

Abstract
Chronic heart failure (HF) is a leading clinical and public problem posing a higher risk of morbidity and mortality in different populations. HF appears to be in both phenotypic forms: HF with reduced left ventricular ejection fraction (HFrEF) and HF with preserved left ventricular ejection fraction (HFpEF). Although both HF phenotypes can be distinguished through clinical features, co-morbidity status, prediction score, and treatment, the clinical outcomes in patients with HFrEF and HFpEF are similar. In this context, investigation of various molecular and cellular mechanisms leading to the development and progression of both HF phenotypes is very important. There is emerging evidence that epigenetic regulation may have a clue in the pathogenesis of HF. This review represents current available evidence regarding the implication of epigenetic modifications in the development of different HF phenotypes and perspectives of epigenetic-based therapies of HF.
Keywords: Heart failure with reduced ejection fraction, Heart failure with preserved ejection fraction, Epigenetic modifications, Chromatin remodeling
Highlights
-
•
The causes of heart failure development with presentation of preserved or reduced ejection fraction are not clear
-
•
The epigenetic modifications are discussed as a phenotypic regulator in the failing heart.
-
•
The phenotypic response in heart failure may regulate by microRNAs via interaction with histone modification or DNA methylation.
-
•
The epigenetic modifications of cardiomyocytes may be a target for personalized management in HF individuals.
1. Introduction
Chronic heart failure (HF) has remained a serious clinical and public problem [1]; despite that the prevalence of chronic HF especially HF with reduced left ventricular ejection fraction (HFrEF) seems to have a tendency to decrease within the last decade in developed countries [2]. In contrast, the frequency of new cases of HF with preserved left ventricular ejection fraction (HFpEF) is on the rise and HFpEF is considered more common than HFrEF predominantly in the older population and among subjects with hypertension, diabetes, and respiratory diseases [3]. Currently, more than 50% of patients with the clinical syndrome of HF might have a preserved left ventricular ejection fraction [4]. Nevertheless, recent clinical studies have shown a sufficient distinction between the presentation of both HF phenotypes (HFrEF or HFpEF) in individuals at risk for HF development in a primary care setting [5], [6]. Although HFpEF patients might have lower levels of predictive biomarkers [7], the clinical outcomes in HFpEF individuals, i.e. cardiovascular (CV) death, HF-related death, sudden death and readmission, are not better than in patients with HFrEF [8], [9]. Current clinical HF guidelines recommend the use of early identification and treatment of the underlying cause of cardiac dysfunction to prevent HF manifestation [10], [11], whereas no evidence exists that HF phenotypes' development is preventable with specific medical care [12]. Taken together, all these data clarify the importance of stratification of individuals at risk of HFrEF or HFpEF development and progression [13], [14].
Various molecular and cellular mechanisms are involved in the development and progression of both HF phenotypes. There is emerging evidence showing that the epigenetic regulation may take an important part in the pathogenesis of HF playing a pivotal role in the phenotypic response of a failing heart [15]. Epigenetic modifications affecting DNA methylation, ATP-dependent chromatin remodeling, histone modifications, and microRNA-related mechanisms are considered sufficient factors contributing to adverse cardiac remodeling and preceding cardiac dysfunction [16]. Probably, in the future epigenetic modifications of cardiomyocytes would be a target for personalized management and much more effective tools for the prevention of HF development [17]. The review summarizes the knowledge regarding the implication of epigenetic modifications in the development of different HF phenotypes.
1.1. Definition of epigenetics
A classical definition of epigenetics was proposed by Conrad Waddington in the 1950s, who believed that an organism's phenotype arises from its genotype through the so called “programmed epigenetic” event. Epigenetics was considered neither as the study of changes in states of any gene activity nor of inherited activity states only [18]. In some definitions, only those activity states that are inherited across cell division were defined [19]. In the Roadmap Epigenomics Project epigenetics is used as an emerging frontier of science that involves the study of changes in the regulation of gene activity and expression that are not dependent on gene sequence [20]. According to the Banbury Conference Center and Cold Spring Harbor Laboratory, a chromatin-based epigenetic trait is defined as a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence [21]. By now, it has been suggested that the basis of epigenetics is non-genetic cellular memory that is expressed or non-expressed in specific developmental and environmental situations [22].
Berger et al. in 2009 [21] have suggested using at least three categories of signals that modulate epigenetic state: “Epigenator”, “Epigenetic Initiator” and “Epigenetic Maintainer”. The authors proposed a signal named the “Epigenator” that might be an attribute of the environment and could be a trigger in an intracellular pathway. The other signal is called the “Epigenetic Initiator”, and corresponds to the “Epigenator” signal and is essential for defining the precise arrangement of the epigenetic chromatin environment. Finally, the “Epigenetic Maintainer” signal is deemed as the chromatin environment contributing to the first and subsequent generations. The role of different classes of potential signals affecting epigenetic modifications of chromatin is not well defined and is widely discussed.
In this context, discrete chemical modifications of DNA and primarily histones might regulate expression or repression of target genes and can be transmitted to the descent via epigenetic memory [23]. It has been suggested that the macro-molecular complex, called the “Epigenetic Code REplication Machinery” (ECREM), is involved in the inheritance of the epigenetic code. The main the members of ECREM could be enzymes involved in epigenetic modification of chromatin, i.e. DNA methyltransferases, histone deacetylases, histone acetyltransferases, and sirtuins. Moreover, deregulated ECREM is considered a clue of cell reprogramming [23], [24]. However, epigenetic events in various cells closely provide control of gene expression and genomic regulation through multiple generations leading to phenotypes' variability.
