

Abstract
Small angle x-ray scattering, SAXS, is a powerful and easily employed experimental technique that provides solution structures of macromolecules. The size and shape parameters derived from SAXS provide global structural information about these molecules in solution and essentially complement data acquired by other biophysical methods. As applied to protein systems, SAXS is a relatively mature technology: sophisticated tools exist to acquire and analyze data, and to create structural models that include dynamically flexible ensembles. Given the expanding appreciation of RNA’s biological roles, there is a need to develop comparable tools to characterize solution structures of RNA, including its interactions with important biological partners. We review the progress towards achieving this goal, focusing on experimental and computational innovations. The use of multiphase modeling, absolute calibration and contrast variation methods, among others, provides new and often unique ways of visualizing this important biological molecule and its essential partners: ions, other RNAs or proteins.
CONTRIBUTIONS OF SAXS TO SOLUTION STUDIES OF RNA
Small angle x-ray scattering, or SAXS, is a powerful tool for measuring solution structure(s) of biological macromolecules1, 2, including RNA3–8. SAXS provides an accurate and diverse set of parameters that describe biomolecules, including global information about macromolecular size and shape, intermolecular association, domain motion and linker flexibility, in addition to more-difficult-to-acquire information about biological partners, ranging from ions9 through proteins2, 10. SAXS data complement those from other biophysical methods including NMR4, 11, 12, single molecule fluorescence resonance energy transfer13, fluorescence correlation spectroscopy14, crystallography15–17, chemical footprinting18–20, SEC and dynamic light scattering21. Readily available computational tools allow modeling of structures from SAXS data22; more recently developed computational methods can be applied to model the complex solution environment around RNA23, 24. This review focuses on how SAXS enhances our understanding of RNA structures and interaction with partners: ions, other RNAs or proteins. We begin with a description of SAXS, including guidelines for acquiring high quality data on samples that contain RNA. This introduction is followed by a brief description of several SAXS-derived parameters and examples of their relevance to understanding RNA and its interaction with partners.
THE ORIGIN OF THE SAXS SIGNAL
Small angle scattering appears when ~ Å wavelength x-rays probe ~nanometer sized solutes, like biomolecules, that are dissolved in a solvent. The random orientations of macromolecules in solution results in an isotropic scattering profile, which is collected on a two-dimensional area detector, as indicated in Figure 1. The effect of averaging many randomly orientated molecules within the volume of x-ray beams is equivalent to averaging all directions of one molecule (Figure 1). Hence, it is difficult to extract directional information from a SAXS measurement without additional information. For example, chirality cannot be determined with SAXS.
Figure 1.

Schematic of a SAXS experiment. The top panel represents a typical SAXS setup at the G1 station of Cornell’s High Energy Synchrotron Source (CHESS). An incident x-ray beam is collimated using 2 (or more) sets of slits, then passes through a quartz capillary that contains a plug of diluted sample solution. The solutes are schematically shown as black dots in the upper panel; they are magnified in the lower left panel. To minimize radiation damage, the sample is oscillated during the exposure. X-rays scattered by the sample are captured by a 2 dimensional area detector. The isotropic scattering pattern arises from the random orientation of the RNA in solution. A beamstop blocks the direct beam to avoid detector damage. The scattering intensity is integrated at each angle to produce a 1 dimensional Intensity vs. q curve. As an example, the scattering profile of tRNA, the structure shown in the lower left panel, is plotted in the lower right panel. As described in the text, its radius of gyration can be readily computed from a linear fit to the low q (or Guinier) region of this curve plotted as ln(I(q)) vs. q2 in the inset.
The scattering angle 2θ is defined relative to the direction of the incident beam; the scattering intensity typically decreases with increasing angle. To analyze the data, an angular average of the intensity is computed at each θ, and is plotted as a function of the momentum transfer, q=4πsinθ/λ, where λ is the x-ray wavelength. In the small angle region of interest, q~sinθ ~ θ. The signal intensity is proportional to the sample concentration, the square of the electron density difference, or contrast, between the biomolecules and their solvent, and other factors25. Both proteins and nucleic acids are more electron dense than the buffer that surrounds them; however nucleic acids are denser than proteins, due to the phosphorous atoms along the backbone. These differences can be exploited to separate the contributions to the overall scattering arising from these distinct biological macromolecules26, 27, enabling new approaches for examining macromolecular complexes in solution7. Finally, the SAXS intensity extrapolated to q=0, I(0) sensitively reports interparticle interactions. Relative to the signal from non-interacting monomers, I(0) decreases when repulsive interactions are present. Increases in I(0) signal association or attraction between monomers; as discussed below (see Calculating molecular weight using RNA standards and absolute calibration), I(0) roughly indicates the molecular weight of the scatterer.
THE EFFECT OF HIGH RNA CHARGE ON SAXS SIGNALS
The large, negative charge of the RNA backbone can lead to strong repulsive forces between different molecules. These intermolecular interactions are easily detected by SAXS. For example, low ionic strength buffers, or buffers that lack Mg2+ ions, are typical starting conditions for RNA folding experiments28, 29. If the inter-particle separation is smaller than the electrostatic screening distance of the backbone charge, electrostatic repulsion exists between particles (and also within one particle30). This effect is easiest to quantify in model systems consisting of short RNA duplexes9. When repulsive interactions dominate, the loose self-organization of the duplexes modifies the scattering profile; the net result is a ‘downturn’ of the measured scattering intensity at the lowest angles9. When the [RNA] is high, the molecules form a loosely ordered array to minimize these repulsive interactions. The inter-particle separation in this array is reflected by a peak in the scattering profile, easily visible in Figure 2A (1 mM Na+ curve). At the other extreme, when attraction dominates, the intensity of SAXS profile increases at the lowest scattering angles25 relative to that of non-interacting monomers. For this system the addition of salt more locally screens the duplexes and they associate end-to-end9. The sign and strength of these interactions depend on the ionic strength and the valence of the ions present in solution. Figure 2A shows the surprisingly strong variations in scattering resulting from changes in electrostatic screening, as MgCl2 is added to a buffered solution containing 25 base pair RNA duplexes.
Figure 2.

