

Abstract
Lung cancer is the most important cause of death among neoplastic diseases worldwide, and cigarette smoke (CS) is the major risk factor for cancer. Complementarily to avoidance of exposure to CS, chemoprevention will lower the risk of cancer in passive smokers, ex-smokers, and addicted current smokers who fail to quit smoking. Unfortunately, chemoprevention clinical trials have produced disappointing results to date and, until recently, a suitable animal model evaluating CS carcinogenicity was not available. We previously demonstrated that mainstream CS induces a potent carcinogenic response when exposure of mice starts at birth. In the present study, neonatal mice (strain H) were exposed to CS for 120 consecutive days, starting at birth. The chemopreventive agents budesonide (2.4 mg/kg diet), phenethyl isothiocyanate (PEITC, 1,000 mg/kg diet), and N-acetyl-l-cysteine (NAC, 1,000 mg/kg body weight) were administered orally according to various protocols. The experiment was stopped after 210 days. Exposure to CS resulted in a high incidence and multiplicity of benign lung tumors and in significant increases of malignant lung tumors and other histopathological alterations. All three chemopreventive agents, administered to current smokers after weanling, were quite effective in protecting both male and female mice from CS pulmonary carcinogenicity. When given to ex-smokers after withdrawal of exposure to CS, the protective capacity of budesonide was unchanged, while PEITC lost part of its cancer chemopreventive activity. In conclusion, the proposed experimental model provides convincing evidence that it is possible to prevent CS-induced lung cancer by means of dietary and pharmacological agents.
Keywords: mainstream cigarette smoke, lung cancer, budesonide, phenethyl isothiocyanate, N-acetyl-l-cysteine
Lung cancer is the most important cause of death among neoplastic diseases worldwide, and tobacco smoking is the dominant risk factor for this disease.1 There is sound epidemiological evidence that the risk of developing lung cancer, cancers at several other sites, and other chronic degenerative diseseas is higher in smokers than in nonsmokers.1 The most obvious way to prevent lung cancer and other cigarette–smoke (CS)–related diseases is either to refrain from smoking or to quit smoking or, in case of passive exposure, not to live in CS–contaminated environments. The risk of developing lung cancer progressively decreases in ex–smokers,2 but the decrease is slow to such an extent that half of lung cancer cases are nowadays diagnosed in ex–smokers.3 While smoking habits are unfortunately increasing in many countries, their decline in the male population of several western countries already had a favorable impact on the epidemiology of lung cancer, which is a milestone in the history of modern medicine.4,5
Another strategy in the prevention of CS–associated diseases, which is obviously complementary and not alternative to avoidance of exposure to CS, is to reinforce the host defense mechanisms by means of pharmacological and dietary agents. This strategy, referred to as chemoprevention, is already a reality in the prevention of cancer and other diseases, such as cardiovascular diseases, and deserves more and more attention. The chemoprevention of CS–associated cancers would save many lives among addicted active smokers, who fail to quit smoking, but especially among ex–smokers and passive smokers, also including transplacentally exposed individuals.