1.2. Epigenetics in failing heart
Epigenetics are referred to as a modification of the non-DNA sequences related heritable changes in gene expression of target cells that is currently recognized as a key to understanding of pathogenesis CV diseases [25]. Epigenetic modifications are based on different molecular mechanisms, which affect DNA methylation and deactylation, ATP-dependent chromatin remodeling, histone modifications, and microRNA regulation (Fig. 1). All these processes coordinate in the modulation of chromatin structure and thereby might affect the modalities and expression of target genes [26]. It has been found that the proliferative capability of human cardiac cells is under tight epigenetic regulation that mediates a dynamic adaptation of the structure and functionality of wide spectrum of cells, i.e. cardiomyocytes, fibroblasts, endothelial cells, and progenitor cells, for environmental challenges and to respond to biochemical stress. It is suggested that dysregulation in epigenetic signals, messengers and molecular targets is a clue for the pathological remodeling of the heart and vessels, progenitor cell dysfunction, worsening of the endogenous repair system, and metabolic memory manifestation [27], [28]. It is now becoming evident that a cumulative effect of these factors may culminate in HF, cardiac hypertrophy, arrhythmia, dyslipidemia, and atherosclerosis/atherothrombosis, and associates with increased risk of CV death [29], [30], [31], [32], [33], [34]. Whether interactions between features of transcriptomics, proteomics and metabolomics are specifically for HFrEF and HFpEF still remains not clear. Moreover, epigenetics might be a clue in the mediated transformation of mid-ranged HF defined as HF with an EF value averaging from 40% to 49% to neither HFrEF nor HFpEF. The different mechanisms of epigenetic modifications may relate to various HF-depended settings, i.e. cardiac hypertrophy, myocardial infarction, atrial fibrillation, and sudden death, and play a controversial role (Fig. 2). The epigenetic pattern of distinguished specific functional classes of target genes, which regulate the phenotypic response of a failing heart, is calculated to be more than 9000 candidate active enhancers [35]. For example, the histone methyltransferase is regulated by histone deacetylase inhibition and associated with pathological cardiac hypertrophy and fibrosis, whereas DNA methylation might directly induce cardiac cavity dilation and reduced pump function. DNA methylation and histone modifications interact with several cell signaling pathways to control target gene expression and might combinatorially affectenforcing or reversing epigenetic marks in chromatin [36]. In this way, none-coding microRNAs that interact with histone modification or DNA methylation is able to emerge as the diverse functions of a failing heart depending on the expression of regulating target genes acting as fundamental regulators of the transcriptional reprogramming [37]. However, the innate mechanisms regarding the causal pathway to HF phenotypes' development are nor fully understood and require more investigations [38].
Fig. 1.

The principal scheme regarding epigenetic regulations in heart failure development.
Abbreviations: α-MHC, the alpha-myosin heavy chain gene, SPR-Ca2 + ATPase, sarcoplasmic reticulum Ca2 + ATPase genes.
Fig. 2.
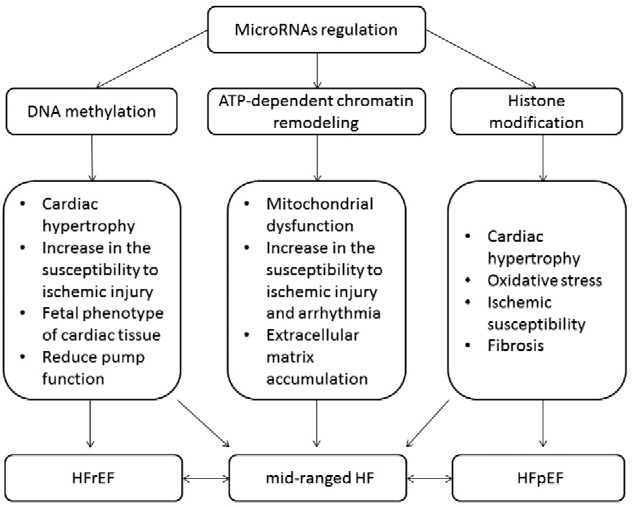
The different mechanisms of epigenetic modifications involved in the HFrEF and HFpEF presentation.
1.3. DNA methylation
DNA methylation is the most common epigenetic chromatin modification [39]. Movassagh et al. in 2011 [40] suggested that there are distinct global patterns of the epigenome leading to the regulation of the expression of underlying genes. The authors investigated genome-wide maps of DNA methylation and histone-3 lysine-36 trimethylation enrichment for cardiomyopathic and normal human hearts. A sufficient difference in DNA methylation in promoters of up-regulated genes has been found, but not down-regulated genes in end-stage cardiomyopathy [40]. Furthermore, the process of DNA methylation was under regulation through several genetic pathways that are modulating by platelet/endothelial cell adhesion molecule 1, hypoxia-inducible factor-1 alpha, angiomotin-like 2, and Rho GTPase activating protein 24 [40], [41]. The authors suggested that epigenetic modifications identified in the failing heart might affect cardiac function directly through regulation of protein structure synthesis and, indirectly via increased activity of cardiac fibroblasts due to prolonged hypoxia contributing to the pro-fibrotic nature of the ischemic milieu.
In contrast, satellite repeat element transcripts, a form of non-coding RNA that is heavily methylated in post-natal tissue, have putative functions in maintaining genomic stability and chromosomal integrity [42]. Moreover, the hypomethylation of satellite elements exhibited a close association with significant up-regulation of satellite transcripts [43]. Finally, there is evidence that methylation-regulated, alternative transcripts might express in a tissue- and cell type-specific manner and they may regulate intragenic promoter activity via enhancing transcription elongation efficiency [39], [43]. Thus, DNA methylations may enhance transcription of the underlying satellite repeat element transcripts.
Xiao et al. in 2014 [44] reported that increased DNA methylation might have a causative role in the programming of heart hypertrophy and reduced global cardiac contractility function. Moreover, impaired contractility of the left ventricle has been associated with an increase in the susceptibility to ischemic injury [44]. In contrast, Haas et al. in 2013 [45] did not find evidence regarding the participation of DNA methylation in genes suspected of HF development. However, DNA methylation has exhibited a causality role in diabetes-induced HFpEF [46]. Unfortunately, the data regarding the role of DNA methylation in the development of both phenotypes of HF beyond inhered forms are very limited, whereas several investigations agree with the hypothesis that chromatin modifications act in a combinatorial manner to specify transcriptional phenotypes in the heart [47]. More investigations are required to understand the underlying mechanisms linked to DNA methylation and HF presentation.
