultured in DMEM ±1 mM sodium formate. Error bars are ±95% confidence interval of the doubling time fit. d. Normalized intracellular levels of water-soluble metabolites in folate pathway deletion cell lines. For each cell line, 3 individual biological replicates are shown, normalized to WT cells analyzed in parallel by LC-MS. e. LC-MS trace of AICAR (m/z 337.055 ±2 ppm) in folate gene deletion cells. f. Metabolite levels of purine biosynthetic intermediates normalized to WT cells as in (d). g. 1 mM formate reverses AICAR accumulation in mutant cell lines. h. Relative levels of folate species in mutant cell lines. THF and methylene-THF interconvert in cell extracts and accordingly are reported together. All results (unless stated) are mean ±SD, n≥3 biologic replicates and were confirmed in independent experiments.
Mitochondrial 10-formyl-THF is needed to make formyl-methionine for mitochondrial protein synthesis (Tucker et al., 2011). The required amount is small, however, and methioninyl-tRNA formyl-transferase is not upregulated in cancer (Nilsson et al., 2014). Mitochondrial 10-formyl-THF may also be used to generate cytosolic 1C units: while carbon bearing THF species do not cross the mitochondrial membrane, mitochondrial 10-formyl-THF can be cleaved by methylenetetrahydrofolate dehydrogenase 1-like (MTHFD1L) into free formate, which can cross the mitochondrial membrane and be assimilated in the cytosol by methylenetetrahydrofolate dehydrogenase (MTHFD1) (Figure 1a). Embryonic defects induced by deletion of MTHFD1L are partially rescued by formate (Momb et al., 2013).
A puzzling aspect of the essentiality of mitochondrial folate metabolism in development and its upregulation in cancer is the existence of a parallel cytosolic pathway that is sufficient to support cell growth in culture (Patel et al., 2003; Tibbetts and Appling, 2010). Cytosolic serine hydroxymethyl transferase (SHMT1) can use serine to make cytosolic 5,10-methylene-tetrahydrofolate (methylene-THF), which is poised to carry out all of the major physiological functions of 1C units: direct use for thymidine synthesis, reduction to 5-methyl-THF to serve as a 1C donor for methionine synthase, or oxidation to 10-formyl-THF for purine biosynthesis. While other substrates, including glycine and choline, can donate 1C units exclusively to the mitochondrial 1C pool, these substrates have not been shown to contribute significantly to the cytosolic 1C pool (Fan et al., 2014; Jain et al., 2012; Labuschagne et al., 2014).
One possible advantage of compartmentalization of 1C metabolism involves redox homeostasis. Folate metabolism can produce mitochondrial NADPH via the enzyme ALDH1L2 and potentially also MTHFD2, (Fan et al., 2014; Lewis et al., 2014) and knockdown of SHMT2 in certain cancer cell lines increases their vulnerability to oxidative stressors (Kim et al., 2015; Ye et al., 2014). The NADPH/NADP+ ratio in turn may play an important role in regulating the cytosolic flux of 1C units through the MTHFD1 dehydrogenase reaction. The relatively high NADPH/NADP+ ratio in the cytosol may favor reduction of 1C units from 10-formyl-THF into methylene-THF, whereas low NADH levels in the mitochondria favor the opposite (Pelletier and MacKenzie, 1995).
To explore the function of compartmentalized 1C metabolism, here we created using CRISPR a panel of isogenic clonal folate enzyme deletion cell lines. We find that the mitochondrial pathway is the default in most but not all cancer cells. Intriguingly, folate metabolism is both plastic and self-regulating: loss of the mitochondrial pathway triggers, through depletion of cytosolic 10-formyl-THF, de novo generation of 1C units in the cytosol. While well tolerated in nutrient-replete conditions, this switch renders cells dependent upon extracellular serine and glycine to support biosynthesis and antioxidant defense. Despite these liabilities, HCT-116 colon cancer cells with engineered mitochondrial 1C enzyme deletions can form xenograft tumors, by generating cytosolic 1C units from serine via the enzyme SHMT1. However, loss of both the mitochondrial folate pathway and SHMT1 inhibits tumorigenesis.
Results
An isogenic library of folate metabolic enzyme CRISPR-deletion mutants
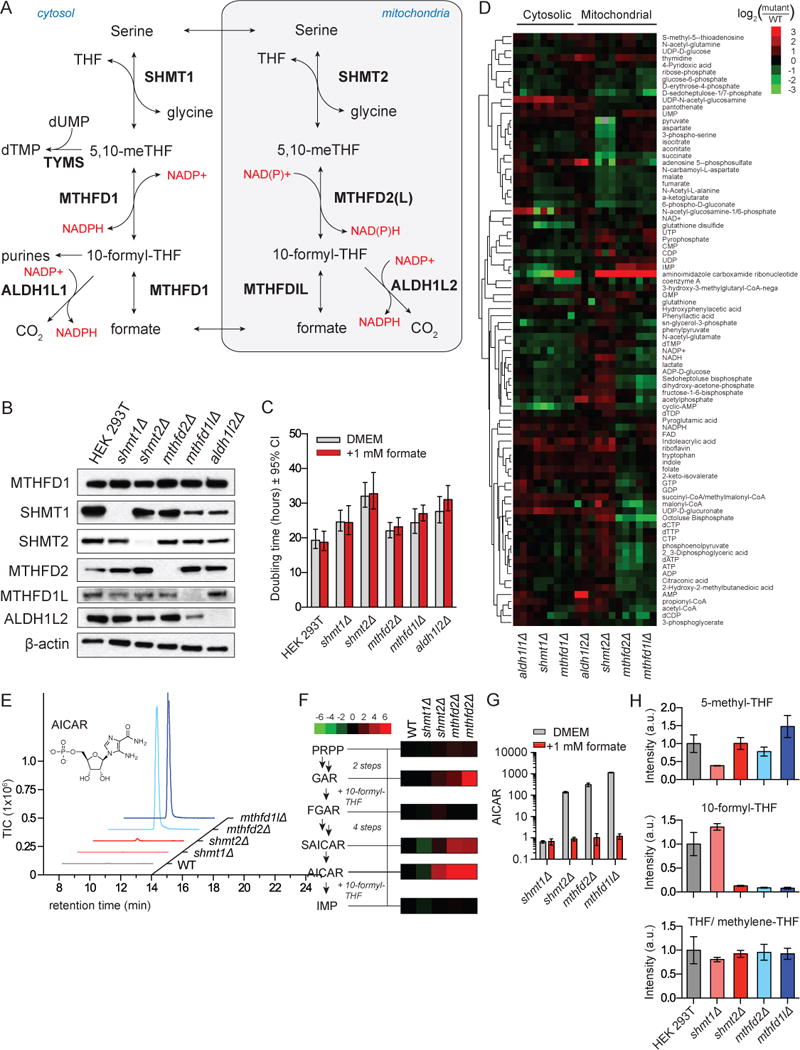
HEK 293T cells were transiently transfected with Cas9 together with guide RNA pairs targeting folate genes (Ran et al., 2013a; 2013b). Single colonies were picked from the transfected cells, and deletions confirmed by Sanger sequencing and western blotting (Figure 1B and Figure S1a–e). We isolated multiple viable clones deleted for each core enzyme of the mitochondrial folate pathway (SHMT2/MTHFD2/MTHFD1L/ALDH1L2) and for the corresponding cytosolic enzymes SHMT1 and ALD1HL1 (Figure S1f–k). We did not detect expression of the adult-specific MTHFD2L isozyme in HEK 293T cells (Bolusani et al., 2011). In the cytosol, the MTHFD1 protein contains separate domains that correspond to the catalytic activities of both MTHFD2 and MTHFD1L. Loss of these activities blocks both known routes of cytosolic 10-formyl-THF synthesis (from either methylene-THF or free formate). Because cytosolic 10-formyl-THF is required for purine synthesis, MTHFD1 deletion is expected to be intolerable. Consistent with this, no MTHFD1 knockout clone was ever isolated, although we did isolate hypomorphic MTHFD1 cell lines with reduced enzyme expression (Figure S1g).