The major limitations in evaluating the efficacy of chemopreventive agents against CS–related cancers are represented by the problems encountered in clinical trials, which have produced only disappointing results to date.6-8 In addition, until recently, an adequate animal model for reproducing CS carcinogenicity was not available. While several chemoprevention studies in rodents have been conducted with typical CS components, such as benzo(a)pyrene [B(a)P], as a prototype of polycyclic aromatic hydrocarbons, and 4–(methylnitrosoamino)–1–(3–pyridyl)–1–butanone (NNK), as a prototype of tobacco–specific nitrosamines,1,9,10 much less attention has been paid to CS, either mainstream (MCS) or sidestream (SCS) or environmental (ECS), as a complex mixture. In spite of the fact that the earliest attempts to study the tumorigenicity of whole CS in mice trace back to 6 or 7 decades ago,11,12 i.e., before the discovery of CS carcinogenicity in humans, for a variety of reasons most carcinogenicity studies with inhaled CS yielded either negative or weakly positive results in experimental animals.1,13-15 The poor carcinogenicity of CS in laboratory animals contrasts with the strong evidence that, in rodents, both MCS and ECS are able to affect a broad variety of intermediate biomarkers, which can be modulated by chemopreventive agents.16
Evidence that MCS is moderately carcinogenic in adult rodents was generated in lifetime studies involving the whole–body exposure, for 30 months, of F344 rats17 and B6C3F1 mice.18 Recently, we showed that MCS becomes a potent carcinogen in mice when exposure starts soon after birth.19 In fact, the whole–body exposure of Swiss albino mice to MCS for 120 days, starting within 12 h after birth, resulted in a potent carcinogenic response, characterized by a short latency time, some tumors being detectable after only 75 days, as well as by a high incidence of preneoplastic lesions and a high yield of benign lung tumors. Moreover, malignant tumors in the lung were detected within 7 months of life, sometimes even within 2–3 months, and malignant tumors also occurred in extrapulmonary organs.19 These results are being confirmed in further studies that are now in progress, using either Swiss albino or H mice or even the poorly sensitive DBA/2 and C57BL mouse strains. In contrast, the yield of “spontaneous” lung tumors is very low in sham–exposed mice belonging to these strains. In fact, only 4 of the 106 sham–exposed mice that we have analyzed so far had lung tumors (3.8%), while as many as 143 of 268 MCS–exposed mice (53.3%) displayed lung tumors of varying histopathological type, more often in lung sections rather than on the lung surface.19
Due to the high potency of the MCS–induced carcinogenic response, this model is expected to be suitable to study the protective effects of chemopreventive agents. Importantly, the chemoprevention studies can be designed to mimic different types of intervention in humans. In fact, in most studies using experimental systems the chemopreventive agents start to be administered before exposure to the carcinogen, which would be inappropriate in the case of CS. In the present model, using mice exposed to MCS since birth, the first option is to administer chemopreventive agents after weanling, as soon as the mice become capable of feeding themselves independently of their dams. This treatment mimics the situation in current smokers, in whom a possible damage to the organism and a possible cancer initiation may already have occurred. The second option is to administer chemopreventive agents after discontinuation of exposure to MCS, thereby mimicking the situation in ex–smokers, when tumors are under development and in some cases are already detectable at histopathological analysis. In this way, it is possible to evaluate whether chemopreventive agents are able to affect both early and advanced stages of the carcinogenesis process and, possibly, to determine the regression of benign tumors.
In order to validate these chemoprevention models, we used three well-known chemopreventive agents: budesonide, phenethyl isothiocyanate (PEITC), and N–acetyl–l–cysteine (NAC). All three agents were tested in current smokers, and the first two were also tested in ex–smokers. Budesonide, an anti-asthmatic drug, belongs to the family of glucocorticoids, which showed chemopreventive properties in animal models.20 PEITC is a naturally occurring compound contained in watercress (Nasturtium officinale), which is an effective inhibitor of NNK carcinogenesis in rodents.9 The aminothiol NAC, used both as a drug and a dietary supplement, is an analogue and precursor of l–cysteine and reduced glutathione (GSH), which exhibited cancer protective effects in rodents and in phase II clinical trials, although it was ineffective in a phase III trial (reviewed in ref. 21).
The results of the present study provide evidence not only that MCS is a potent inducer of lung tumors and other histopathological alterations in mice exposed since birth but also that this model is quite suitable to detect the protective effects of chemopreventive agents towards CS.
Material and methods
Mice
A total of 40 pregnant mice (strain H), originated from Swiss albino mice, were obtained from the Animal Laboratory of the National Center of Oncology (Sofia, Bulgaria). The animals were housed in Makrolon cages on sawdust bedding, and maintained on standard rodent chow. The animal room temperature was 23 ± 2°C, with a relative humidity of 55% and a 12 h day–night cycle. Housing, breeding and treatment of mice were in accordance with national and institutional guidelines.