1.4. ATP-dependent enzymes in chromatin remodeling
The ATP-dependent chromatin remodeling complexes are not able to directly modify DNA or histones, whereas they may use the energy of ATP hydrolysis in processes regarding the destabilization, ejection or restructuring of nucleosomes, which are a functional unit of chromatin [48]. Because nucleosomes actively participate in transcription, chromosome segregation, DNA replication, and DNA repair, the chromosomal DNA packaging by nucleosomes is crucial for the regulation of these processes [49]. The dynamic access to DNA packaging is performed by specialized chromatin remodeling complexes [50]. There are four different families of ATP-dependent chromatin remodeling complexes: switching defective/sucrose non-fermenting complexes (SWI/SNF), imitation switch complexes, chromodomain–helicase–DNA-binding complexes, and inositol-requiring eighty complexes [51], [52]. All members of each family exhibit distinct unique domains that are served to histone–DNA contacts for DNA movement and chromatin restructuring [53]. The covalently modified histones are recognized by these domains of ATP-dependent chromatin remodelers and thereby regulate an expression of distinct gene programs tailored to specificity and biological functions of each family of chromatin remodeling complexes [54].
Recently it has been found that the mitochondrial perturbations might play a central role in human HF and that epigenetic mechanisms are crucial for mitochondrial quality control mechanisms [55]. Indeed, cardiac dysfunction associated with volume overload correlates to mitochondrial damage via increased reactive oxygen species production and free calcium within cardiomyocytes [56]. Evidence for perturbed cardiac mitochondrial dynamics included decreased mitochondria size, reduced numbers of mitochondria, and an altered expression of genes regulating fusion and fission [57]. SWI/SNF that is considered a major regulator of gene expression might facilitate the shifting and exposure of DNA segments within the promoter and several domains to transcription factors and other essential cellular proteins [58]. Bultman et al. in 2016 [59] reported the SWI/SNF ATP-dependent chromatin remodeling complexes in the metabolic homeostasis of the adult cardiac myocytes through cardiac myocyte-specific and inducible deletion of the SWI/SNF ATPases BRG1 and BRM. Moreover, the authors found that epigenetic mechanisms like SWI/SNF chromatin remodeling have been intimately linked to cardiac function and mitochondrial quality control mechanisms. How several factors including metabolic milieu, aging, and hemodynamic stress impact on the epigenetic landscape of the myocardium is still not clear, whereas exploitation of the epigenetic machinery therapeutically may emerge with clinical relevance [60].
1.5. Histone modifications
Histone modification represents a dynamic process affecting histone proteins (H2A, H2B, H3, and H4) that are composed of nucleosomes and mediated by several enzymes [61]. As a result, distinct histone amino-terminal modifications appear to be able to induce synergistic or antagonistic interaction affinities for chromatin-associated proteins, which in turn dictate dynamic transitions between transcriptionally active or transcriptionally silent chromatin states [62]. By now more than 150 post-translation modifications of histones have been reported, including methylations, acetylations, sumoylation, ubiquitinations, ADP-ribosylation, proline isomerization, and phosphorylations [63]. The appearance of conformational changes in chromatin resulting in the alteration due to histone modifications regulates gene expression depending on whether DNA has been became accessible (defined as euchromatin) or inaccessible (called as heterochromatin) for further transcription process [64].
Recent studies have shown that histone modification predominantly methylation closely regulates inflammatory and metabolic disorders, as well as links CV disease and vascular homeostasis [65], [66], [67]. There is evidence that altered redox signaling might mediate trimethylation of histones H3K4 and H3K9 and thereby links an oxidative stress pathway with biochemical mechanisms underlying HFrEF development [68]. Probably, aging might affect the development of either HFpEF or HFrEF through the so-called “epigenetic drift” via marked regulation of DNA and histone methylation [69]. However, further investigations are needed to explain in detail the role of histone modification in the impairment of cardiac structure and functionality [70].
1.6. MicroRNA-depending mechanisms
MicroRNAs (miRNAs) are small non-coding RNAs that exert their function by both transcript degradation and translational inhibition, resulting in changes in target genes and proteins' expression [71]. It has been suggested that reactivation of a fetal microRNA program substantially contributes to alterations of gene expression in the failing human heart. Indeed, recent studies have shown the increased expression of miRNA-1, miRNA-21, miRNA-29b, miRNA-129, miRNA-133, miRNA-208, miRNA-210, miRNA-211, miRNA-212, and miRNA-423, and miRNA-499 miRNA503 [72], [73], [74]. The reduced expression of miRNA-30, miRNA-182, and miRNA-526 are associated with HF development and progression [74]. Therefore, altered microRNA expression in the human heart was found in ischemic cardiomyopathy, dilated cardiomyopathy, and aortic stenosis [74], [75], [76]. There is evidence that the disproportion between myocardial expressions of alpha- and beta-myosin heavy chain (MHC) might be associated with over-expression of heart-specific miRNA-208a leading to arrhythmia, fibrosis, and cardiac hypertrophy [77]. Nevertheless, miRNA-208a expression is under negative control of MED13, a subunit of the mediator complex, which controls transcription by thyroid hormone and other nuclear hormone receptors [78]. The down-regulation of miRNA-1 and up-regulation of miRNA-195 are necessary for cardiac hypertrophy and HFpEF development, while the underlying molecular mechanisms of these effects are still not clear [73], [79]. Finally, cardiac hypertrophy may associate with the activation of Nppa, Nppb, and Acta1 (skeletal α-actin) fetal gene program [80], [81]. It has been deemed that the key components of a cardiomyocyte hypertrophy mediator might be a miRNA-dependent regulator of calcium signaling pathways [79]. miRNA-24 has exhibited a regulatory role in failing heart through excitation–contraction uncoupling of the sarcoplasmic reticulum and T-tubules via the junctophilin-2 protein [82]. Up-regulated miRNA-24 may suppress cardiac remodeling associated with the accumulation of collagen and fibronectin in a TGF-β-dependent manner [83]. Zhou et al. in 2016 [84] have found that up-regulated miR-503 in the failing heart might promote cardiac fibrosis via the miR-503–Apelin-13–TGF-β–CTGF–collagen production pathway. Other types of miRNAs, i.e. miRNA-21 and miRNA-29 contribute to the development of HF via enhancing fibrosis by stimulating the extracellular signal-regulated kinase (ERK)–mitogen-activated protein (MAP) kinase signaling pathway [85], [86]. Moreover, miRNA-29 has been positively associated with cardiac hypertrophy and fibrosis [87]. In contrast, over-expressed cardiac-specific miRNA-133 may attenuate cardiac fibrosis in an animal model [88]. miRNA-210 and miRNA-211 are none-cardiac specific mediators of mitochondrial function, cell differentiation and growth. Although it has been found that down-regulation of miRNA-210 has inhibited cell growth, cell proliferation and induced apoptosis [89], the causal role of miRNA-210 and miRNA-211 in cardiac remodeling is under investigation. Hinkel et al. in 2013 [90] have reported that over-expressed miRNA-92a in the failing heart is a clue of tissue damage and that the inhibition of miRNA-92a might protect cardiac myocytes from ischemia/reperfusion injury.