Effects of counterions on RNA. (A) SAXS profiles of 25 bp RNA duplexes at different Mg2+ concentrations. These curves have been normalized by [RNA] to enable a comparison of their shapes. The profile measured at low salt without Mg2+ yields a sharp ‘downturn’ at the lowest q, indicating strong repulsive inter-particle interactions. The peak reflects the mean separation between highly charged duplexes in solution. As the [Mg2+] increases, the intensity rises at the lowest q. At first this increase signals decreasing repulsion; at higher [Mg2+] the continued increase reflects end-to-end association of RNA duplexes. (B) Side view of a pRNA pentameric ring superimposed with the cryo-EM envelopes of the pRNA and the pRNA-bound gp16 ATPase 85, in a functional structure. The construction of the pentamer is mediated by base pairing between the Lce and Ld loops of adjacent pRNA mononers, forming 5 superhelix scaffolds. (C) SAXS profiles of pRNA in solutions with varying salts. In Na+, SAXS profiles of dilute solutions of pRNA demonstrate weak repulsive interactions between monomers. The dramatic increase in the signal at low q, when 2mM Mg2+ is added, suggests strong intermolecular interactions between pRNA monomers.
Although the effect of these repulsive and attractive interactions is to modify the overall scattering profiles of the isolated molecules by introducing a structure factor9, the latter term can be used to extract a ‘potential of mean force’ (e.g. the second virial coefficient A2), enhancing our understanding of interparticle interactions. The second virial coefficient is a measure of the intermolecular interaction potential, and is easily derived from SAXS data measured at a series of RNA concentrations9. Knowledge of these potentials can be exploited in problems ranging from RNA complex formation through crystal growth (e.g. Ref.31). It is interesting to note that for RNA, salt affects both the conformations present in solution (which can be readily assessed by SAXS) as well as the conditions that successfully produce crystals.
Salt-dependent association can also be critical for biological function. Consider the case of prohead RNA (pRNA) that is essential for packaging dsDNA in the bacteriophage ϕ29. Five pRNA monomers associate to form a ring that sits at the base of the ϕ29 capsid (Figure 2B); the pRNA molecules assemble into a functional, supramolecular ring32, 33. Even in the absence of prohead proteins, pRNA monomers have a high binding affinity; they dimerize through loop-loop base stacking in the presence of Mg2+34. Figure 2C shows SAXS profiles of a pRNA-containing sample. These curves are concentration normalized to enable a direct shape comparison. In the presence of added monovalent ions (Na+), but no Mg2+ in the buffer, the pRNA exist as monomers. These SAXS profiles reveal the electrostatic repulsion between individual monomers (especially the no added salt, or 0 mM Na+ curve), which manifests as a ‘downturn’ at the lowest angles. The [pRNA] is lower than that of the duplexes shown in the top panel; hence the repulsion is weaker; the strong peak is absent. Increasing [Na+] decreases the screening length, hence the repulsion between monomers. These changes are evident from the rising signal at the lowest angles. When Mg2+ is added, the dramatic increase in intensity at low angles signals the intermolecular association of pRNA monomers, consistent with other biochemical studies34. This example illustrates one of the challenge/features of SAXS studies of RNA: RNAs may be stable and monomeric in solutions of moderate, monovalent, ionic strength, yet may rapidly associate when Mg2+ is added. For pRNA, the association is biologically important.
Clearly, the counterion atmosphere around RNA (e.g.35) strongly affects interactions between RNAs, and the resulting SAXS profiles. Counterions also scatter. Those localized around the RNA contribute to the SAXS signal, primarily at the lowest scattering angles. The magnitude of counterion associated scattering depends on the number of excess counterions, relative to the buffer, as well as on ion identity (atomic number). It comprises about 10% of the RNA signal for low molecular weight counterions such as K+ or Na+36. As we discuss below, (see RNA and counterions) SAXS offers unique opportunities for learning about RNA’s ion partners.
PROTOCOLS FOR RNA SAXS EXPERIMENTS
Although SAXS data can be used to quantify interparticle interactions (see above), or to characterize ensembles of structures present at any time (see below), small angle scattering from homogenous and monodisperse samples is the easiest to interpret. Here, we discuss protocols for acquiring SAXS data, including some methods for determining the homogeneity of the sample. Additional, detailed information about protocols for preparing RNA samples for SAXS and for acquiring high quality SAXS data can be found in two recent reviews6, 8.
We begin with the simplest case of a homogeneous and monodisperse population. Many RNAs are synthesized in vitro through PCR amplification followed by denaturing gel purification. A good annealing protocol is critical for obtaining correctly folded RNA structures. For designed sequences, web based programs like Mfold37 can be used to predict secondary contacts. Melting curve analysis based on absorbance hyperchromicity or fluorescence is also useful for measuring RNA thermodynamics38. However, at the relatively high RNA concentrations used for SAXS experiments, some unplanned association may occur. Because the low q part of the signal is roughly proportional to molecular weight, undesired dimers or higher order oligomers can dominate, rendering the measurements difficult to interpret. Separation techniques, like liquid chromatography (e.g. size exclusion (SEC)), can be implemented to isolate different species before or during a SAXS experiment39, 40.
Scattering from the background, which includes the buffer as well as parasitic scattering from collimating slits, curved capillary sample holders and windows along the beamline, can contribute a large fraction of the measured signal. The small angle scattering from RNA itself can be orders of magnitude weaker than crystal diffraction or powder scattering. In some cases it is equal in magnitude to the background contribution. Thus, it is critical to measure scattering from a matching buffer: an RNA-absent sample that is, in all other respects, identical to the RNA-present sample, in the same sample holder. We commonly use either the SEC buffer or the dialysis buffer for this match, to ensure that it is as similar as possible to the buffer in the RNA-containing solution. We measure the scattering of the buffer before and after each sample measurement41. Comparison of these pre and post buffer scattering profiles also allows ready evaluation of the sample chamber cleaning protocol and beam stability.
Measurements of purified RNAs at various concentrations are essential to either characterize or eliminate inter-particle association. High sample concentration favors strong repulsion at low salt, or inter-particle attraction and dimer/oligomer formation at higher salt concentrations. The use of a low concentration sample can minimize these effects, but leads to a reduction in signal relative to the background. It is advisable to conduct a series of SAXS measurements at different sample concentrations42. Programs, like Primus in the ATSAS package43, can extrapolate this concentration series to the case of infinite dilution. If in situ chromatography is available at the SAXS beamline, the “infinitely diluted” sample can be readily measured at the tail of the elution peak. At these low concentrations, inter-particle interactions are minimized.
With the intense growth of interest in SAXS by the structural biology community, it is now routine to acquire SAXS data at synchrotron facilities; most have dedicated beamline staff to assist with both data acquisition and preliminary processing. Many biological SAXS beamlines provide in situ liquid chromatography purification to improve sample monodispersity and hence data quality44–46. Beamlines, such as the SSRL BL4-2, SIBYLS, Cornell MacCHESS and the bioSAXS beamline P12 of EMBL at DESY, are equipped with automated pipelines for high throughput SAXS data collection44, 45, 47–50. Some SAXS beamlines employ a mail-in service48.