Compensatory changes in the expression of folate pathway enzymes in deletion clones were limited. Cytosolic pathway protein levels and MTHFD2 levels were largely unaffected. Across multiple clones, however, deletion of SHMT1 decreased SHMT2, whereas deletion of SHMT2 increased SHMT1 (Figure S1h–i). In addition, deletion of MTHFD1L decreased the abundance of ALDH1L2 (Figure S1k). Overall, each of the single gene deletions grew well, without a significant decrease in growth rate for SHMT1, MTHFD2 or MTHFD1L mutant cells and a modest reduction in growth rate for SHMT2 and ALDH1L2 mutant cells (Figure 1c).
Mitochondrial folate pathway knockout depletes 10-formyl-THF and elevates AICAR
To investigate how cellular metabolism responds to targeted deletion of folate enzymes, we analyzed the water soluble metabolome of the deletion mutants using liquid chromatography mass spectrometry (LC-MS) (Figure 1d). A variety of changes occurred that were not connected to 1C metabolism by known mechanisms, including increased UDP-sugars upon deletion of the cytosolic enzymes SHMT1 and ALDH1L1, decreased sugar bisphosphates upon deletion of the mitochondrial enzymes MTHFD2 and MTHFD1L, and decreased TCA cycle intermediates upon depletion of SHMT2. However, by far the strongest metabolic change occurred in 5-aminoimidazole carboxamide ribonucleotide (AICAR, also known as ZMP).
AICAR is the final intermediate in the de novo purine biosynthesis pathway, and its conversion to inosine monophosphate (IMP) requires 10-formyl-THF (Figure 1f). While loss of the cytosolic enzyme SHMT1 resulted in 5-fold decreased AICAR, deletion mutants for the core mitochondrial folate metabolism enzymes SHMT2, MTHFD2, and MTHFD1L manifested between 50- and 500-fold increases (Figure 1e). The other purine biosynthetic intermediate immediately upstream of a step requiring 10-formyl-THF, glycineamide ribonucleotide (GAR), was also elevated in these cell lines (Figure 1f). Moreover, AICAR was strongly elevated in the MTHFD1 hypomorphic cells, which are defective in both routes of making cytosolic 10-formyl-THF (Figure 1d). Collectively, these observations suggest that wild-type HEK 293T cells make most cytosolic 10-formyl-THF from mitochondrial 1C units which are excreted as formate and reassimilated by MTHFD1.
To prove the link between AICAR and formyl unit availability, we fed cells 1 mM formate, which is present in human serum at 20–30 μM (Hanzlik et al., 2005; Lamarre et al., 2014). Although addition did not rescue the modest growth defects of the SHMT2 and ALDH1L2 knockout cells (Figure 1c), it fully reversed the AICAR elevation in each of the mitochondrial folate pathway deletion lines (Figure 1g), but not in the MTHFD1 hypomorphic line (Figure S1l). To prove that loss of the mitochondrial pathway depletes 10-formyl-THF, we directly measured cellular folate species using LC-MS. These measurements, which cannot distinguish between THF and methylene-THF due to abiotic exchange (Jägerstad and Jastrebova, 2014), revealed selective 10-formyl-THF depletion in the mitochondrial folate pathway enzyme knockouts (Figure 1h). Thus, AICAR reports on cytosolic formyl units, with the mitochondrial folate pathway required to maintain normal levels of cytosolic 10-formyl-THF.
1C units are generated exclusively in the mitochondria in most but not all cultured cells
To directly track mitochondrial versus cytosolic 1C unit production, we employed an isotopic tracer assay. Cells were fed [2,3,3-2H]serine and monitored for subsequent labeling of thymidine triphosphate (dTTP) (Herbig et al., 2002). Serine has been reported to be the predominant 1C donor in cultured cells (Fu et al., 2001; Labuschagne et al., 2014) and we confirmed that in HEK 293T cells it accounted for essentially all (> 98%) of 1C units incorporated into biomass building blocks (thymidine and purines; there was no detectable synthesis of methionine; Figure S2a–b). Thymidine is made from cytosolic methylene-THF. Direct production of methylene-THF by SHMT1 will generate M+2 deoxythymidine trisphosphate (dTTP), whereas either cytosolic exchange flux between methylene-THF and 10-formyl-THF or transfer of 1C units from the mitochondrion to cytosol will result in M+1 dTTP (Figure 2a). In wild type (WT) or SHMT1 deletion HEK 293T cells, after correction for natural isotope abundances (see Methods), no M+2 dTTP was detected. In contrast, knockout of any of the mitochondrial pathway enzymes resulted in majority M+2 dTTP (Figure 2b). Consistent with a prior report using this tracer in engineered MEFs (Pike et al., 2010), this result validates the tracing strategy and confirms that the M+1 form arises primarily from mitochondrial 1C production rather than MTHFD1 reversibility. Furthermore, it definitively demonstrates that HEK 293T cells normally process 1C units oxidatively in the mitochondria followed by reductive reincorporation of formate in the cytosol. When mitochondrial folate metabolism is inhibited, cells can adapt by reversing cytosolic flux.
Figure 2. Cytosolic 1C flux is reversed in mitochondrial mutants due to 10-formyl-THF depletion.
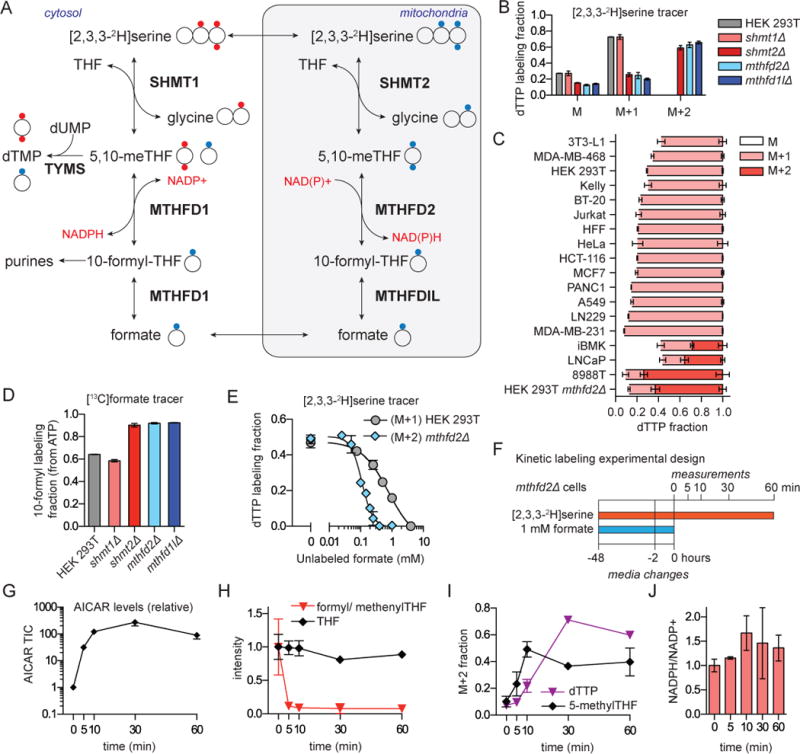
a. Schematic of [2,3,3-2H]serine labeling into thymidine (dTMP). Cytosolic production of methylene-THF directly from serine (red circles) results in doubly 2H-labeled dTMP, whereas its production from 1C units initially generated in mitochondria (blue circles) produces singly 2H-labeled dTMP. Reversibility of MTHFD1 could potentially produce singly labeled dTMP from cytosol-derived 1C units. b. Thymidine (dTTP) labeling in WT and mutant HEK 293T cells reveals cytosolic 1C unit generation (dTTP M+2) solely in the mitochondrial pathway mutants. Labeling fractions are corrected for natural isotope abundances. c. [2,3,3-2H]serine labeling into dTTP in different proliferating cell lines. Cytosolic 1C metabolism (dTTP M+2) is evident only in the HEK 293T MTHFD2 knockout cells and the 8988T pancreatic cancer cell line. d. Exogenous formate (1 mM) is incorporated into ATP via 10-formyl-THF, whose labeling fraction was calculated from the ATP mass isomer distribution. e. Formate competes with [2,3,3-2H]serine for labeling of dTTP. f. Design of experiment probing cytosolic pathway reversal mechanism. Cells lacking the mitochondrial pathway are initially fed formate, resulting in flux from 10-formyl-THF towards methylene-THF, i.e., upwards in (a). Formate is then suddenly removed and metabolic changes monitored. Cytosolic flux directly from [2,3,3-2H]-serine to methylene-THF was tracked as dTTP M+2 and 5-methyl-THF M+2. g. AICAR increases immediately upon formate withdrawal. Signal normalized to time 0. h. 10-formyl-THF but not THF levels drop upon formate withdrawal. i. M+2 dTTP and 5-methyl-THF appear immediately upon formate withdrawal, indicating a switch to cytosolic production of methylene-THF directly from serine (mean ±SD, n≥3) j. NADPH/NADP+ ratio after formate withdrawal from mthfd2Δ cells.