Theoretically, there is a well-described signature of cardiac-specific miRNAs, which may be involved in cardiac remodeling forming HF phenotypes. Indeed, down-regulated miRNA-1, miRNA-29, miRNA-133 and miRNA-208 in response to cardiac injury or overload correspond to cardiac hypertrophy, whereas predominantly over-expression of these miRNAs contributes to cardiac fibrosis. An imbalance between regulatory mechanisms affecting expression of miRNA-1, miRNA-29, miRNA-133 and miRNA-208 might represent specific HF phenotypes, i.e. HFrEF or HFpEF [91]. Whether the altered signature of miRNA is considered a clue for cardiac hypertrophy and dysfunction [81], the low number of direct clinical evidence regarding specifically HF phenotypes' development relating to miRNA signature remains a part of scientific discussion [66].
In clinical settings, low circulating levels of miRNA-103, miRNA-142-3p, miRNA-30b, miRNA-342-3p, and miRNA-652-3p and high levels of miRNA-499 and miRNA-508-5p were found as diagnostic biomarkers of advanced HF [92], [93], [94], [95]. Moreover, in advance HF patients, low circulating levels of miRNA-423-5p are associated with poor long-term outcomes [96].
Recently studies have shown that miRNAs' deregulation of the endothelial progenitor cells (EPCs) and circulating angiogenic cells involved in endothelial function modulation might play a key role in HF progression [93], [97], [98]. Qiang et al. in 2013 [98] have found that low levels of miRNA-126 and high levels of miRNA-508a-5p in endothelial progenitor cells were associated with cardiovascular death in ischemia-induced HF and non-ischemia-induced HF respectively. Furthermore, decreased miRNA-18a-5p and miRNA-652-3p during a hospitalization for HF was predictive for 180-day mortality [93]. Whether we can use the signature of miRNAs measured in EPCs as predictive biomarkers for HFrEF and HFpEF is not still understood [99].
2. Future direction: epigenetic-based therapies of heart failure?
The impact on molecular targets regulating epigenetic mechanisms appears to be promised. Several drugs may inhibit DNA methyltransferase directly or by reducing its gene expression, i.e. hydralazine and procainamide, might have anti-proliferative and anti-inflammatory properties [100]. Therefore, nutrients are currently under investigation as potent modulators of DNA methyltransferase activity [101]. There are expectations regarding cardio-protective effects of histone deacetylase inhibitors, such as valproic acid [100], [101]. There is evidence that histone deacetylase inhibitors may attenuate pathological cardiac remodeling and hypertrophic gene expression [102]. Nural-Guvener et al. in 2015 [103] reported that histone deacetylase inhibitor mocetinostat in HF has exhibited anti-fibrotic effects associated with the IL-6/STAT3 signaling pathway. Additionally, the regulation of the expression of microRNA targets is deemed a pharmacological tool for the prevention of cardiac remodeling and advance of HFpHF and HFrEF [104], [105], [106]. However, no epigenetically active drugs that have actually entered clinical trials for HFpHF and HFrEF have been approved by the FDA [107]. Yet, there is uncertainty regarding perspectives of therapeutic strategy based on the inhibition of cardiac hypertrophy in clinical settings [108], [109].
In conclusion, the current available data preliminarily clarify that epigenetic modifications might be discussed as a clue of forming phenotypes of HF, whereas there is no strong evidence regarding epigenetic signatures representing causal pathways leading to specific forms of cardiac remodeling associated with HFrEF or HFpEF. Although epigenetic-based therapies of HF are promised, no large clinical trials supported the hypothesis regarding the capabilities of DNA methyltransferase inhibitors, histone deacetylase inhibitors and miRNAs to prevent both HF phenotypes from forming. More investigations are required to discover epigenetic regulation features, because of progress in this setting which appears to be much more promised for translation to individualized medical care.
Abbreviations
- α-MHC
the alpha-myosin heavy chain gene
- BRG1
Brahma-related gene 1
- CV
cardiovascular
- DNA
deoxyribonucleic acid
- ECREM
Epigenetic Code REplication Machinery
- HFrEF
heart failure with reduced ejection fraction
- HFpEF
heart failure with preserved ejection fraction
- miRNA
micro ribonucleic acid
- MAP
mitogen-activated protein
- MHC
myosin heavy chain
- SPR-Ca2 + ATPase
sarcoplasmic reticulum Ca2 + ATPase genes
- SWI/SNF
SWItch/Sucrose NonFermen
- TGF-β
transforming growth factor beta
Conflicting interests
None of the authors has any conflict of interest to disclose related to the content of this study.