For SAXS data reduction and processing, most beamlines have their own dedicated software (e.g. ATSAS at DESY, Blu-ICE at SSRL BL4-2 and RAW at Cornell MacCHESS)51–53. These packages are usually portable and can be downloaded and installed on off-site computers. In addition, a number of useful online resources allow further analysis and interpretation of data (e.g. ATSAS and BioIsis)53, 54. ATSAS provides a comprehensive collection of tools for SAXS data manipulation and interpretation53. While the ATSAS package is constantly updated for improved performance, and downloadable from the EMBL website, some major programs, such as CRYSOL55, DAMMIN56, MONSA56 and EOM57, are also accessible through online services. BioIsis is an open source for SAXS data deposition and distribution54. It also contains useful tutorials for SAXS data analysis.
SAXS REPORTS THE SIZE AND SHAPE OF RNA IN SOLUTION
Measurements of global structural parameters, such as molecular size and shape, can help guide our understanding of RNA and its interaction with partners. SAXS data readily report the radius of gyration (Rg) of macromolecules, which is related to their overall size. The Rg is easily extracted from SAXS profiles of monodisperse samples, using the Guinier approximation1: at the lowest scattering angles or q, the intensity I(q) follows approximately a Gaussian shape, centered at q=0, which can be described as
| (1) |
A plot of ln(I(q)) as a function of q2, yields a straight line in the low q or Guinier regime (Figure 1). In this approximation, the slope of the curve is proportional to Rg2. This method works best for globular structures, and for small q values such that qRg < 1.31. Accordingly, the q range (proportional to the number of data points can be used for fitting) is limited for larger molecules. A curved line in the Guinier region is a strong indicator of association or aggregation. The Rg values of non-aggregated, moderately sized molecules can be determined with statistical accuracy down to sub-angstrom length scales. Most beamline data processing software can quickly create Guinier plots, calculating Rg values on the fly49–51 to provide frequent checks of molecular size and molecular weight during SAXS data acquisition. This useful information can guide experiments in real time.
A particularly nice example of the use of radius of gyration in studies of RNA structure can be found in Ref.40. Riboswitches are regulatory RNAs that change conformation following the addition of a particular ligand58. Figure 3 shows the Rg’s of several riboswitches as a function of their length40. These data show an almost monotonic relationship between the size and length of these riboswitches in both bound and unbound state. They also illustrate that most riboswitches form more compact conformations when bound to ligands: Rg can change upon ligand binding, in some cases by more than 4 Å. Figure 3 illustrates the use of SAXS to detect a 0.5 Å change in Rg for the FMN riboswitch.
Figure 3.

SAXS analysis of Riboswitches. The radius of gyration (Rg) of free (red) and ligand-bound riboswitches (blue) is plotted as a function of RNA length. In all samples, Mg2+ is present. A linear least square fit to these two data sets shows a nearly monotonic relationship between the size and length of the riboswitches. The two separated straight lines also show that the ligand-bound riboswitches are in general more compact than the free riboswitches. The Rgs of T box stem I was measured in our lab and reported in Ref. (60). The Rgs of other riboswitches are from Table 1 in Ref. (40).
The pair-wise distance distribution function (P(R)), is another useful tool for evaluating molecular global structural features in real space. This function is derived from I(q) through Eqn. 2. The radius of gyration can also be computed from P(R) curves using Eqn. 3.
| (2) |
| (3) |
The GNOM program in the ATSAS package calculates P(R)’s using an indirect Fourier transform59. As input, this method requires knowledge of the maximum dimension of the molecule (Dmax), which can be estimated from a known structure, and/or based on the goodness of fit of the GNOM regularized intensity to the raw SAXS data. In addition to Rg, P(R) provides insight into molecular shape1. For example, a peak in a P(R) curve represents a prominent length scale within the molecule.
An example of the insights that can be gained from P(R) analysis of RNA structures can be found in a study of T box RNA60. This RNA is a non-coding regulatory RNA in bacteria that binds tRNA, sensing its aminoacylation state60, 61. Figure 4A shows the distinctly different shapes of P(R)’s for a T box RNA, a tRNA and their complex. The “rod-like” T box RNA (Figure 4B) has a skewed P(R) distribution while the complex P(R) has a broader peak indicating a disk-like conformation. Additional discussion of T box RNA – tRNA interactions revealed by SAXS will be provided below.
Figure 4.

SAXS analysis of T box RNA – tRNA complex. (A) Pair distance distribution functions P(R) of tRNA, T box–stem I86, and stem I86–tRNA complex, computed as described in the text. (B) Docking of the T box stem I86 model into the single phase SAXS reconstructed envelope from DAMMIF (average NSD = 0.77 ± 0.12). (C) Docking of the stem I86–tRNA complex model into the SAXS reconstructed envelope from DAMMIF, the single phase approximation treats the two components as part of a single particle (average NSD = 0.66 ± 0.09). (D and E) Experimental SAXS profiles of tRNA, T box–stem I86, and stem I86–tRNA complex in linear (D) and Kratky representations (E). The dashed curve (black) is the sum of tRNA (red) and T box RNA (blue). (F) Docking of the stem I86–tRNA complex model into the averaged two-phase MONSAreconstructed envelope. This model shows the relative placement of the two RNA components in the reconstruction
RECONSTRUCTING RNA STRUCTURE FROM SAXS PROFILES
A popular method for ‘viewing’ SAXS data in real space involves determining low resolution 3-D molecular shapes of RNAs. The most popular programs for ab initio SAXS reconstruction are DAMMIN56 and DAMMIF62, developed by the Svergun lab. These programs generate bead models to represent the shape of biomolecules in solution. The beads inside a search volume are randomly selected using an algorithm to form an interconnected model, until the calculated scattering profile from the model matches the measured profile. DAMMIF improves on DAMMIN by utilizing a new algorithm to speed up the simulated annealing process, and by introducing an expandable search volume to avoid boundary artifacts. These bead models are useful when no prior global structural information is available; they can also validate hypothetical atomic structures20, 60. Monodisperse samples are required for accurate envelope generation by these programs. As examples of SAXS reconstructions applied to RNA, Figures 4B and 4C show low-resolution envelopes reconstructed from I vs. q for T box RNA and its complex with a tRNA, respectively, using DAMMIF.