We next investigated mitochondrial versus cytosolic 1C unit production across a spectrum of proliferating cultured cell lines (quiescent cells do not synthesize dTTP and thus cannot be probed by this assay). These cell lines included a diversity of cancers, as well as primary human foreskin fibroblasts (HFF) and proliferating 3T3-L1 preadipocytes. In the majority of cells only M+1 dTTP was observed, demonstrating that most cytosolic 1C units come from mitochondrial folate metabolism (Figure 2c). In some cell lines however, including the transformed renal epithelial line iBMK, the prostate cancer line LNCaP, and pancreatic cancer line 8988T, we observed a strong M+2 peak in dTTP. In the iBMK cells and LNCaP cells, we observed a labeling pattern intermediate between WT and mitochondrial pathway knockout HEK 239T cells, suggesting 1C unit generation in both the cytosol and mitochondria. In 8988T cells, the pattern matched the engineered mitochondrial knockout cells, suggesting a sole reliance on cytosolic 1C generation. Thus, while most cultured cells produce 1C units exclusively in the mitochondria, some cells show mixed usage or a sole reliance on the cytosolic pathway.
Formyl-THF depletion enables cytosolic 1C unit production
To explore the mechanism underlying reversal of cytosolic 1C unit flux in the mitochondrial folate enzyme knockout cells, we introduced [13C]formate and monitored its incorporation via formyl-THF into ATP. WT or SHMT1 knockout cells incorporated a mixture of the exogenously provided [13C]formate and endogenously generated 1C units exported from the mitochondria as unlabeled formate (Figure 2d), with the extent of ATP and dTTP labeling dependent on the added formate concentration (Figure S2c–d). Added formate further labeled serine in an SHMT dependent manner (Figure S2e). In contrast, cells with the mitochondrial pathway deletion assimilated almost solely the exogenously provided formate (Figure 2d), which alleviated their chronic cytosolic 10-formyl-THF deficiency as indicated by normalization of AICAR (Figure 1g).
We then evaluated the exogenous formate concentration required to outcompete endogenous mitochondrial formate production (in WT cells) or to drive reductive 1C flux in the cytosol (in MTHFD2 mutant cells). To this end, cells were cultured in a fixed concentration of [2,3,3-2H]serine and dTTP labeling was monitored upon formate titration (Figure 2e). In WT cells, the IC50 for inhibition of serine dTTP labeling by formate was 800 μM, suggesting that endogenous mitochondrial production yields cytosolic formate concentrations in this range. In the MTHFD2 deletion cells, the IC50 for inhibiting cytosolic derived serine dTTP labeling was 140 μM. Thus, mitochondrial folate metabolism produces cytosolic formate concentrations ~40-fold higher than circulating formate and ~6-fold higher than required to drive cytosolic flux in the direction of formyl-THF reduction.
Building from the observation that mitochondrial deletion cells readily used formate, we examined the effects of sudden formate removal on 1C metabolism (Figure 2f). Within 5 minutes, AICAR rose 50-fold and 10-formyl-THF fell by roughly 10-fold (Figure 2g–h). Using the [2,3,3-2H]serine tracer, we simultaneously monitored the appearance of M+2 labeling in 5-methyl-THF and dTTP (Figure 2i). Such M+2 labeling was absent in the presence of formate and rose rapidly over the first 30 minutes after formate removal, indicating immediate cytosolic pathway activation.
Reversal of the cytosolic pathway from 10-formyl-THF reduction to methylene-THF oxidation requires switching the thermodynamically favored direction of the MTHFD1 methylene-THF dehydrogenase-cyclohydrolase reaction sequence (Equation 1):
| (Eq. 1) |
In the default reductive direction, the major substrates are 10-formyl-THF and NADPH, which make methylene-THF and NADP+. Upon formate removal, the NADPH/NADP+ ratio increased slightly (Figure 2j), consistent with this reaction now making rather than consuming NADPH; this change, however, works against the necessary change in reaction free energy. Thus, the large decrease in 10-formyl-THF upon formate removal is sufficient to switch the thermodynamically favored direction of these reactions from consuming to making 10-formyl-THF. This mechanism, mediated by the concentration of a single metabolite, renders folate metabolism, at least under favorable nutrient conditions, robust to loss of the mitochondrial pathway.
Folate metabolism can produce either mitochondrial or cytosolic NADPH
Oxidation of methylene-THF to 10-formyl-THF is coupled to reduction of NAD(P)+ to NAD(P)H depending upon the compartment in which it occurs. The associated redox-active hydrogen can be labeled by feeding with [2,3,3-2H]serine. Compartmentalization and cofactor usage can then be explored based on passage of the 2H-label into downstream NAD(P)H products. Fatty acid synthesis specifically uses cytosolic NADPH, with 14 NADPH consumed per palmitate (Figure 3a). Consistent with 1C unit oxidation occurring in mitochondria, no labeling of palmitate was observed in WT HEK 293T cells (Figure 3b). Labeling was, however, observed in HEK 293T mutants defective in mitochondrial 1C metabolism and in the 8988T pancreatic cancer cell line with an apparent intrinsic defect in mitochondrial 1C metabolism (Figure 3b–c). This change in labeling was not due to changes in cellular NADPH levels (Figure S3a). The quantitative contribution of folate metabolism to cytosolic NADPH, after correcting for the 2H-kinetic isotope effect (KIE) (see Methods), ranged from 5% to 16%.
Figure 3. Folate metabolism generates NADPH in a compartment-specific manner.
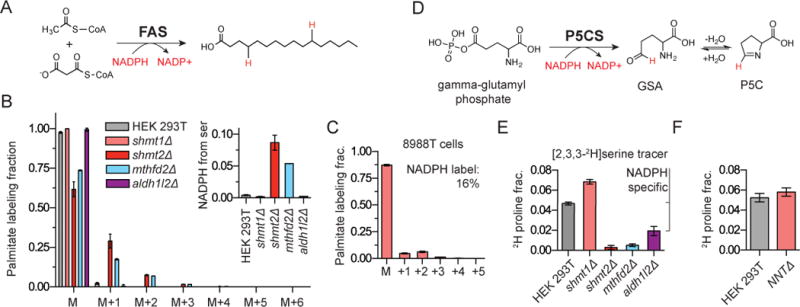
a. Fatty acid synthase combines malonyl-CoA and acetyl-CoA to synthesize saturated fatty acid chains. Deuterons from labeled NADPH will be incorporated throughout the fatty acid chain in an amount depending on the extent of cytosolic NADPH labeling. b. Palmitate isotope labeling from [2,3,3-2H]serine. Data are corrected for natural isotope abundances. Inset shows calculated NADPH labeling fraction after correction for the extent of serine labeling and the 2H-kinetic isotope effect. c. Palmitate labeling from [2,3,3-2H]serine in pancreatic cell line 8988T. d. The committed step of proline biosynthesis is the NADPH-dependent reduction of gamma-glutamyl phosphate to pyrolline-5-carboxylate (P5C). e. 2H-labeling of proline in selected folate mutant cell lines. The difference in 2H labeling between the WT and aldh1l2Δ is highlighted as the ALDH1L2 reaction is known to make mitochondrial NADPH. f. 2H-proline labeling fraction in WT and nntΔ cells confirms that proline labeling does not require passage of 2H from NADH to NADPH (mean ±SD, n≥3).