Transparency Document
Transparency document.
Footnotes
Funding and grants: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The Transparency document associated with this article can be found, in the online version.
References
- 1.Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M. American Heart Association statistics committee and stroke statistics subcommittee. Executive summary: heart disease and stroke statistics—2016 update: A report from the American Heart Association. Circulation. 2016;133(4):447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Güder G., Gelbrich G., Edelmann F., Wachter R., Pieske B., Pankuweit S., Competence Network Heart Failure Germany Reverse epidemiology in different stages of heart failure. Int. J. Cardiol. 2015;184:216–224. doi: 10.1016/j.ijcard.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 3.van Riet E.E., Hoes A.W., Wagenaar K.P., Limburg A., Landman M.A., Rutten F.H. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016;18(3):242–252. doi: 10.1002/ejhf.483. [DOI] [PubMed] [Google Scholar]
- 4.Edelmann F. Epidemiology and prognosis of heart failure. Herz. 2015;40(2):176–184. doi: 10.1007/s00059-015-4215-5. [DOI] [PubMed] [Google Scholar]
- 5.Jorge A.L., Rosa M.L., Martins W.A., Correia D.M., Fernandes L.C., Costa J.A. The prevalence of stages of heart failure in primary care: a population-based study. J. Card. Fail. 2016;22(2):153–157. doi: 10.1016/j.cardfail.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 6.van Veldhuisen D.J., Linssen G.C., Jaarsma T., van Gilst W.H., Hoes A.W., Tijssen J.G. B-type natriuretic peptide and prognosis in heart failure patients with preserved and reduced ejection fraction. J. Am. Coll. Cardiol. 2013;61(14):1498–1506. doi: 10.1016/j.jacc.2012.12.044. [DOI] [PubMed] [Google Scholar]
- 7.Bettencourt P., Azevedo A., Fonseca L., Araújo J.P., Ferreira S., Almeida R. Prognosis of decompensated heart failure patients with preserved systolic function is predicted by NT-proBNP variations during hospitalization. Int. J. Cardiol. 2007;117(1):75–79. doi: 10.1016/j.ijcard.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Kang S.H., Park J.J., Choi D.J., Yoon C.H., Oh I.Y., Kang S.M., KorHF Registry Prognostic value of NT-proBNP in heart failure with preserved versus reduced EF. Heart. 2015;101(23):1881–1888. doi: 10.1136/heartjnl-2015-307782. [DOI] [PubMed] [Google Scholar]
- 9.Nichols G.A., Reynolds K., Kimes T.M., Rosales A.G., Chan W.W. Comparison of risk of re-hospitalization, all-cause mortality, and medical care resource utilization in patients with heart failure and preserved versus reduced ejection fraction. Am. J. Cardiol. 2015;116(7):1088–1092. doi: 10.1016/j.amjcard.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 10.McMurray J.J., Adamopoulos S., Anker S.D., Auricchio A., Böhm M., Dickstein K., ESC Committee for Practice Guidelines ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012;33(14):1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 11.Yancy C.W., Jessup M., Bozkurt B., Butler J., Casey D.E., Jr., Drazner M.H., American College of Cardiology Foundation, American Heart Association Task Force on Practice Guidelines AHA/AHA guideline for the management of heart failure: report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013;62(16):e147–e239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 12.Fischer C., Steyerberg E.W., Fonarow G.C., Ganiats T.G., Lingsma H.F. A systematic review and meta-analysis on the association between quality of hospital care and readmission rates in patients with heart failure. Am. Heart J. 2015;170(5) doi: 10.1016/j.ahj.2015.06.026. (1005-1017.e2) [DOI] [PubMed] [Google Scholar]
- 13.Berezin A.E. Predicting heart failure phenotypes using cardiac biomarkers: hype and hope. J. Dis. Markers. 2015;2(4):1035–1041. [Google Scholar]
- 14.Berezin A.E. Prognostication in different heart failure phenotypes: the role of circulating biomarkers. J. Circulating Biomark. 2016;5 doi: 10.5772/62797. (ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Napoli C., Grimaldi V., De Pascale M.R., Sommese L., Infante T., Soricelli A. Novel epigenetic-based therapies useful in cardiovascular medicine. World J. Cardiol. 2016;8(2):211–219. doi: 10.4330/wjc.v8.i2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Salvo T.G., Haldar S.M. Epigenetic mechanisms in heart failure pathogenesis. Circ. Heart Fail. 2014;7(5):850–863. doi: 10.1161/CIRCHEARTFAILURE.114.001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mollova M.Y., Katus H.A., Backs J. Regulation of CaMKII signaling in cardiovascular disease. Front. Pharmacol. 2015;6:178. doi: 10.3389/fphar.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann J.R. Epigenetics and memigenetics. Cell. Mol. Life Sci. 2014;71(7):1117–1122. doi: 10.1007/s00018-014-1560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roloff T.C., Nuber U.A. Chromatin, epigenetics and stem cells. Eur. J. Cell Biol. 2005;84(2–3):123–135. doi: 10.1016/j.ejcb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 20.Romanoski C.E., Glass C.K., Stunnenberg H.G., Wilson L., Almouzni G. Epigenomics: roadmap for regulation. Nature. 2015;518(7539):314–316. doi: 10.1038/518314a. [DOI] [PubMed] [Google Scholar]
- 21.Berger S.L., Kouzarides T., Shiekhattar R., Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Y., Lu L., Liu M., Li X.C., Sun R.R., Zheng Y., Zhang P.Y. Impact of epigenetics in the management of cardiovascular disease: a review. Eur. Rev. Med. Pharmacol. Sci. 2014;18(20):3097–3104. [PubMed] [Google Scholar]
- 23.Bronner C., Chataigneau T., Schini-Kerth V.B., Landry Y. The “Epigenetic Code Replication Machinery”, ECREM: a promising drugable target of the epigenetic cell memory. Curr. Med. Chem. 2007;14(25):2629–2641. doi: 10.2174/092986707782023244. [DOI] [PubMed] [Google Scholar]
- 24.Swaminathan V., Reddy B.A., Ruthrotha Selvi B., Sukanya M.S., Kundu T.K. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell. Biochem. 2007;41:397–428. [PubMed] [Google Scholar]