CALCULATING MOLECULAR WEIGHT USING RNA STANDARDS AND ABSOLUTE CALIBRATION
Although the intensity at zero scattering angle, I(0) cannot be directly measured because the beamstop blocks the direct incident beam (Figure 1), it can be extrapolated from low q data using equation 1. I(0) is very useful for indicating association of macromolecules: if all of the RNAs in a sample convert from monomers to dimers, the number of excess electrons per molecule doubles, but the concentration is halved. The net effect is a factor of two increase in I(0). Thus, I(0) roughly indicates the molecular weight of an RNA sample. Using an RNA standard, the molecular weight of an unknown RNA (including its tightly associated counterions) can be readily calculated using the following relationship63.
| (4) |
where MW stands for molecular weight, and MaC represents mass concentration. The MW here must be viewed as approximate as a result of many factors, including uncertainty in sample concentration and differential ion contributions. However, the level of accuracy is sufficient to determine the oligomerization state of a sample in some cases, for example when pure monomers or pure dimers are known to exist63. As an example, Table 1 presents data showing that tRNA with known molecular weight and concentration can be used as a standard to estimate the molecular weight of T box RNA. At high concentration (2.6 mg/ml), the molecular weight of T box RNA is almost twice that measured at low concentration (0.45 mg/ml), indicating intermolecular association. When compared to the theoretical value of its molecular weight (28.2 kDa), this result suggests that, at 2.6 mg/ml, most T box RNAs dimerize. For RNA with unknown concentration, Rambo et. al. recently described a method of extracting mass concentration from direct SAXS measurements64. In addition to I(0) and Rg, this method requires calculating another SAXS invariant, the volume of correlation Vc. Details of using this method can be found in the BioIsis tutorials.
Table 1.
Calculating Molecular Weight of T box RNA using tRNA as standard
| RNA sample | Concentration (mg/ml) |
I(0) (a.u) |
Theoretical MW (kDa) |
Calculated MW (kDa) |
|---|---|---|---|---|
| tRNA | 2.3 | 38.92 | 24.5 | N/A |
| T box High | 2.6 | 96.74 | 28.2 | 53.8 |
| T box Low | 0.45 | 9.12 | 28.2 | 29.3 |
It is also possible to calibrate the x-ray scattering intensity in absolute units of electrons2 using pure water as a standard. A detailed description of the procedure for absolute calibration can be found in65–67. Significantly, water calibrated x-ray scattering data enables quantitative study of the ion cloud around RNAs (see below).
SAXS INFORMS ABOUT RNA BINDING PARTNERS
RNA-RNA complexes
When combined with multiphase modeling techniques, SAXS provides useful information about the overall structure of RNA-RNA or RNA-protein complexes, including the geometrical configuration or arrangement of different binding components. This topic was recently reviewed in Ref.7. To illustrate, we return to the system shown in Figure 4. T box RNA and tRNA form a stable complex through base stacking interactions at two distant positions on the stem I region of the former RNA60. The SAXS profiles of the T box, tRNA and the complex are plotted in both linear (I vs q) and Kratky (Iq2 vs q) representations in Figures 4D and 4E. Kratky plots emphasize the scattering at larger q, which reflects smaller spatial length scales. The sum of the T box RNA and tRNA scattering profiles is also plotted as a dashed line in both figures. As expected this sum looks very different from the scattering profile of the complex. This difference arises because SAXS measures intensity, not amplitude. The total scattering amplitude includes contributions from both RNAs, Fcomplex = FT box + FtRNA; the intensity is the square of the amplitude, I = FcomplexF*complex, where the second term is the complex conjugate of the first. Significant, additional contributions to the overall scattering arise from the cross terms (Figure 4D).
Insight into the structure of the T box – tRNA complex was gained through ab initio shape reconstruction using DAMMIF to generate the SAXS envelope. This single-phase (all RNA) model is in good agreement with the hypothetical complex structure; however, it lacks critical information that can distinguish the two RNA components (Figure 4C). To resolve this docking ambiguity, the multiphase modeling technique MONSA56, can instead be used to generate 2-phase models for the T box-tRNA complex. The resulting reconstruction is shown in Figure 4F. Here, the contribution from the different RNA components is distinguished within the envelope of the complex. To accomplish this rigid body multiphase modeling, under the assumption that the components do not change conformation upon binding, MONSA requires the scattering profiles of three species: the two RNA components and the complex.
RNA-protein complexes
A similar modeling approach can also be used to characterize RNA-protein interactions using SAXS. In a recent study of DEAD-box RNA chaperones by Mallam et al.68, the SAXS structures of the full length DEAD-box helicase protein CYT-19, a deletion mutant with the C-terminal tail removed, and the protein-RNA complex were obtained to assess the functionalities of two core protein domains and the C-terminal tail.
In cases when the high resolution structures of individual subunits in a complex are known, rigid body modeling techniques, like BUNCH69, can dock these subunits into the complex envelope. These manipulations are guided by a comparison of theoretical and experimental SAXS profiles and provide insight into the spatial configuration of different subunits68, 70. Figures 5B and 5C present the DAMMIN reconstructed envelopes docked against the BUNCH atomic models, showing that the two protein domains adopt an open conformation without RNA substrates.