To analyze folate-mediated mitochondrial NAPDH production, we employed proline as a reporter metabolite. The first step in proline biosynthesis is catalyzed by the mitochondrial enzyme P5CS which reduces gamma-glutamyl-phosphate to pyrroline-5-carboxylate (P5C) using NADPH (Figure 3d). The subsequent downstream steps in proline synthesis also involve NAD(P)H-driven reduction, with compartmentalization depending on the isozyme involved. In WT HEK 293T cells fed [2,3,3-2H]serine, after correction for the extent of serine labeling (but not the 2H-KIE, which has not been measured for the ALDH1L2 reaction), we determined that 5% of proline contains a hydrogen from serine. Deletion of either SHMT2 or MTHFD2 blocks this labeling, confirming that it occurs via mitochondrial folate metabolism (Figure 3e). The labeling was decreased in the ALDH1L2 deletion clone, indicating dedicated mitochondrial NADPH production by this enzyme (Figure 3e). To confirm that the labeling does not involve conversion of NADH to NADPH by nicotinamide nucleotide transhydrogenase (NNT) (Leung et al., 2015), we knocked out NNT and observed no impact on proline labeling (Figure 3f). The results with endogenous fatty acid and proline reporters were confirmed with engineered 2-hydroxygluturate reporters based on overexpression of mutant IDH1 and IDH2 (Figure S3b) (Lewis et al., 2014). Consistent with earlier results (Fan et al., 2014), cell lines deficient in MTHFD2 or ALDH1L2 were sensitized to oxidative stress in the form of hydrogen peroxide (Figure S3c). Thus, folate metabolism, substantially via ALDH1L2, contributes to physiologically significant mitochondrial NADPH production.
Folate metabolism produces mitochondrial NADH and excess formate
To measure the contribution of mitochondrial folate metabolism to NADH production, we used malate as a reporter metabolite. In the TCA cycle, malate dehydrogenase (MDH) catalyzes the reversible conversion of NAD+ and malate into NADH and oxaloacetate (OAA) (Figure 4a). In HEK 293T cells fed [2,3,3-2H]serine, we observed substantial malate labeling. Quantitative analysis, correcting for the extent of serine labeling and the 2H-KIE, revealed that 7% of the NADH pool that reduces OAA to malate incorporates a hydride from serine (Figure 4b). Labeling was abolished in SHMT2 and MTHFD2 deletion cell lines, diminished in the MTHFD1L deletion cell, and only modestly affected in the ALDH1L2 deletion. Thus, serine-driven mitochondrial flux through MTHFD2 is a significant NADH source.
Figure 4. Mitochondrial folate flux exceeds 1C demand.
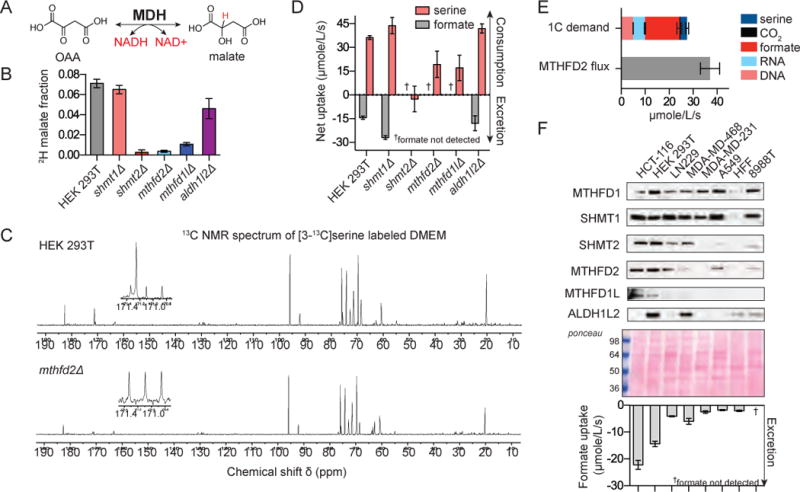
a. Malate dehydrogenase incorporates a hydride from NADH into malate. b. Calculated fraction of malate redox-active hydrogen derived from serine. 2H-labeling fraction of malate was corrected for the extent of [2,3,3-2H]serine labeling and the 2H-kinetic isotope effect. c. 13C-NMR spectra of spent media (8 h incubation) in cells fed [3-13C]serine. Samples were spiked with [2H-13C]formate standard (a triplet at 171.09 ppm). 13C formate is a singlet at 171.24 ppm. d. Net uptake of serine and excretion of formate. e. Fluxes consuming 1C units in HEK 293T cells. 1C units are consumed by DNA, RNA, and serine synthesis and excreted as CO2 and formate. MTHFD2 flux is calculated from malate labeling in (b) and O2 consumption (Methods). f. Expression of 1C metabolism enzymes and excretion of formate for a set of proliferating human cell lines (mean ±SD, n≥3).
The magnitude of the 1C metabolism contribution to mitochondrial NADH was remarkable, given the high rate of NADH production from the TCA cycle. Total mitochondrial NADH turnover can be estimated based on the cellular O2 uptake rate, with ~ 80% of O2 uptake consumed by oxidative phosphorylation and 75% of oxidative phosphorylation driven by NADH (Fan et al., 2013; Rolfe and Brown, 1997). Under these assumptions, a 7% contribution of 1C metabolism to mitochondrial NADH requires MTHFD2 flux of ~35 μmol/L/s (L of cell volume) (Figure S4a–b and Methods), which is roughly triple the 1C unit flux required to support HEK 293T cell biosynthetic reactions (10 μmol/L/s) (See Methods). This raises the possibility that 1C metabolism, under these nutrient replete conditions, runs in excess of 1C unit demand.
To look directly for excess 1C units coming from mitochondrial serine catabolism, we measured cellular excretion of serine-derived CO2 and formate. Using [3-14C]serine, we measured flux of serine-derived carbon through the ALDH1L enzymes into CO2, but the magnitude was small, consistent with the extent of mitochondrial ALDH1L2-mediated NADPH production (Figure S4b). In contrast, feeding cells [3-13C]serine and monitoring the production of [13C]formate by NMR, revealed a large formate excretion flux. Serine-derived [13C]formate was evident as a singlet NMR peak at 171.24 ppm, which was absent in MTHFD2 deletion cells, against a background triplet peak resulting from [2H-13C]formate internal standard (Figure 4c). Total formate excretion was equal to nearly half of serine uptake in WT HEK 293T cells, increased yet more in the SHMT1 deletion cells, and was absent in deletion mutants for the core mitochondrial folate metabolism pathway (Figure 4d). The total measured 1C unit production, combining biosynthetic demand, serine synthesis from glycine (Figure S4c–d), CO2 release, and formate excretion, was 28±3 μmol/L/s, in close agreement with the estimated MTHFD2 flux from NADH labeling (37±4 μmol/L/s) (Figure 4e). Changes to media serine concentration did not alter WT growth rates but did reduce formate excretion (Figure S4e).
We then measured formate excretion across a set of proliferating human cell lines. Excretion rates varied considerably, and generally mirrored expression of the mitochondrial folate enzymes MTHFD2, SHMT2 and MTHFD1L (Figure 4f). Thus, 1C unit production is not closely regulated to match biosynthetic demand, but instead depends on the expression level of the mitochondrial pathway enzymes. The exception was the pancreatic cell line 8988T, which despite expression of SHMT2, MTHFD2 and the mitochondrial folate transporter, (Figure S4f) produced no measurable formate (low levels of MTHFD1L expression were difficult to ascertain by our antibody). Consistent with the 2H-tracing data, this result suggested that 8988T cells contain a mutation in a mitochondrial folate metabolism gene. We reverse transcribed and sequenced the mRNAs of 1C metabolism genes from this cell line and identified a 10 bp insertion between exons 8 and 9 of the MTHFD1L mRNA transcript (Figure S4g). This change reflects the introduction of an intronic de novo exon 9 splice acceptor site at position -11 (NG_029185.1: g.57888A>G) (Figure S4h). The variant was confirmed in 8988S, a sister cell line to 8988T. We identified this MTHFD1L variant in the ExAC database as a rare allele in the normal population, suggesting that certain individuals harbor heterozygous mutations in MTHFD1L, which may, due to loss of heterozygosity, result in tumors defective in mitochondrial folate metabolism (Figure S4i) (Consortium et al., 2015).