Design of the study
The 433 neonatal mice born from 40 dams, which received no treatment during pregnancy, were divided into 7 groups. The number of mice composing each group and their subdivision by gender are shown in Tables 1 and 2. One group of neonatal mice was kept in filtered air (sham-exposed mice), while all other mice were exposed to MCS (MCS-exposed mice), starting within 12 h after birth and continuing daily for 120 consecutive days. After weanling (about 35 days), the mice were divided by gender and kept in separate cages, and 3 groups of MCS-exposed mice started to be treated with either NAC, budesonide or PEITC. Two other groups started to be treated with either budesonide or PEITC after discontinuing exposure to MCS (120 days). Treatment with the chemopreventive agents continued until the end of the experiment (210 days).
TABLE I. Survival and Body Weights of Variously Treated Neonatal H Mice.
| Survival after 210 days | Body weights (g) | |||
|---|---|---|---|---|
| Treatment | Gender | 120 days | 210 days | |
| Sham | M | 22/27 (81.4%) | 31.4 ± 0.83 | 32.9 ± 1.24 |
| F | 20/25 (80.0%) | 27.8 ± 0.73 | 29.3 ± 0.84 | |
| MCS* | M | 24/30 (80.0%) | 27.3 ± 0.782 | 30.9 ± 0.78 |
| F | 18/25 (72.0%) | 23.5 ± 0.862 | 26.8 ± 1.11 | |
| MCS* + NAC (current smokers)** | M | 16/22 (72.7%) | 27.9 ± 0.692 | 30.7 ± 1.36 |
| F | 16/23 (69.6%) | 24.6 ± 0.832 | 27.8 ± 1.24 | |
| MCS* + Budesonide (current smokers)** | M | 32/43 (74.4%) | 25.6 ± 0.892 | 32.8 ± 0.72 |
| F | 36/44 (81.8%) | 21.8 ± 0.972 | 26.4 ± 0.751 | |
| MCS* + PEITC (current smokers)** | M | 16/21 (76.2%) | 26.1 ± 0.832 | 30.1 ± 1.54 |
| F | 22/29 (75.9%) | 23.1 ± 1.112 | 27.2 ± 1.38 | |
| MCS* + Budesonide (ex-smokers)*** | M | 24/31 (77.4%) | 26.4 ± 0.712 | 31.7 ± 0.80 |
| F | 37/45 (82.2%) | 22.1 ± 1.072 | 24.7 ± 0.791 | |
| MCS* + PEITC (ex-smokers)*** | M | 25/29 (86.2%) | 23.4 ± 0.703,4 | 31.0 ± 0.92 |
| F | 32/39 (82.1%) | 21.7 ± 0.703 | 24.9 ± 0931 | |
Body weight values are means ± SE within each experimental group.
Exposure to MCS started immediately after birth and continued for 120 days.
Treatment with chemopreventive agents started after weaning (30-35 days) and continued until the end of the experiment (210 days).
Treatment with chemopreventive agents started after discontinuing exposure to MCS (120 days) and continued until the end of the experiment (210 days).
p < 0.05,
p < 0.01, and
p < 0.001 as compared with the corresponding Sham;
p < 0.001, as compared with the corresponding MCS.
Table II. Incidence and Multiplicity of Lung Tumors in Variously Treated Neonatal H Mice.