Figure 5.
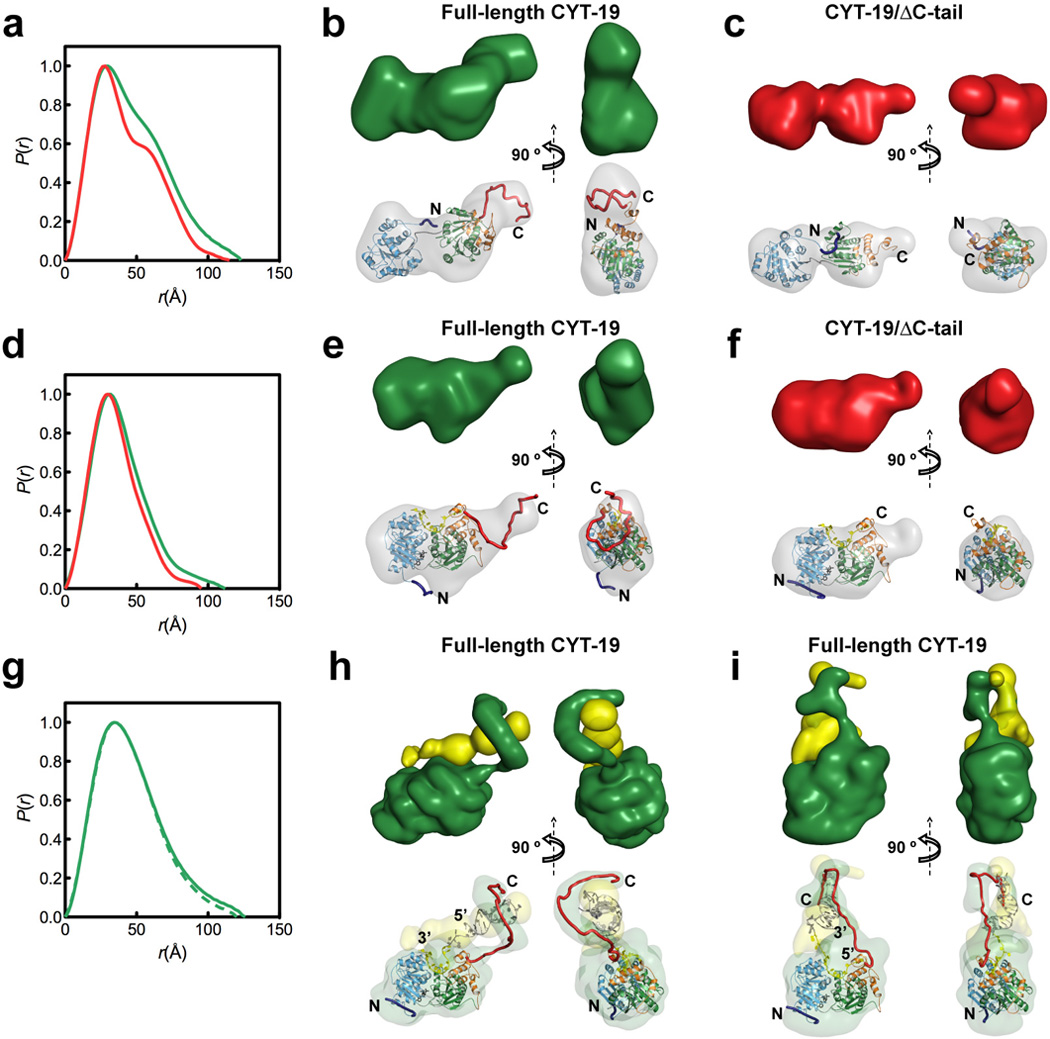
SAXS analysis of DEAD-box helicase protein CYT-19. (A–C) SAXS data for full-length CYT-19 (green) and CYT-19/ΔC-tail (red) in the absence of ligands. (A) Normalized distance distribution functions. (B) and (C) low-resolution envelopes calculated by DAMMIN (Upper) and BUNCH atomic models (Lower), which are aligned inside the DAMMIN envelope (gray). BUNCH models were generated using a homology model of CYT-19 that is based upon its sequence similarity to Mss116p (see SI Methods in Ref. 68). (D–F) SAXS data for CYT-19 bound to U10–RNA and ADP-BeFx, shown in the same arrangement as in (A–C). For the minimal CYT-19/ΔC-tail complex, the DAMMIN envelope in (F) is aligned to the homology model for CYT-19. (G–I) SAXS data for full-length CYT-19 bound to large nucleic acid substrates.* (G) Normalized distribution functions for CYT-19-ADP-BeFx bound to RNA-DNA-duplex 1 (solid green line) and RNA-DNAduplex RNA–DNA duplex 1 (substrate 1) and RNA–DNA duplex 2 (substrate 2), respectively. Two-phase models of protein (green) and nucleic acid (yellow) were constructed by MONSA (Upper) and atomic models for protein and nucleic acid were manually placed inside the corresponding SAXS envelopes (Lower). This figure and caption originally appeared in the article Mallam et al., Proceedings of the National Academy of Sciences USA, 108(30):12254–12259, (2011).
Reconstruction of an RNA-protein complex from SAXS data is more challenging than the RNA-RNA complexes described above, because of the different electron densities of nucleic acids and proteins. This example provides an excellent illustration of the appropriate use of modeling tools that suit different circumstances. For CYT-19 complexed with a single strand U10-RNA, single phase DAMMIN modeling is sufficient to reconstruct the complex envelope (Figures 5E and 5F), because the scattering from the small RNA is negligible. However, when CYT-19 is bound to the large nucleic acid substrate, both components contribute significantly to the scattering profile. MONSA multiphase modeling was applied to characterize the interactions between DEAD-box protein and the large nucleic acid substrate (Figures 5H and 5I). Comparison with the docked atomic structures reveals that the DEAD-box protein transits into a compact closed state upon binding to RNA and ADP-BeFx, and the basic C-terminal tail is positioned to tether the RNA and the helicase core.
Finally, small angle x-ray and neutron scattering studies of the ribosome show the great potential of contrast variation methods to study RNA-protein complexes71. Although contrast variation methods for neutron scattering are perhaps more effective to implement as they involve replacing H2O with D2O and enable ‘blanking’ of either the protein or nucleic acid components of the sample1, contrast variation SAXS presents unique opportunities for studying the dynamics of RNA-protein complexes. In SAXS experiments, high concentrations of electron dense sucrose are added to the buffer to increase its electron density to equal that of the protein. The scattering of the RNA, with higher density, remains significant, even above this electron dense background. The contrast variation method has recently been applied to study DNA-protein complexes, where the use of high intensity x-ray sources enables time resolved studies27.
RNA and counterions
Highly negatively charged RNAs attract cations, which form a diffuse counterion cloud around the macromolecule72. These condensed counterions greatly affect RNA’s conformation, stability, binding affinity and folding73. Anomalous small angle x-ray scattering (ASAXS) effectively quantifies the monovalent (Rb+) and divalent (Sr2+) ion atmospheres around RNA and DNA. It has been applied to study the ion atmosphere around short 25bp RNA duplexes74, revealing both the number and spatial distribution of these ions around their RNA partners. This information is critical in developing an atomically detailed picture of how RNA folds and interacts with binding partners. The ASAXS technique requires an energy-tunable beamline; SAXS profiles are acquired at several x-ray energies close to but below the absorption edge of the ions. Near the absorption edge of Rb+ and Sr2+, the atomic scattering factor fion(E) is given by
| (5) |
where fo is the solvent corrected, energy independent scattering factor for the ion and the energy dependent scattering factors f’ and f” account for changes in ion scattering near the respective absorption edges. The former term is negative and real, reducing the ion’s contribution to the overall scattering near the edge; the latter term is imaginary and represents sample absorption. Changes in f’ for both Rb+ and Sr2+ near their absorption edges are shown in Figures 6A and 6B. ASAXS experiments are carried out at energies below the absorption edge where changes in sample absorption (or f”) are negligible. As shown in Ref.66, the energy dependent scattering intensity, I(q,E), can be approximately described as
| (6) |
Figure 6.

Probing the ion atmosphere around RNA using ASAXS. (A and B) Real part of the anomalous scattering factor f’ for Rb+ ions (A) and Sr2+ ions (B), respectively. The f’ values at the energies used in the multiple energy ASAXS experiment are circled in red. Measurements at five energies are used to extract the number of ions from Eqn. 7. As described in the text, measurements at only two energies are used to generate the anomalous difference signals that report the spatial distribution of ions around the macromolecule. (C and D) ASAXS difference signals, Ianom (q) ~ b(q), for 0.1 M Rb+ ions (C) and 0.01 M Sr2+ ions (D), respectively. The ASAXS signal is calibrated on an absolute scale using water as a standard.