Mitochondrial folate metabolism mutants require exogenous serine and glycine
WT HEK 293T cells grow rapidly even in the absence of exogenous serine and glycine (Figure 5a). However, MTHFD2 deletion cells are dependent upon both exogenous serine and glycine for growth (Figure 5a), as were the 8988T cells naturally defective in MTHFD1L (Figure S5a). When mitochondrial folate metabolism mutant cells were starved for serine, growth was restored by 1 mM formate (Figure 5b and S5b), indicating that extracellular serine is required for the SHMT1 reaction to meet 1C unit demand. Indeed, even though it did not block growth, serine deprivation also led to 1C stress in WT cells with the intact mitochondrial pathway, as indicated by markedly enhanced intracellular AICAR (Figure 5c). This AICAR increase, despite no change in the AMP/ATP ratio, was sufficient to modestly activate AMP-activated protein kinase (AMPK), as indicated by increased phosphorylation of both AMPK itself and the classical AMPK substrate acetyl-CoA carboxylase (ACC) (Figure 5d). Thus, 1C unit sufficiency depends on both extracellular serine and the mitochondrial 1C pathway. Absence of both results in a severe enough 1C deficiency to impair growth.
Figure 5. Mitochondrial folate metabolism mutants are dependent upon exogenous serine and glycine.
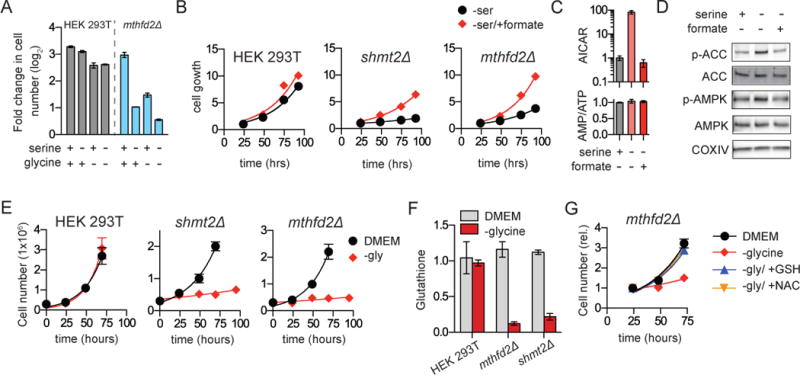
a. Growth of HEK 293T and mthfd2Δ cell lines (plotted as log2[fold change in cell number]) in glycine- and serine-free media after 4 days. b. Growth of mthfd2Δ and shmt2Δ cells in serine-free media is rescued by 1 mM sodium formate. c. AICAR signal and AMP/ATP ratio after 1 h serine withdrawal in WT HEK 293T cells with and without formate (1 mM). d. Western blot of cells treated as in (c) shows phosphorylation of AMPK (T172) and ACC (S79) in response to serine withdrawal. e. Growth of mthfd2Δ and shmt2Δ cells requires glycine. f. Glutathione is depleted in mthfd2Δ and shmt2Δ cells grown without glycine. g. Growth of mthfd2Δ cells in glycine-free media is restored by addition of reduced glutathione (GSH; 250 μM) or N-acetylcysteine (NAC; 250 μM) (mean ±SD, n≥3).
Similar to serine, and consistent with prior reports in mouse embryonic fibroblasts and Chinese hamster ovary cells (Patel et al., 2003; Stover et al., 1997), glycine was required to support growth of MTHFD2 and SHMT2, but not SHMT1 knockout cells (Figure 5e and S5c). Interestingly, formate did not rescue but rather worsened growth (Figure S5d). Thus, glycine is not needed to provide 1C units. To understand the metabolic contribution of glycine, we performed metabolomic analysis on glycine-deprived SHMT2 cells (Figure S5e). Among the strongest metabolic changes was depletion of glutathione (γ-glutamyl-cysteinyl-glycine) (Figure 5f). This suggested that the growth-impairment arose from antioxidant deficiency. Consistent with this, either glutathione or N-acetylcysteine supplementation in the media rescued growth (Figure 5g). We conservatively calculated cellular glycine demand (protein, purines, and glutathione) and found it to be comparable to and likely in excess of 1C demand (purines, dTTP). Thus, when it cannot be imported, glycine is a critical product of folate metabolism (Figure S5f), with folate-mediated mitochondrial glycine production supporting glutathione synthesis and associated redox homeostasis.
MTHFD2 knockout impairs colon cancer xenograft tumor growth
Given the overexpression of mitochondrial folate metabolism genes in cancer, we sought to test if cells lacking this pathway could establish tumors in vivo. We generated MTHFD2 deletion clones in the human colorectal cancer cell line HCT-116 (Figure 6a), which highly expresses mitochondrial folate metabolism genes and rapidly excretes formate (Figure 4f). In standard DMEM media, growth of the MTHFD2 deletion clones was similar to the parental cell line (Figure 6b). As in the HEK 293T background, the HCT-116 MTHFD2 deletion cells had massively elevated AICAR (Figure 6c), indicating 1C unit deficiency, and switched to cytosolic 1C unit production as indicated by M+2 labeling of dTTP and palmitate labeling from [2,3,3-2H]serine (but not malate labeling) (Figure 6d–f). The HCT-116 MTHFD2 deletion cells also shared the dependency of the HEK 293T MTHFD2 knockout cells on exogenous glycine and serine (Figure 6g–i). Thus, in vitro, the requirement for mitochondrial 1C metabolism depends on the nutrient environment.
Figure 6. Tumor growth is attenuated in HCT-116 cells lacking MTHFD2.
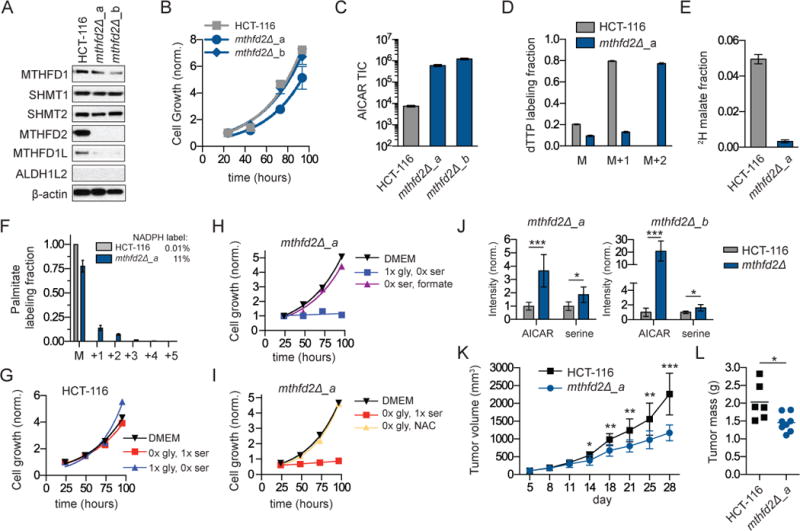
a. Western blot of folate metabolism genes in HCT-116 cells and two MTHFD2 deletion clones. b. Cell growth in complete media. c. Steady-state AICAR levels. d. dTTP labeling from [2,3,3-2H]serine (48 h). e. Fraction of malate redox-active hydrogen derived from serine as per figure 4b. f. 2H-labeling into palmitate from [2,3,3-2H]serine. Inset shows associated NADPH labeling fraction. g. Growth of HCT-116 cells in media without serine or glycine. h. Growth of HCT-116 mthfd2Δ_a cells require either serine or formate (1 mM). i. Growth of HCT-116 mthfd2Δ_a cells require either glycine of NAC (1 mM). j. Intratumor metabolite levels (AICAR and serine) from WT and deletion tumors implanted in CD-1 nude mice and measured by LC-MS (n=8–10). k. Tumor growth (volume ±SEM as measured by calipers) in nude mice containing subcutaneous tumors from HCT-116 WT and mthfd2Δ cells (n=10). l. Final tumor weights from tumor growth experiment in (k). * p<0.05; ** p<0.01; *** p<0.001 by unpaired t-test.