| Treatment | Gender | No. of mice | Tumor-bearing mice | Microadenomas | Adenomas | Carcinomas | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. (%) | Mean ± SE | No. (%) | Mean ± SE | No. (%) | Mean ± SE | No. (%) | Mean ± SE | |||
| Sham | M | 27 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| F | 25 | 1 (4.0%) | 0.05 ± 0.05 | 0 | 0 | 1 (4.0%) | 0.05 ± 0.05 | 0 | 0 | |
| M + F | 52 | 1 (1.9%) | 0.02 ± 0.02 | 0 | 0 | 1 (1.9%) | 0.02 ± 0.02 | 0 | 0 | |
| MCS* | M | 30 | 20 (66.7%) | 3.8 ± 0.76 | 13 (43.3%) | 2.3 ± 0.64 | 11 (36.7%) | 2.1 ± 0.68 | 3 (10.0%) | 0.10 ± 0.06 |
| F | 25 | 15 (60.0%) | 2.6 ± 0.72 | 9 (36.0%) | 2.2 ± 0.73 | 5 (20.0%) | 0.2 ± 0.08 | 3 (12.0%) | 0.12 ± 0.06 | |
| M + F | 55 | 35 (63.6%) | 3.3 ± 0.53 | 22 (40.0%) | 2.3 ± 0.48 | 16 (29.1%) | 1.2 ± 0.39 | 6 (10.9%) | 0.11 ± 0.04 | |
| MCS* + NAC (current smokers)** | M | 22 | 2 (9.1%)4 | 0.5 ± 0.463 | 0 (0%)4 | 03 | 2 (9.1%)2 | 0.6 ±0.462 | 1 (4.5%) | 0.05 ± 0.05 |
| F | 23 | 6 (26.1%)2 | 2.0 ± 0.80 | 2 (8.7%)2 | 0.7 ± 0.481 | 3 (13.0%) | 1.0 ± 0.61 | 0 (0%)1 | 01 | |
| M + F | 45 | 8 (17.8%)4 | 1.3 ± 0.473 | 2 (4.4%)4 | 0.3 ± 0.254 | 5 (11.1%)2 | 0.8 ± 0.38 | 1 (2.3%)1 | 0.02 ± 0.021 | |
| MCS* +Budesonide (current smokers)** | M | 43 | 10 (23.3%)4 | 1.3 ± 0.423 | 7 (16.3%)3 | 1.0 ± 0.411 | 2 (4.7%)4 | 0.2 ±0.133 | 3 (7.0%) | 0.07 ± 0.04 |
| F | 44 | 9 (20.5%)4 | 0.8 ± 0.333 | 3 (6.8%)3 | 0.5 ± 0.322 | 4 (9.1%) | 0.2 ± 0.09 | 0 (0%)2 | 02 | |
| M + F | 87 | 19 (21.8%)4 | 1.0 ± 0.274 | 10 (11.5%)4 | 0.7 ± 0.263 | 6 (6.9%)4 | 0.2 ±0.083 | 3 (3.4%)1 | 0.03 ± 0.021 | |
| MCS* + PEITC (current smokers)** | M | 21 | 4 (19.0%)4 | 1.1 ± 0.603 | 4 (19.0%)1 | 1.2 ± 0.58 | 3 (14.3%)1 | 0.6 ± 0.48 | 3 (14.3%) | 0.14 ± 0.08 |
| F | 29 | 10 (34.5%)1 | 1.4 ± 0.50 | 0 (0%)4 | 02 | 5 (17.2%) | 0.6 ± 0.37 | 2 (6.7%) | 0.07 ± 0.05 | |
| M + F | 50 | 14 (28.0%)4 | 1.3 ±0.383 | 4 (8.0%)4 | 0.5 ± 0.263 | 8 (16.0%) | 0.6 ± 0.29 | 5 (10.0%) | 0.10 ± 0.04 | |
| MCS*+ Budesonide (ex-smokers)*** | M | 31 | 8 (26.8%)3 | 0.7 ± 0.264 | 7 (22.6%)1 | 1.0 ± 0.232 | 5 (16.1%)1 | 0.4 ± 0.173 | 2 (6.5%) | 0.06 ± 0.04 |
| F | 45 | 10 (22.2%)3 | 1.1 ± 0.382 | 4 (8.9%)1 | 0.2 ± 0.163 | 5 (11.1%) | 0.7± 0.36 | 0 (0%)2 | 02 | |
| M + F | 76 | 18 (23.7%)3 | 0.9 ± 0.254 | 11 (14.5%)4 | 0.3 ± 0.124 | 10 (13.2%)2 | 0.5 ± 0.21 | 2 (2.6%)2 | 0.03 ± 0.022 | |
| MCS* + PEITC (ex-smokers)*** | M | 29 | 14 (48.3%) | 1.6 ± 0.492 | 16 (55.2%) | 1.6 ± 0.43 | 4 (13.8%)2 | 0.2 ± 0.133 | 1 (3.4%) | 0.03 ± 0.03 |
| F | 39 | 14 (35.9%)1 | 1.5 ± 0.47 | 6 (15.4%)1 | 0.8 ± 0.371 | 10 (25.6%) | 0.7 ± 0.32 | 1 (2.6%) | 0.03 ± 0.03 | |
| M + F | 68 | 28 (41.2%)2 | 1.6 ± 0.343 | 22 (32.4%) | 1.1 ± 0.292 | 14 (20.6%) | 0.5 ± 0.192 | 2 (2.9%)1 | 0.03 ± 0.02 | |
Exposure to MCS started immediately after birth and continued for 120 days.