- 25.Marín-García J., Akhmedov A.T. Epigenetics of the failing heart. Heart Fail. Rev. 2015;20(4):435–459. doi: 10.1007/s10741-015-9483-x. [DOI] [PubMed] [Google Scholar]
- 26.Yang J., Xu W.W., Hu S.J. Heart failure: advanced development in genetics and epigenetics. Biomed Res. Int. 2015;2015:352734. doi: 10.1155/2015/352734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiano C., Vietri M.T., Grimaldi V., Picascia A., Pascale M.R., Napoli C. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol. Sci. 2015;36(4):226–235. doi: 10.1016/j.tips.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Berezin A.E. Circulating cell-free mitochondrial DNA as biomarker of cardiovascular risk: new challenges of old findings. Angiology. 2015;3:161–163. [Google Scholar]
- 29.Berezin A. Metabolic memory phenomenon in diabetes mellitus: Achieving and perspectives. Diabetes Metab. Syndr. 2016 doi: 10.1016/j.dsx.2016.03.016. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 30.Berezin A.E. Impaired phenotype of circulating endothelial-derived microparticles: novel marker of cardiovascular risk. J. Cardiol. Ther. 2015;2(4):273–278. [Google Scholar]
- 31.Berezin A.E. Endothelial derived micro particles: biomarkers for heart failure diagnosis and management. J. Clin. Trials Cardiol. 2015;2(3):1–3. [Google Scholar]
- 32.Berezin A., Kremzer A., Berezina T., Yu M., Gromenko O. Pattern of endothelial progenitor cells and apoptotic endothelial cell-derived microparticles in chronic heart failure patients with preserved and reduced left ventricular ejection fraction. EBioMedicine. 2016;4:86–94. doi: 10.1016/j.ebiom.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berezin A., Kremzer A., Berezina T., Martovotskaya Y. Pattern of circulating microparticles in chronic heart failure patients with metabolic syndrome: relevance to neurohumoral and inflammatory activation. BBA Clin. 2015;4:69–75. doi: 10.1016/j.bbacli.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duygu B., Poels E.M., da Costa Martins P.A. Genetics and epigenetics of arrhythmia and heart failure. Front. Genet. 2013;4:219. doi: 10.3389/fgene.2013.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papait R., Cattaneo P., Kunderfranco P., Greco C., Carullo P., Guffanti A. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 2013;110(50):20164–20169. doi: 10.1073/pnas.1315155110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suganuma T., Workman J.L. Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 37.Tao H., Yang J.J., Shi K.H. Non-coding RNAs as direct and indirect modulators of epigenetic mechanism regulation of cardiac fibrosis. Expert Opin. Ther. Targets. 2015;19(5):707–716. doi: 10.1517/14728222.2014.1001740. [DOI] [PubMed] [Google Scholar]
- 38.Piran S., Liu P., Morales A., Hershberger R.E. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J. Am. Coll. Cardiol. 2012;60(4):283–289. doi: 10.1016/j.jacc.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Maunakea A.K., Nagarajan R.P., Bilenky M., Ballinger T.J., D'Souza C., Fouse S.D. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Movassagh M., Choy M.K., Knowles D.A., Cordeddu L., Haider S., Down T. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124(22):2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson C.J., Collier P., Tea I., Neary R., Watson J.A., Robinson C. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014;23(8):2176–2188. doi: 10.1093/hmg/ddt614. [DOI] [PubMed] [Google Scholar]
- 42.Haider S., Cordeddu L., Robinson E., Movassagh M., Siggens L., Vujic A., Choy M.K., Goddard M., Lio P., Foo R. The landscape of DNA repeat elements in human heart failure. Genome Biol. 2012;13(10):R90. doi: 10.1186/gb-2012-13-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones P.A. The DNA methylation paradox. Trends Genet. 1999;15(1):34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 44.Xiao D., Dasgupta C., Chen M., Zhang K., Buchholz J., Xu Z., Zhang L. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc. Res. 2014;101(3):373–382. doi: 10.1093/cvr/cvt264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haas J., Frese K.S., Park Y.J., Keller A., Vogel B., Lindroth A.M. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013;5(3):413–429. doi: 10.1002/emmm.201201553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konig A., Bode C., Bugger H. Diabetes mellitus and myocardial mitochondrial dysfunction: bench to bedside. Heart Fail. Clin. 2012;8(4):551–561. doi: 10.1016/j.hfc.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Chen H., Orozco L.D., Wang J., Rau C.D., Rubbi L., Ren S. DNA methylation indicates susceptibility to isoproterenol-induced cardiac pathology and is associated with chromatin states. Circ. Res. 2016;118(5):786–797. doi: 10.1161/CIRCRESAHA.115.305298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varga-Weisz P. Chromatin remodeling factors and DNA replication. Prog. Mol. Subcell. Biol. 2005;38:1–30. doi: 10.1007/3-540-27310-7_1. [DOI] [PubMed] [Google Scholar]
- 49.Clapier C.R., Cairns B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 50.Saha A., Wittmeyer J., Cairns B.R. Mechanisms for nucleosome movement by ATP-dependent chromatin remodeling complexes. Results Probl. Cell Differ. 2006;41:127–148. doi: 10.1007/400_005. [DOI] [PubMed] [Google Scholar]
- 51.Lange M., Demajo S., Jain P., Di Croce L. Combinatorial assembly and function of chromatin regulatory complexes. Epigenomics. 2011;3(5):567–580. doi: 10.2217/epi.11.83. [DOI] [PubMed] [Google Scholar]
- 52.Ho L., Crabtree G.R. Chromatin remodelling during development. Nature. 2010;463(7280):474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newell-Price J., Clark A.J., King P. DNA methylation and silencing of gene expression. Trends Endocrinol. Metab. 2000;11(4):142–148. doi: 10.1016/s1043-2760(00)00248-4. [DOI] [PubMed] [Google Scholar]