To evaluate growth in a tumor environment in vivo, we implanted WT and mutant cells onto the hind flanks of CD-1 nude mice. Both WT and MTHFD2 mutant cells established tumors, which we collected at small size (~100 mgs) to obtain samples for metabolomics prior to substantial tumor necrosis. As in cultured cells, AICAR levels were consistently high in the mutant tumors (Figure 6j). Additionally, intratumor serine levels were elevated, consistent with defective serine catabolism (Figure 6j). The same changes in tumor metabolites were observed when these cells were implanted in NOD scid gamma (NSG) mice (Figure S6a). We next evaluated the effect of this mutation on tumor growth. MTHFD2 deletion tumors grew slower than WT controls and attained lower final tumor weights in both nude and NSG mouse models (Figure 6k–l, S6b–c); however, progressive tumor growth was observed in all cases. Thus, mitochondrial 1C metabolism is not required for either HCT-116 xenograft tumor initiation or growth, but does accelerate the tumor growth rate.
Reversal of SHMT1 flux enables growth of MTHFD2 deletion xenografts
The growth of MTHFD2 mutant tumors in vivo led us to ask whether the mechanisms underlying this escape were the same as we had observed in cell culture. Cells may overcome the 1C deficit in vivo by reversing SHMT1 flux, or by scavenging nutrients such as purines, folates and formate from the serum or surrounding tissues. To directly measure the source of 1C units in vivo, we infused [2,3,3-2H]serine via jugular venous catheter into nude mice bearing bilateral HCT-116 xenografts, one WT and the other deleted for MTHFD2 (Figure 7a). Steady-state circulating serine labeling was achieved by 4 h (Figure S7a), with substantially greater cardiac than venous labeling indicating tissue uptake of labeled serine and release of unlabeled serine (Figure 7a). The serine infusion resulted in labeling of circulating formate, demonstrating that circulating formate can originate from tissue serine catabolism (as opposed to exclusively dietary sources) (Figure 7b). Total enriched serine was nearly identical between the mutant and WT tumors (Figure 7a). However, when we analyzed dTTP labeling from tumors, large differences were apparent. Nearly 20% of mutant tumor dTTP was M+2 labeled, indicating methylene-THF production from serine via the cytosolic SHMT1 reaction (Figure 7b). In contrast, only M+1 dTTP was observed in the WT tumors, indicating mitochondrial generation of the 1C units from serine (Figure 7b). Thus MTHFD2 mutant tumors in vivo acquire 1C units via cytosolic serine catabolism.
Figure 7. Growth of HCT-116 MTHFD2 deletion tumors requires SHMT1.
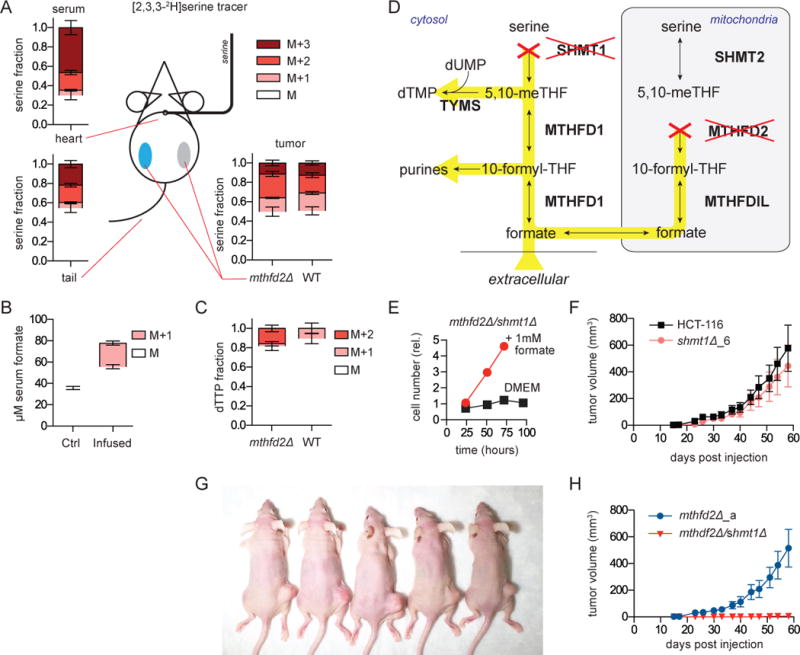
a. Serine enrichment from steady-state infusion (130 mg/kg/h) of [2,3,3-2H]serine. Serum serine isotope labeling obtained from cardiac puncture (upper left) or tail vein draw (bottom left) or from tumor metabolite extraction (bottom right) (n=7). b. Serum formate concentration and labeling in mice that did or did not receive [2,3,3-2H]serine infusion (n=4–8). c. dTTP 2H-labeling fraction from extracted tumors. M+2 dTTP is from cytosolic (SHMT1-mediated) serine-driven methylene-THF production; M+1 is from mitochondrial serine catabolism. d. Schematic showing blocked reactions and potential for formate rescue (yellow highlighting) in SHMT1/MTHFD2 double deletion cells. e. Growth of HCT-116 mthfd2Δ/shmt1Δ cells ±1 mM formate. f. Growth of shmt1Δ xenografts (n=10). g. Representative mice 8 weeks post injection of mthfd2Δ (right) and mthfd2Δ/shmt1Δ (left) cells. h. Associated tumor growth curves (mean ±SEM, n=12).
We next tested whether SHMT1 flux was essential for tumor growth in MTHFD2 deletion tumors. We generated a HCT-116 SHMT1 deletion cell line and a double MTHFD2/SHMT1 deletion cell line (Figure 7d and S7b–d). As expected, the double mutant line was auxotrophic for formate (Figure 7e, S7e–f). In vivo, the SHMT1 deletion tumors engrafted and grew comparably to the WT tumors (Figure 7f). In contrast, no tumors formed from two separate MTHFD2/SHMT1 double deletion cell lines (Figure 7g–h and S7g–i). While there is compartmental flexibility with respect to tumor 1C metabolism, intracellular generation of 1C units from serine is essential for HCT-116 tumorigenesis.
Discussion
The mammalian metabolic network is complex with multiple potential redundancies available to overcome nearly any single disruption. Our work demonstrates the capability, using modern genome editing methods and isotope tracing, to comprehensively dissect a complex multi-compartment pathway in mammalian cells. The system we targeted, folate metabolism, plays a central role in supporting cell growth in fetal development, immune activation, and tumorigenesis. We quantitate the mitochondrial folate metabolic fluxes that supply both 1C units and glycine, and determine the compensatory mechanisms invoked when this metabolism is genetically ablated.
In addition, we re-examine the contribution of folate pathways to redox metabolism. We previously established that deuterium tracers could be used to probe both the sources and sinks of NADPH (Fan et al., 2014). We showed that most cytosolic NADPH is consumed by reductive biosynthesis. We further proposed that NADPH could be produced from both cytosolic and mitochondrial folate pathways and validated this in part using shRNA against folate enzymes. Here, using CRISPR knockout cell lines, we confirm the role of folate metabolism in generating mitochondrial NADPH. These findings are consistent with additional recent work demonstrating a role for mitochondrial folate-mediated NADPH production in melanoma metastasis (Piskounova et al., 2015). We find, however, that, consistent with most prior literature on this topic, cytosolic 1C pathway fluxes in most proliferating cells are typically running in the direction from 10-formyl-THF to methylene-THF, and therefore are consuming, not producing NADPH (Tibbetts and Appling, 2010). Our prior assertion that most cells growing in culture showed limited flux from mitochondrially derived formate to purines, and accordingly generated formyl units in the cytosol with concomitant NADPH production, was based on the observation that, even in transformed cells with active glycine cleavage, glycine does not contribute substantially to cytosolic 1C units. However, in many of the cultured cell lines we have examined, absolute glycine cleavage flux is low relative to mitochondrial serine catabolism, diluting the glycine-derived signal in isotope labeling experiments and limiting the utility of this as a compartmental tracer. While the metabolic role of glycine cleavage in cell culture remains unclear, the integrative genetic and tracer analysis here, in support of the related prior literature, effectively proves the predominance of mitochondrial 1C generation from serine in most cultured cells.