Treatment with chemopreventive agents started after weaning (30-35 days) and continued until the end of the experiment (210 days).
Treatment with chemopreventive agents started after discontinuing exposure to MCS (120 days) and continued until the end of the experiment (210 days).
p < 0.1 (borderline to significance),
p < 0.05,
p < 0.01, and
p < 0.001, as compared with the corresponding MCS.
Exposure to MCS
A whole-body exposure of mice to MCS was achieved by placing the mice of each litter and their dam or, after weanling, groups of 9-12 mice of the same gender in 22.5 l sealed Makrolon chambers that were subsequently filled with MCS.22 Filter-tipped commercial cigarettes (Sredetz, Bulgartabac), having a declared content of 9 mg tar and 0.6 mg nicotine each and delivering 10 mg CO in the MCS of each cigarette, were used. MCS was generated by drawing 15 consecutive puffs, each of 60 ml and lasting 6 s, by means of a syringe connected with the exposure chamber. Each daily session of treatment with MCS involved 6 consecutive exposures, lasting 10 min each, with 1 min intervals, during which a total air change was made.19 The average concentration of total particulate matter in the exposure chambers was 723 mg/m3 air. In previous studies, this exposure method proved to be successful in inducing a variety of alterations in rodents, such as early histopathological changes,23 cytogenetic damage,24 biochemical alterations,25 adducts to both nuclear DNA and mitochondrial DNA,26 and hyperproliferation and apoptosis in pulmonary alveolar macrophages and bronchial epithelial cells.27
Treatment with chemopreventive agents
Budesonide and PEITC were purchased from Sigma Chemical Co. (St. Louis, MO) and were added to the diet at the concentrations of 2.4 and 1,000 mg/kg, respectively. These doses were selected based on a previous subchronic toxicity study, which showed that they did not affect body weight and did not cause any apparent sufference or alteration of behavior in Swiss albino mice treated for 6 weeks.28 NAC, in the form of a pharmaceutical preparation (Fluimucil, Zambon, Vicenza, Italy), was added to the drinking water at a concentration accounting for a calculated daily intake of 1,000 mg/kg body weight. This dose has been used in a number of previous studies.21 The chemopreventive agents were administered according to two different schedules. The first one, used for all three agents, involved their administration starting after weanling (approximately 4 weeks) and continuing until the end of the experiment at 210 days. The second schedule, used for budesonide and PEITC, involved their administration starting after having discontinued exposure to MCS at 120 days and continuing until the end of the experiment at 210 days.
Histopathological analyses
Moribund mice and all surviving mice after 210 days were deeply anesthesized with diethyl ether and killed by cervical dislocation. A complete necropsy was performed. Lungs, liver, stomach, kidney, urinary bladder, and all organs with suspected macroscopical lesions were fixed in 10% formalin, cut into standardized sections and subjected to standard histopathological analysis. In particular, the left lung was cut into 3 pieces. The accessory, cranial and middle lobes of the right lung were cut into 2 pieces each, and an additional section was obtained from the right lung caudal lobe. This accounted for a total of 10 sections to be analyzed microscopically. Six sections were analyzed per each kidney, and 4 sections per liver.
Statistical analyses
The yield of tumors and other lesions was expressed in terms of incidence and, in case of multiple tumors, of multiplicity. Multiplicity was calculated for all mice, both tumor bearing and non-tumor bearing, within each experimental group. Body weights and multiplicity data were expressed as means ± SE of the mice composing each experimental group, and comparisons between groups were made by Student's t-test for unpaired data. Comparisons between groups regarding survival and incidence were made by χ2 analysis.