- 54.Chodavarapu R.K., Feng S., Bernatavichute Y.V., Chen P.Y., Stroud H., Yu Y. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466(7304):388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kunkel G.H., Chaturvedi P., Tyagi S.C. Resuscitation of a dead cardiomyocyte. Heart Fail. Rev. 2015;20(6):709–719. doi: 10.1007/s10741-015-9501-z. [DOI] [PubMed] [Google Scholar]
- 56.Kunkel G.H., Chaturvedi P., Tyagi S.C. Epigenetic revival of a dead cardiomyocyte through mitochondrial interventions. Biomol. Concepts. 2015;6(4):303–319. doi: 10.1515/bmc-2015-0011. [DOI] [PubMed] [Google Scholar]
- 57.Wiley M.M., Muthukumar V., Griffin T.M., Griffin C.T. SWI/SNF chromatin-remodeling enzymes Brahma-related gene 1 (BRG1) and Brahma (BRM) are dispensable in multiple models of postnatal angiogenesis but are required for vascular integrity in infant mice. J. Am. Heart Assoc. 2015;4(4) doi: 10.1161/JAHA.115.001972. (pii: e001972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marquez S.B., Thompson K.W., Lu L., Reisman D. Beyond mutations: additional mechanisms and implications of SWI/SNF complex inactivation. Front. Oncol. 2015;4:372. doi: 10.3389/fonc.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bultman S.J., Holley D.W., de Ridder G., Pizzo S.V., Sidorova T.N., Murray K.T. BRG1 and BRM SWI/SNF ATPases redundantly maintain cardiomyocyte homeostasis by regulating cardiomyocyte mitophagy and mitochondrial dynamics in vivo. Cardiovasc. Pathol. 2016;25(3):258–269. doi: 10.1016/j.carpath.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S.Y., Morales C.R., Gillette T.G., Hill J.A. Epigenetic regulation in heart failure. Curr. Opin. Cardiol. 2016 doi: 10.1097/HCO.0000000000000276. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y., Fatima N., Dufau M.L. Coordinated changes in DNA methylation and histone modifications regulate silencing/derepression of luteinizing hormone receptor gene transcription. Mol. Cell. Biol. 2005;25(18):7929–7939. doi: 10.1128/MCB.25.18.7929-7939.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 63.Handy D.E., Castro R., Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Margueron R., Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11(4):285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robb G.B., Carson A.R., Tai S.C., Fish J.E., Singh S., Yamada T. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J. Biol. Chem. 2004;279(36):37982–37996. doi: 10.1074/jbc.M400271200. [DOI] [PubMed] [Google Scholar]
- 66.Tai S.C., Robb G.B., Marsden P.A. Endothelial nitric oxide synthase: a new paradigm for gene regulation in the injured blood vessel. Arterioscler. Thromb. Vasc. Biol. 2004;24(3):405–412. doi: 10.1161/01.ATV.0000109171.50229.33. [DOI] [PubMed] [Google Scholar]
- 67.Shi Y., Whetstine J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell. 2007;25(1):1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 68.Kim G.H., Ryan J.J., Archer S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal. 2013;18(15):1920–1936. doi: 10.1089/ars.2012.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Illi B., Ciarapica R., Capogrossi M.C. Chromatin methylation and cardiovascular aging. J. Mol. Cell. Cardiol. 2015;83:21–31. doi: 10.1016/j.yjmcc.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 70.Berezin A. “Impaired immune phenotype” of endothelial cell-derived microparticles: the missed link between diabetes-related states and cardiovascular complications? J. Data Min. Genomics Proteomics. 2016;7(2):195–197. [Google Scholar]
- 71.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 72.Thum T., Galuppo P., Wolf C., Fiedler J., Kneitz S., van Laake L.W. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116(3):258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 73.van Rooij E., Sutherland L.B., Liu N., Williams A.H., McAnally J., Gerard R.D., Richardson J.A., Olson E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sayed D., Hong C., Chen I.Y., Lypowy J., Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 75.Ikeda S., Kong S.W., Lu J., Bisping E., Zhang H., Allen P.D., Golub T.R., Pieske B., Pu W.T. Altered microRNA expression in human heart disease. Physiol. Genomics. 2007;31(3):367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 76.Sucharov C., Bristow M.R., Port J.D. miRNA expression in the failing human heart: functional correlates. J. Mol. Cell. Cardiol. 2008;45(2):185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Udali S., Guarini P., Moruzzi S., Choi S.W., Friso S. Cardiovascular epigenetics: from DNA methylation to microRNAs. Mol. Asp. Med. 2013;34(4):883–901. doi: 10.1016/j.mam.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 78.Grueter C.E., van Rooij E., Johnson B.A., DeLeon S.M., Sutherland L.B., Qi X. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149(3):671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ikeda S., He A., Kong S.W., Lu J., Bejar R., Bodyaket N. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell. Biol. 2009;29(8):2193–2204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Izumo S., Nadal-Ginard B., Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc. Natl. Acad. Sci. U. S. A. 1988;85:339–343. doi: 10.1073/pnas.85.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bagnall R.D., Tsoutsman T., Shephard R.E., Ritchie W., Semsarian C. Global microRNA profiling of the mouse ventricles during development of severe hypertrophic cardiomyopathy and heart failure. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo C., Deng Y., Liu J., Qian L. Cardiomyocyte-specific role of miR-24 in promoting cell survival. J. Cell. Mol. Med. 2015;19:103–112. doi: 10.1111/jcmm.12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J., Huang W., Xu R., Nie Y., Cao X., Meng J. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012;16:2150–2160. doi: 10.1111/j.1582-4934.2012.