Importantly, while essentially all 1C units in many cell lines are generated via mitochondrial serine catabolism, other cell lines (the transformed renal epithelial cell line iBMK and the prostate cancer cell line LNCaP) showed mixed cytosolic and mitochondrial 1C generation. The active utilization of the cytosolic 1C pathway in these cells may relate to their tissue of origin; for example, kidney expresses high levels of SHMT1. In yet other cell lines, mitochondrial folate metabolism was overtly defective, either due to endogenous mutations (8988T pancreatic cancer cells) or engineered enzyme loss. In such cases, all 1C units for nucleotide synthesis are derived in the cytosol and folate metabolism becomes a major cytosolic NADPH source.
The relative ease of CRISPR-mediated gene knockout enabled us to evaluate the impact of losing each of the core enzymes of the mitochondrial 1C pathway: SHMT2, MTHFD2, and MTHFD1L. Common effects include high levels of AICAR, glycine auxotrophy and cytosolic 1C flux reversal. While the MTHFD2 and MTHFD1L knockout cell lines grew normally, loss of SHMT2 impaired cell growth in a manner that was not rescued by formate. In addition, loss of SHMT2 was unique in depleting mitochondrial metabolites. This suggests that SHMT2 plays a role in mitochondrial health independent of 1C cycling between the cytosol and mitochondria, likely related to its ability to generate localized glycine and/or methylene-THF, with the latter potentially needed to support mitochondrial thymidine production (Anderson et al., 2011).
We find that, in HEK 293T and HCT-116 cells, the cytosolic enzyme SHMT1 runs in the direction of serine synthesis, consuming glycine and 1C units. Indeed, one function of SHMT1 may be to avoid toxicity arising from inappropriate accumulation of these species (Kim et al., 2015). Intriguingly, the rate of formate elimination predicts resistance across species to the dietary poison methanol (Sweeting et al., 2010), suggesting such a detoxification role could be evolutionarily significant. Under cytosolic 1C stress, however, SHMT1 can flip flux direction to enable 1C unit generation. This may be important in contexts such as hypoxia or electron transport chain inhibition, where an increased NADH/NAD+ ratio could impair mitochondrial 1C flux. We find that, in the context of mitochondrial pathway deletion, such serine catabolic flux by SHMT1 is the primary mode of tumor 1C acquisition and essential for xenograft formation.
The absolute requirement for either SHMT1 or MTHFD2 activity in vivo, while a logical example of synthetic lethality, was nevertheless somewhat surprising. The potential compensatory mechanisms to circumvent MTHFD2 deletion in vivo are many and involve both metabolic and genetic mechanisms. Metabolically, 1C unit demand could have been met by circulating formate, methyl-THF, purines and pyrimidines. Genetically, an isozyme of the MTHFD2 gene, MTHFD2L, encodes another functional mitochondrial methylene-THF dehydrogenase, which if expressed, would be expected to rescue mitochondrial 1C metabolism. Finally, SHMT2 is constitutively found not only in mitochondria but also the nucleus (Anderson and Stover, 2009). Given that the nucleus is thought to be in free metabolic exchange with the cytosol, one would have anticipated that nuclear SHMT2 could substitute for SHMT1. The lack of rescue by any of these mechanisms highlights the absolute dependence of growing cells in vivo on intrinsic 1C metabolism, and the potential, despite the pathway’s flexibility, for its rational therapeutic targeting.
Experimental Procedures
Generation of CRISPR knockouts
Clonal CRISPR knockout cell lines were generated using the double nicking variant of the published protocol of Ran and colleagues (Ran et al., 2013b). Briefly, guide RNAs against exons of target genes (Table S1) were cloned into a Cas9 nickase expression vector. Cells were transiently transfected using Lipofectamine 2000 (Life Technologies) (HEK 293T) or Fugene HD (Promega) (HCT-116) and selected for 48 h with 2 μg/mL puromycin. After selection, cells were serially diluted in 96 well plates. SHMT1/MTHFD2 double deletion cell lines were selected for in media with 1 mM formate. Functional gene deletion was confirmed by western blotting followed by targeted genomic sequencing.
Metabolite Profiling
Cells were grown in 6 cm tissue culture dishes for at least 2 doublings, during which the medium was replaced every day (DMEM supplemented with 10% dialyzed fetal bovine serum (F0392, Sigma-Aldrich)), and additionally 2 hours before extraction. Metabolism was quenched and metabolites were extracted by aspirating media and immediately adding 800 μL −80°C 80:20 methanol:water extraction solution. F or amino acid labeling experiments, cells were washed 3× with warm PBS before quenching with extraction solution. The resulting mixture was incubated on dry ice, scraped, collected into a microfuge tube, and centrifuged at 5000 × g for 5 min. Insoluble pellets were re-extracted with 250 μL extraction solution. The supernatants were combined and dried under N2, and finally resuspended in water to a dilution of 50 times the packed cell volume. Samples were analyzed by reversed-phase ion-pairing chromatography coupled with negative-mode electrospray-ionization high-resolution MS on a stand-alone orbitrap (ThermoFisher Exactive) (Lu et al., 2010).
Proliferation Assays
Standard proliferation assays were conducted in 24 well plates and relative cell number measured using resazurin sodium salt. 2×104 cells were plated in each well with 1 mL DMEM supplemented with 10% dialyzed fetal bovine serum. For serine and glycine drop-out media, DMEM (4.5 g/L glucose, 4 mM glutamine) was formulated from scratch following standard procedures (pH 7.4). Cell growth at each day was read as fluorescence intensity using a Synergy HT plate reader (BioTek).
Mouse xenografts
All animal studies were approved and conducted under the supervision of the Princeton University Institutional Animal Care and Use Committee. For tumor growth studies 6–7 week old female CD1/nude mice were injected on the rear flank with HCT-116 cells (1×106 cells in 100 μL). For the comparison between WT and MTHFD2 mutant growth, cells were implanted in individual animals using Matrigel (50%), whereas for studies using SHMT1 deletions, knockout and control cells were implanted bilaterally on the same animal in growth media. Tumor growth measurements were taken biweekly using two caliper measurements (volume= ½[L × W2]). Animals were euthanized when tumors reached 2500 mm3 or if they displayed any signs of distress or morbidity. For tumor metabolomic studies 6–7 week old female CD1/nude or male NOD scid gamma (NSG) mice were bilaterally injected on the rear flank with HCT-116 wild type (right) and MTHFD2 mutant cells (left) (1×106 cells in 100 μL PBS). Mice were sacrificed when control tumors had achieved ~100 mg (approximately 5 mm) in average size to avoid the development of tumor necrosis. Tumors were removed and immediately frozen in liquid nitrogen for LC-MS analysis. Isolated tumors were weighed and then 50 mg of tissue was disrupted using a cryomill, and lysed in 1mL ice cold 40:40:20 acetonitrile:methanol:water. Solids were precipitated, spun down and re-extracted with 1 mL lysis buffer. Combined supernatants were dried down and resuspended in water to a concentration of 50 mg/mL (original tumor mass) before analysis by LC-MS.
Mouse infusion studies
6–7 week old female CD1/nude mice (Charles River Laboratories) were subcutaneously implanted with 1×106 HCT-116 cells (100 μL total volume) bilaterally on hind flanks. At an average tumor size of 50 mm3, jugular venous catheters were placed following standard procedures (Kmiotek et al., 2012). After surgery, mice were housed individually and allowed to recover for a one-week period before infusion of [2,3,3-2H]serine (400 mM in PBS) at a rate of 130 mg/kg/h over 4 hours. During the course of the experiment mice were allowed to move freely using a tethering apparatus (Instech Laboratories, Plymouth Meeting, PA) with no access to food. To assess venous tracer enrichment, blood was sampled from tail vein 30 min before the end of the experiment. Tumors were harvested under anesthesia and a terminal cardiac draw was performed to obtain serine enrichment in the cardiac serum.
Statistical Analysis
Samples sizes, error bars, and p-values are defined in each figure legend. Results for technical replicates are presented as mean ± SD, for calculated growth rates as mean ± 95% CI and for tumor volumes mean ± SEM. Growth rates were calculated by fitting to an exponential. Statistical significance between conditions was calculated using an unpaired two-tailed Student’s t-test. All statistical calculations were performed using the software package Prism 6.0g.
Supplementary Material
Highlights.