Difference signals, Ianom, computed from scattering profiles acquired at two (or more) energies can be used to extract the term b(q) which contains information about the spatial distribution of ions around the RNA (Figure 6C and 6D). These measurements provide additional ion-specific information for comparison with simulations of ion atmospheres. Details of ASAXS measurements and interpretation can be found in Ref36, 66, 67.
Absolute calibration of SAXS profiles enables ion counting via ASAXS67. Measurements at multiple energies allow extraction of the energy independent terms c(q) and b(q). Extrapolation to q=0 provides an experimental measurement of b(0) and c(0). As shown in Ref.67, the number of excess ions around the RNA is given by:
| (7) |
In summary, SAXS provides unique structural information when RNAs interact with ion clouds, nucleic acids and proteins. State-of-the-art multiphase modeling techniques enable the characterization of different components within a RNA complex. ASAXS experiments are designed to characterize the counterion atmosphere around RNA.
COMPUTATIONAL METHODS FOR SAXS MODELING OF STRUCTURAL ENSEMBLES AND HYDRATION SHELL
The information derived from many of the techniques described above is simplest to interpret when the structural ensemble is uniform and static; however many of the most biologically interesting problems involve dynamically changing RNA conformations. New methods for interpreting SAXS data are being applied to characterize ensembles containing multiple structures. Many of these methods have already been very successfully applied to modeling flexible protein structures57. For flexible single stranded RNAs or RNAs with dynamics structures, flexible modeling methods, like Ensemble Optimization Method (EOM)57, can be used to sample the conformational space of the RNA, and to identify the ensemble of structures whose calculated SAXS profiles best recapitulates the experimental SAXS data. A recent use of EOM in modeling the solution structures of full-length HCV IRES RNA revealed that this large RNA is highly flexible, requiring an ensemble of five conformations to fit the experimental data75.
An alternative approach to SAXS-guided structural modeling is to directly compare the experimental and theoretical scattering curves in reciprocal space. CRYSOL, also developed by Svergun et. al., is the most commonly used program for computing theoretical scattering profiles from a given atomic structure55. There have been new developments in improving the accuracy of prediction76–79. In particular, a program called fast-SAXS-RNA (later integrated into fast-SAXS-pro) successfully computes the contribution from the hydration layer and condensed ions around RNA molecules through explicit solvent treatment80, 81. Methods have been developed to improve the structural determination of RNA molecules using experimental SAXS data as a constraint82, 83.
CONCLUSIONS AND OUTLOOK
Clearly, SAXS provides important information, not only about the structures of isolated RNA monomers in solution, but about RNA in complex with biologically significant partners. A unique strength of SAXS is its ability to selectively highlight the scattering of counterions, or of the RNA component of an RNA protein complex, using methods described here. As modeling methods continue to improve, with the development of methods to analyze ensembles of structures, or new tools that provide a more accurate depiction of the water and ions around RNAs, solution studies will greatly expand our understanding of these important systems.
Finally, we mention the great potential of x-ray free electron laser sources that offer new approaches for small angle scattering experiments, based on recovering intensity transforms from solution scattering data. As pointed out in Ref.84, RNA may be the ideal target for experiments at these next generation sources.
Acknowledgments
We thank the following individuals for graciously sharing data or images with us: Ailong Ke (Figure 2), Adrian Ferré-d’Amare (Figure 3), Rick Russell (Figure 5) and Suzette Pabit (Figure 6). We gratefully acknowledge support through NIH GM085062 and the NSF-STC “Biology with X-ray Lasers” (NSF-1231306).
References
- 1.Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Reports on Progress in Physics. 2003;66:1735–1782. [Google Scholar]
- 2.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annual Review of Biophysics and Biomolecular Structure. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- 3.Doniach S, Lipfert J. Use of Small Angle X-Ray Scattering (Saxs) to Characterize Conformational States of Functional RNAs. Methods in Enzymology, Vol 469: Biophysical, Chemical, and Functional Probes of RNA Structure, Interactions and Folding, Pt B. 2009;469:237–251. doi: 10.1016/S0076-6879(09)69011-X. [DOI] [PubMed] [Google Scholar]
- 4.Fang XY, Stagno JR, Bhandari YR, Zuo XB, Wang YX. Small-angle X-ray scattering: a bridge between RNA secondary structures and three-dimensional topological structures. Current Opinion in Structural Biology. 2015;30:147–160. doi: 10.1016/j.sbi.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baird NJ, West J, Sosnick TR. RNA Structure and Folding Analyzed Using Small-Angle X-Ray Scattering. Handbook of RNA Biochemistry, Vols 1 and 2, 2nd Edition. 2014:407–425. [Google Scholar]
- 6.Burke JE, Butcher SE. Nucleic acid structure characterization by small angle X-ray scattering (SAXS) Current protocols in nucleic acid chemistry / edited by Serge L. Beaucage … [et al.] 2012;CHAPTER:Unit7.18–Unit17.18. doi: 10.1002/0471142700.nc0718s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rambo RP. Chapter Twelve - Resolving Individual Components in Protein–RNA Complexes Using Small-Angle X-ray Scattering Experiments. In: Woodson Sarah A, Allain Frédéric HT., editors. Methods in Enzymology. Vol. 558. Academic Press; 2015. pp. 363–390. [DOI] [PubMed] [Google Scholar]
- 8.Reyes FE, Schwartz CR, Tainer JA, Rambo RP. Chapter Eleven - Methods for Using New Conceptual Tools and Parameters to Assess RNA Structure by Small-Angle X-Ray Scattering. In: Burke-Aguero Donald H., editor. Methods in Enzymology. Vol. 549. Academic Press; 2014. pp. 235–263. [DOI] [PubMed] [Google Scholar]
- 9.Pollack L. SAXS Studies of Ion-Nucleic Acid Interactions. Annual Review of Biophysics, Vol 40. 2011;40:225–242. doi: 10.1146/annurev-biophys-042910-155349. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhuri BN. Emerging applications of small angle solution scattering in structural biology. Protein Science. 2015;24:267–276. doi: 10.1002/pro.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salmon L, Yang S, Al-Hashimi HM. Advances in the Determination of Nucleic Acid Conformational Ensembles. Annual Review of Physical Chemistry, Vol 65. 2014;65:293–316. doi: 10.1146/annurev-physchem-040412-110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen B, Zuo XB, Wang YX, Dayie TK. Multiple conformations of SAM-II riboswitch detected with SAXS and NMR spectroscopy. Nucleic Acids Research. 2012;40:3117–3130. doi: 10.1093/nar/gkr1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo TY, Meisburger SP, Hinshaw J, Pollack L, Haran G, Sosnick TR, Plaxco K. Small-Angle X-ray Scattering and Single-Molecule FRET Spectroscopy Produce Highly Divergent Views of the Low-Denaturant Unfolded State. Journal of Molecular Biology. 2012;418:226–236. doi: 10.1016/j.jmb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magbanua E, Konarev PV, Svergun DI, Hahn U. Exploring RNA Oligomerization and Ligand Binding by Fluorescence Correlation Spectroscopy and Small Angle X-Ray Scattering. RNA Folding: Methods and Protocols. 2014;1086:321–334. doi: 10.1007/978-1-62703-667-2_19. [DOI] [PubMed] [Google Scholar]