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou Y., Deng L., Zhao D., Chen L., Yao Z., Guo X. MicroRNA-503 promotes angiotensin II-induced cardiac fibrosis by targeting Apelin-13. J. Cell. Mol. Med. 2016;20(3):495–505. doi: 10.1111/jcmm.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thum T., Gross C., Fiedler J., Fischer T., Kissler S., Bussen M. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 86.WS, Hill J.A., Olson E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Derda A.A., Thum S., Lorenzen J.M., Bavendiek U., Heineke J., Keyser B. Blood-based microRNA signatures differentiate various forms of cardiac hypertrophy. Int. J. Cardiol. 2015;196:115–122. doi: 10.1016/j.ijcard.2015.05.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen S., Puthanveetil P., Feng B., Matkovich S.J., Dorn G.W., 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 2014;18(3):415–421. doi: 10.1111/jcmm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sen C.K. MicroRNAs as new maestro conducting the expanding symphony orchestra of regenerative and reparative medicine. Physiol. Genomics. 2011;43(10):517–520. doi: 10.1152/physiolgenomics.00037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hinkel R., Penzkofer D., Zuhlke S., Fischer A., Husada W., Xu Q.F., Baloch E., van Rooij E., Zeiher A.M., Kupatt C., Dimmeler S. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation. 2013;128:1066–1075. doi: 10.1161/CIRCULATIONAHA.113.001904. [DOI] [PubMed] [Google Scholar]
- 91.Vegter E.L., van der Meer P., de Windt L.J., Pinto Y.M., Voors A.A. MicroRNAs in heart failure: from biomarker to target for therapy. Eur. J. Heart Fail. 2016 doi: 10.1002/ejhf.495. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 92.van Rooij E., Sutherland L.B., Thatcher J.E., DiMaio J.M., Naseem R.H., Marshall Ellis K.L. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur. J. Heart Fail. 2013;15:1138–1147. doi: 10.1093/eurjhf/hft078. [DOI] [PubMed] [Google Scholar]
- 93.Ovchinnikova E.S., Schmitter D., Vegter E.L., Ter Maaten J.M., Valente M.A., Liu L.C. Signature of circulating microRNAs in patients with acute heart failure. Eur. J. Heart Fail. 2015 doi: 10.1002/ejhf.332. [DOI] [PubMed] [Google Scholar]
- 94.Corsten M.F., Dennert R., Jochems S., Kuznetsova T., Devaux Y., Hofstra L. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010;3:499–506. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 95.Qiang L., Hong L., Ningfu W., Huaihong C., Jing W. Expression of miR-126 and miR-508-5p in endothelial progenitor cells is associated with the prognosis of chronic heart failure patients. Int. J. Cardiol. 2013;168:2082–2088. doi: 10.1016/j.ijcard.2013.01.160. [DOI] [PubMed] [Google Scholar]
- 96.Seronde M.F., Vausort M., Gayat E., Goretti E., Ng L.L., Squire I.B., GREAT network Circulating microRNAs and outcome in patients with acute heart failure. PLoS One. 2015;10(11) doi: 10.1371/journal.pone.0142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Recchioni R., Marcheselli F., Antonicelli R., Lazzarini R., Mensà E., Testa R. Physical activity and progenitor cell-mediated endothelial repair in chronic heart failure: is there a role for epigenetics? Mech. Ageing Dev. Mar 24 2016 doi: 10.1016/j.mad.2016.03.008. (pii: S0047–6374(16)30029-X, Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 98.Suzuki T., Lyon A., Saggar R., Heaney L.M., Aizawa K., Cittadini A. Biomarkers of acute cardiovascular and pulmonary diseases. Eur Heart J Acute Cardiovasc Care. 2016 doi: 10.1177/2048872616652309. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 99.Berezin A. Impaired phenotype of endothelial cell-derived micro particles: the missed link in heart failure development? Biomark. J. 2016;2(2):14–19. [Google Scholar]
- 100.Napoli C., Grimaldi V., De Pascale M.R., Sommese L., Infante T., Soricelli A. Novel epigenetic-based therapies useful in cardiovascular medicine. World J. Cardiol. 2016;8(2):211–219. doi: 10.4330/wjc.v8.i2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aune S.E., Herr D.J., Kutz C.J., Menick D.R. Histone deacetylases exert class-specific roles in conditioning the brain and heart against acute ischemic injury. Front. Neurol. 2015;6:145. doi: 10.3389/fneur.2015.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ooi J.Y., Tuano N.K., Rafehi H., Gao X.M., Ziemann M., Du X.J. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. 2015;10(5):418–430. doi: 10.1080/15592294.2015.1024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nural-Guvener H., Zakharova L., Feehery L., Sljukic S., Gaballa M. Anti-fibrotic effects of class I HDAC inhibitor, mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int. J. Mol. Sci. 2015;16(5):11482–11499. doi: 10.3390/ijms160511482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Montgomery R.L., Hullinger T.G., Semus H.M., Dickinson B.A., Seto A.G., Lynch J.M. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124:1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y., Huang X.R., Wei L.H., Chung A.C., Yu C.M., Lan H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 2014;22:974–985. doi: 10.1038/mt.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Rooij E., Olson E.N. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat. Rev. Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Voelter-Mahlknecht S. Epigenetic associations in relation to cardiovascular prevention and therapeutics. Clin. Epigenetics. 2016;8:4. doi: 10.1186/s13148-016-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crozatier B., Ventura-Clapier R. Inhibition of hypertrophy, per se, may not be a good therapeutic strategy in ventricular pressure overload: other approaches could be more beneficial. Circulation. 2015;131(16):1448–1457. doi: 10.1161/CIRCULATIONAHA.114.013895. [DOI] [PubMed] [Google Scholar]
- 109.Berezin A. Progenitor endothelial cell dysfunction in heart failure: clinical implication and therapeutic target? Transl. Med. (Sunnyvale) 2016;6(3):176–177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.