In most growing cells, mitochondrial 1C metabolism maintains cytosolic formyl-THF
Mitochondrial 1C metabolism also supports redox homeostasis by making glycine and NADPH
Depletion of formyl-THF induces cytosolic flux reversal to make 1C from serine
Cytosolic folate metabolism is sufficient to support tumor growth
Acknowledgments
We thank all members of the Rabinowitz lab, in particular W. Lu, S. Zhang and Z. Zhang for assistance in mass spectrometry methods and X. Su for isotope correction calculations. We thank C. Lewis for the IDH reporter construct. We acknowledge V. Suri and M. Manfredi of Raze Therapeutics for assistance with tumor growth data. This research was supported in part by funding to J.D.R. from the U.S. National Institutes of Health (NIH) (R01CA16359-01A1), Stand Up to Cancer (SU2C-AACR-DT0509) and to Y.K from the Brewster Foundation, the NIH (R01CA141062) and the U. S. Department of Defense (BC123187). G.S.D. is supported by a postdoctoral fellowship (PF-15-190-01- TBE) from the American Cancer Society and received prior assistance from a NJCCR postdoctoral fellowship (DFHS15PPC044). M.E. is supported by a predoctoral fellowship from the NIH (F31CA192461). R.J.M. is supported by a PMU-FFF fellowship (L-15/03/004-MOR). J.D.R. is a founder and member of the scientific advisory board of Raze Therapeutics, which seeks to target 1C metabolism for cancer therapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: G.S.D. and J.D.R. conceived of the project and designed the research plan. G.S.D., L.C., J.M.G. and X.T. performed biochemical experiments. Y.K., M.E., R.J.M. and G.S.D. planned and designed mouse experiments. R.J.M., M.E. and G.S.D. performed mouse experiments. G.S.D. analyzed the data with assistance from X.T. and L.C. G.S.D. and J.D.R. wrote the manuscript. All authors reviewed and edited the manuscript before submission.
References
- Anderson DD, Stover PJ. SHMT1 and SHMT2 Are Functionally Redundant in Nuclear De novo Thymidylate Biosynthesis. PLoS ONE. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108:15163–15168. doi: 10.1073/pnas.1103623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult S, Asara JM. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–732. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolusani S, Young BA, Cole NA, Tibbetts AS, Momb J, Bryant JD, Solmonson A, Appling DR. Mammalian MTHFD2L Encodes a Mitochondrial Methylenetetrahydrofolate Dehydrogenase Isozyme Expressed in Adult Tissues. Journal of Biological Chemistry. 2011;286:5166–5174. doi: 10.1074/jbc.M110.196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EA, Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O’Donnell-Luria A, Ware J, Hill A, et al. Analysis of protein-coding genetic variation in 60, 706 humans. 2015 doi: 10.1038/nature19057. bioRxiv 030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015 doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro E, Sirois J, Tremblay ML, MacKenzie RE. Mitochondrial NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase is essential for embryonic development. Molecular and Cellular Biology. 2002;22:4158–4166. doi: 10.1128/MCB.22.12.4158-4166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Molecular Systems Biology. 2013;9:1–11. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TF, Rife JP, Schirch V. The role of serine hydroxymethyltransferase isozymes in one-carbon metabolism in MCF-7 cells as determined by (13)C NMR. Archives of Biochemistry and Biophysics. 2001;393:42–50. doi: 10.1006/abbi.2001.2471. [DOI] [PubMed] [Google Scholar]
- Hanzlik RP, Fowler SC, Eells JT. Absorption and elimination of formate following oral administration of calcium formate in female human subjects. Drug Metabolism and Disposition. 2005;33:282–286. doi: 10.1124/dmd.104.001289. [DOI] [PubMed] [Google Scholar]
- Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem. 2002;277:38381–38389. doi: 10.1074/jbc.M205000200. [DOI] [PubMed] [Google Scholar]
- Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul. 1994;34:397–419. doi: 10.1016/0065-2571(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jägerstad M, Jastrebova J. 5,10-Methylene-tetrahydrofolate dissociates into tetrahydrofolate and formaldehyde at physiological pH and acidic pH, typical conditions used during sample extraction and LC-MS/MS analysis of biological samples. Biomed Chromatogr. 2014;28:1041–1042. doi: 10.1002/bmc.3214. [DOI] [PubMed] [Google Scholar]
- Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiotek EK, Baimel C, Gill KJ. Methods for Intravenous Self Administration in a Mouse Model. JoVE. 2012:1–5. doi: 10.3791/3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, Maddocks ODK. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. CellReports. 2014:1–11. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- Lamarre SG, MacMillan L, Morrow GP, Randell E, Pongnopparat T, Brosnan ME, Brosnan JT. An isotope-dilution, GC–MS assay for formate and its application to human and animal metabolism. Amino Acids. 2014;46:1885–1891. doi: 10.1007/s00726-014-1738-7. [DOI] [PubMed] [Google Scholar]
- Lee GY, Haverty PM, Li L, Kljavin NM, Bourgon R, Lee J, Stern H, Modrusan Z, Seshagiri S, Zhang Z, et al. Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes. Cancer Res. 2014;74:3114–3126. doi: 10.1158/0008-5472.CAN-13-2683. [DOI] [PubMed] [Google Scholar]
- Leung JH, Schurig-Briccio LA, Yamaguchi M, Moeller A, Speir JA, Gennis RB, Stout CD. Structural biology. Division of labor in transhydrogenase by alternating proton translocation and hydride transfer. Science. 2015;347:178–181. doi: 10.1126/science.1260451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, Vokes NI, Feist AM, Vander Heiden MG, Metallo CM. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of Mammalian cells. Mol Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic Analysis via Reversed-Phase Ion-Pairing Liquid Chromatography Coupled to a Stand Alone Orbitrap Mass Spectrometer. Anal Chem. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momb J, Lewandowski JP, Bryant JD, Fitch R, Surman DR, Vokes SA, Appling DR. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc Natl Acad Sci USA. 2013;110:549–554. doi: 10.1073/pnas.1211199110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, Mootha VK. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nature Communications. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Pietro ED, MacKenzie RE. Mammalian fibroblasts lacking mitochondrial NAD+-dependent methylenetetrahydrofolate dehydrogenase-cyclohydrolase are glycine auxotrophs. J Biol Chem. 2003;278:19436–19441. doi: 10.1074/jbc.M301718200. [DOI] [PubMed] [Google Scholar]
- Pelletier JN, MacKenzie RE. Binding and interconversion of tetrahydrofolates at a single site in the bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase. Biochemistry. 1995;34:12673–12680. doi: 10.1021/bi00039a025. [DOI] [PubMed] [Google Scholar]
- Pike ST, Rajendra R, Artzt K, Appling DR. Mitochondrial C1-Tetrahydrofolate Synthase (MTHFD1L) Supports the Flow of Mitochondrial One-carbon Units into the Methyl Cycle in Embryos. Journal of Biological Chemistry. 2010;285:4612–4620. doi: 10.1074/jbc.M109.079855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, Deberardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013a;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013b;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- Stover PJ, Chen LH, Suh JR, Stover DM, Keyomarsi K, Shane B. Molecular Cloning, Characterization, and Regulation of the Human Mitochondrial Serine Hydroxymethyltransferase Gene. J Biol Chem. 1997;272:1842–1848. doi: 10.1074/jbc.272.3.1842. [DOI] [PubMed] [Google Scholar]
- Sweeting JN, Siu M, McCallum GP, Miller L, Wells PG. Species differences in methanol and formic acid pharmacokinetics in mice, rabbits and primates. Toxicol Appl Pharmacol. 2010;247:28–35. doi: 10.1016/j.taap.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Tibbetts AS, Appling DR. Compartmentalization of Mammalian Folate-Mediated One-Carbon Metabolism. Annu Rev Nutr. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- Tucker EJ, Hershman SG, Köhrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, et al. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;14:428–434. doi: 10.1016/j.cmet.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LWS, Lu C, Lindsten T, Cross JR, et al. Serine Catabolism Regulates Mitochondrial Redox Control during Hypoxia. Cancer Discovery. 2014;4:1406–1417. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. The global burden of disease: 2004 update. Geneva: WHO Press; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.