- 15.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 16.Nowak E, Potrzebowski W, Konarev PV, Rausch JW, Bona MK, Svergun DI, Bujnicki JM, Le Grice SFJ, Nowotny M. Structural analysis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Research. 2013;41:3874–3887. doi: 10.1093/nar/gkt053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason AC, Rambo RP, Greer B, Pritchett M, Tainer JA, Cortez D, Eichman BF. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7618–7623. doi: 10.1073/pnas.1324143111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenowitz M, Pollack L. Following RNA Folding From Local and Global Perspectives. In: Russell R, editor. Biophysics of RNA folding. 2012. [Google Scholar]
- 19.Kazantsev AV, Rambo RP, Karimpour S, Santalucia J, Tainer JA, Pace NR. Solution structure of RNase P RNA. RNA-α Publication of the RNA Society. 2011;17:1159–1171. doi: 10.1261/rna.2563511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Launer-Felty K, Wong CJ, Cole JL. Structural Analysis of Adenovirus VAI RNA Defines the Mechanism of Inhibition of PKR. Biophysical Journal. 2015;108:748–757. doi: 10.1016/j.bpj.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuukkanen AT, Svergun DI. Weak protein-ligand interactions studied by small-angle X-ray scattering. FEBS Journal. 2014;281:1974–1987. doi: 10.1111/febs.12772. [DOI] [PubMed] [Google Scholar]
- 22.Graewert MA, Svergun DI. Impact and progress in small and wide angle X-ray scattering (SAXS and WAXS) Current Opinion in Structural Biology. 2013;23:748–754. doi: 10.1016/j.sbi.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Yang SC. Methods for SAXS-Based Structure Determination of Biomolecular Complexes. Advanced Materials. 2014;26:7902–7910. doi: 10.1002/adma.201304475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen HT, Pabit SA, Meisburger SP, Pollack L, Case DA. Accurate small and wide angle x-ray scattering profiles from atomic models of proteins and nucleic acids. Journal of Chemical Physics. 2014:141. doi: 10.1063/1.4896220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eliezer D, Chiba K, Tsuruta H, Doniach S, Hodgson KO, Kihara H. Evidence of an Associative Intermediate on the Myoglobin Refolding Pathway. Biophysical Journal. 1993;65:912–917. doi: 10.1016/S0006-3495(93)81124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacques DA, Trewhella J. Small-angle scattering for structural biology--expanding the frontier while avoiding the pitfalls. Protein Science. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Tokuda JM, Topping T, Sutton JL, Meisburger SP, Pabit SA, Gloss LM, Pollack L. Revealing transient structures of nucleosomes as DNA unwinds. Nucleic Acids Research. 2014;42:8767–8776. doi: 10.1093/nar/gku562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollack L. Time Resolved SAXS and RNA Folding. Biopolymers. 2011;95:543–549. doi: 10.1002/bip.21604. [DOI] [PubMed] [Google Scholar]
- 29.Pollack L, Doniach S. Time-Resolved X-Ray Scattering and RNA Folding. Methods in Enzymology, Vol 469: Biophysical, Chemical, and Functional Probes of RNA Structure, Interactions and Folding, Pt B. 2009;469:253–268. doi: 10.1016/S0076-6879(09)69012-1. [DOI] [PubMed] [Google Scholar]
- 30.Bai Y, Das R, Millett IS, Herschlag D, Doniach S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1035–1040. doi: 10.1073/pnas.0404448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnete F, Vivares D. Interest of the normalized second virial coefficient and interaction potentials for crystallizing large macromolecules. Acta Crystallographica Section D. 2002;58:1571–1575. doi: 10.1107/s090744490201418x. [DOI] [PubMed] [Google Scholar]
- 32.Ding F, Lu CR, Zhao W, Rajashankar KR, Anderson DL, Jardine PJ, Grimes S, Ke AL. Structure and assembly of the essential RNA ring component of a viral DNA packaging motor. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7357–7362. doi: 10.1073/pnas.1016690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408:745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trottier M, Mat-Arip Y, Zhang C, Chen C, Sheng S, Shao Z, Guo P. Probing the structure of monomers and dimers of the bacterial virus phi29 hexamer RNA complex by chemical modification. RNA. 2000;6:1257–1266. doi: 10.1017/s1355838200992501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manning GS. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. The Journal of Chemical Physics. 1969;51:924–933. [Google Scholar]
- 36.Meisburger SP, Pabit SA, Pollack L. Determining the Locations of Ions and Water around DNA from X-Ray Scattering Measurements. Biophysical Journal. 2015;108:2886–2895. doi: 10.1016/j.bpj.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puglisi JD, Tinoco I., Jr Absorbance melting curves of RNA. Methods in Enzymology. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 39.Rambo RP, Tainer JA. Improving small-angle X-ray scattering data for structural analyses of the RNA world. RNA. 2010;16:638–646. doi: 10.1261/rna.1946310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Jones CP, Ferre-D'Amare AR. Global analysis of riboswitches by small-angle X-ray scattering and calorimetry. Biochimica et Biophysica Acta. 2014;1839:1020–1029. doi: 10.1016/j.bbagrm.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skou S, Gillilan RE, Ando N. Synchrotron-based small-angle X-ray scattering of proteins in solution. Nature Protocols. 2014;9:1727–1739. doi: 10.1038/nprot.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacques DA, Guss JM, Svergun DI, Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallographica D Biological Crystallography. 2012;68:620–626. doi: 10.1107/S0907444912012073. [DOI] [PubMed] [Google Scholar]
- 43.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. [Google Scholar]
- 44.David G, Perez J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. Journal of Applied Crystallography. 2009;42:892–900. [Google Scholar]
- 45.Martel A, Liu P, Weiss TM, Niebuhr M, Tsuruta H. An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. Journal of Synchrotron Radiation. 2012;19:431–434. doi: 10.1107/S0909049512008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathew E, Mirza A, Menhart N. Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. Journal of Synchrotron Radiation. 2004;11:314–318. doi: 10.1107/S0909049504014086. [DOI] [PubMed] [Google Scholar]
- 47.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, Jenney FE, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nature Methods. 2009;6:606–U683. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods in Molecular Biology. 2014;1091:245–258. doi: 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franke D, Kikhney AG, Svergun DI. Automated acquisition and analysis of small angle X-ray scattering data. Nuclear Instruments & Methods in Physics Research Section A-Accelerators Spectrometers Detectors and Associated Equipment. 2012;689:52–59. [Google Scholar]
- 50.Nielsen SS, Moller M, Gillilan RE. High-throughput biological small-angle X-ray scattering with a robotically loaded capillary cell. Journal of Applied Crystallography. 2012;45:213–223. doi: 10.1107/S0021889812000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Classen S, Rodic I, Holton J, Hura GL, Hammel M, Tainer JA. Software for the high-throughput collection of SAXS data using an enhanced Blu-Ice/DCS control system. Journal of Synchrotron Radiation. 2010;17:774–781. doi: 10.1107/S0909049510028566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nielsen SS, Møller M, Gillilan RE. High-throughput biological small-angle X-ray scattering with a robotically loaded capillary cell. Journal of Applied Crystallography. 2012;45:213–223. doi: 10.1107/S0021889812000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. New developments in the ATSAS program package for small-angle scattering data analysis. Journal of Applied Crystallography. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, Jenney FE, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nature Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Svergun D, Barberato C, Koch MHJ. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography. 1995;28:768–773. [Google Scholar]
- 56.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing (vol 76, pg 2879, 1999) Biophysical Journal. 1999;77:2896–2896. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. Journal of the American Chemical Society. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 58.Garst AD, Edwards AL, Batey RT. Riboswitches: Structures and mechanisms. Cold Spring Harbor perspectives in biology. 2011;3:a003533. doi: 10.1101/cshperspect.a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Svergun DI. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- 60.Grigg JC, Chen YJ, Grundy FJ, Henkin TM, Pollack L, Ke AL. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7240–7245. doi: 10.1073/pnas.1222214110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Green NJ, Grundy FJ, Henkin TM. The T box mechanism: tRNA as a regulatory molecule. FEBS Letters. 2010;584:318–324. doi: 10.1016/j.febslet.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mylonas E, Svergun DI. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. Journal of Applied Crystallography. 2007;40:s245–s249. [Google Scholar]
- 64.Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orthaber D, Bergmann A, Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. Journal of Applied Crystallography. 2000;33:218–225. [Google Scholar]
- 66.Pabit SA, Finkelstein KD, Pollack L. Using Anomalous Small Angle X-Ray Scattering to Probe the Ion Atmosphere around Nucleic Acids. Methods in Enzymology, Vol 469: Biophysical, Chemical, and Functional Probes of RNA Structure, Interactions and Folding, Pt B. 2009;469:391–410. doi: 10.1016/S0076-6879(09)69019-4. [DOI] [PubMed] [Google Scholar]
- 67.Pabit SA, Meisburger SP, Li L, Blose JM, Jones CD, Pollack L. Counting Ions around DNA with Anomalous Small-Angle X-ray Scattering. Journal of the American Chemical Society. 2010;132:16334–16336. doi: 10.1021/ja107259y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mallam AL, Jarmoskaite I, Tijerina P, Del Campo M, Seifert S, Guo L, Russell R, Lambowitz AM. Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12254–12259. doi: 10.1073/pnas.1109566108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophysical Journal. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lamech LT, Mallam AL, Lambowitz AM. Evolution of RNA-Protein Interactions: Non-Specific Binding Led to RNA Splicing Activity of Fungal Mitochondrial Tyrosyl-tRNA Synthetases. PLOS Biology. 2014:12. doi: 10.1371/journal.pbio.1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Svergun DI, Pedersen JS, Serdyuk IN, Koch MH. Solution scattering from 50S ribosomal subunit resolves inconsistency between electron microscopic models. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:11826–11830. doi: 10.1073/pnas.91.25.11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Woodson SA. Metal ions and RNA folding: a highly charged topic with a dynamic future. Current Opinion in Chemical Biology. 2005;9:104–109. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 74.Kirmizialtin S, Pabit SA, Meisburger SP, Pollack L, Elber R. RNA and Its Ionic Cloud: Solution Scattering Experiments and Atomically Detailed Simulations. Biophysical Journal. 2012;102:819–828. doi: 10.1016/j.bpj.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perard J, Leyrat C, Baudin F, Drouet E, Jamin M. Structure of the full-length HCV IRES in solution. Nature Communications. 2013:4. doi: 10.1038/ncomms2611. [DOI] [PubMed] [Google Scholar]
- 76.Watson MC, Curtis JE. Rapid and accurate calculation of small-angle scattering profiles using the golden ratio. Journal of Applied Crystallography. 2013;46:1171–1177. [Google Scholar]
- 77.Knight CJ, Hub JS. WAXSiS: a web server for the calculation of SAXS/WAXS curves based on explicit-solvent molecular dynamics. Nucleic Acids Research. 2015;43:W225–W230. doi: 10.1093/nar/gkv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Research. 2010;38:W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grishaev A, Guo L, Irving T, Bax A. Improved Fitting of Solution X-ray Scattering Data to Macromolecular Structures and Structural Ensembles by Explicit Water Modeling. Journal of the American Chemical Society. 2010;132:15484–15486. doi: 10.1021/ja106173n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ravikumar KM, Huang W, Yang SC. Fast-SAXS-pro: A unified approach to computing SAXS profiles of DNA, RNA, protein, and their complexes. Journal of Chemical Physics. 2013:138. doi: 10.1063/1.4774148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang SC, Parisien M, Major F, Roux B. RNA Structure Determination Using SAXS Data. Journal of Physical Chemistry B. 2010;114:10039–10048. doi: 10.1021/jp1057308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parisien M, Major F. Determining RNA three-dimensional structures using low-resolution data. Journal of Structural Biology. 2012;179:252–260. doi: 10.1016/j.jsb.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gajda MJ, Zapien DM, Uchikawa E, Dock-Bregeon AC. Modeling the Structure of RNA Molecules with Small-Angle X-Ray Scattering Data. PLOS One. 2013:8. doi: 10.1371/journal.pone.0078007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gruner SM, Lattman EE. Biostructural Science Inspired by Next-Generation X-Ray Sources. Annual Review of Biophysics. 2015;44:33–51. doi: 10.1146/annurev-biophys-060414-033813. [DOI] [PubMed] [Google Scholar]
- 85.Morais MC, Koti JS, Bowman VD, Reyes-Aldrete E, Anderson DL, Rossmann MG. Defining Molecular and Domain Boundaries in the Bacteriophage ∏29 DNA Packaging Motor. Structure. 16:1267–1274. doi: 10.1016/j.str.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]