Results
Survival and body weights
Survival and body weights of mice exposed to MCS and treated with chemopreventive agents are shown in Table I. At the end of the experiment (210 days), survival of the mice ranged between 72.0% and 86.2%, without any significant difference among the 7 experimental groups. Premature deaths were mainly due to pneumonias and leukemias, whose prevalence was not affected by treatments. After 120 days, when exposure to MCS was discontinued, the body weights were significantly decreased in both males and females exposed to MCS, irrespective of treatment with chemopreventive agents. A further significant decrease, as compared with MCS–exposed mice in the absence of chemopreventive agents, was observed in male ex–smokers treated with PEITC. After 210 days, the body weights were similar in the various experimental groups, excepting slight but significant decreases in female current smokers treated with budesonide and in female ex-smokers treated with either PEITC or budesonide, as compared with sham-exposed females.
Lung tumors
A total of 123 mice (58 males and 65 females) had lung tumors. In particular, 71 mice (47 males and 24 females) had microscopically detectable microadenomas, 60 mice (27 males and 33 females) had adenomas, and 19 mice (13 males and 6 females) had carcinomas, all of them diagnosed as bronchoalveolar carcinomas except one carcinoma in situ. In most cases the tumors were multiple, sometimes tending to confluence, while some adenomas and all carcinomas were large, invading a lobe or a whole lung. We refer to our previous papers (19, 33) for the microscopic appearance of the various histopathological types.
Fig. 1 shows at a glance the incidence of lung tumor-bearing mice and the multiplicity of lung tumors in the 7 experimental groups. Table II reports detailed incidence and multiplicity data and their statistical analysis. It should be noted that, within each group, the data for tumor bearing mice do not necessarily coincide with the sum of data for the individual tumor types because several mice had tumors of mixed histopathological nature. Exposure to MCS induced an impressive carcinogenic response. In fact, the 63.6% of MCS-exposed mice had lung tumors, and the 10.9% of them had carcinomas, while in sham-exposed mice only 1 adenoma was detected.
Fig. 1.
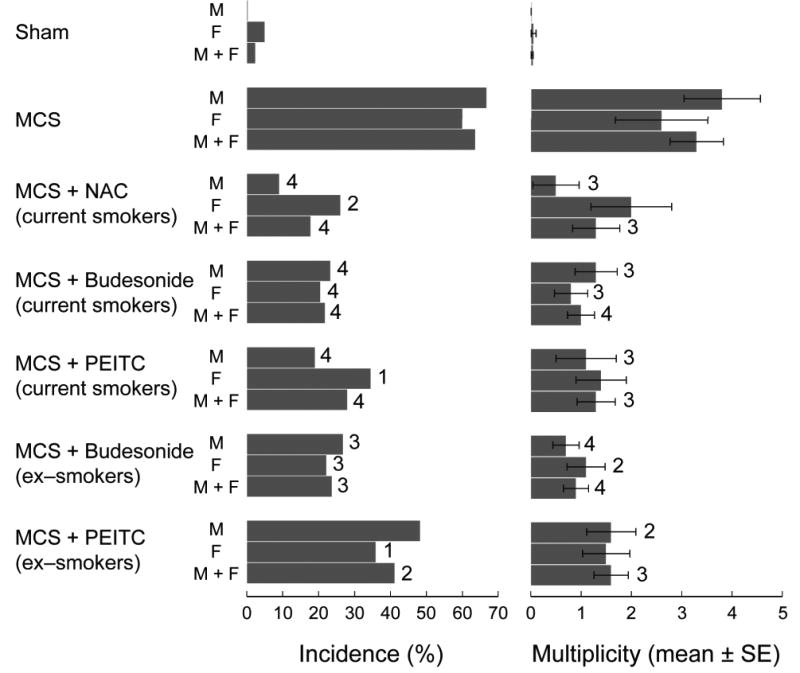
Incidence of lung tumor-bearing mice and multiplicity of total lung tumors as related to exposure of H mice for 120 days, starting within 12 h after birth, and treatment with the chemopreventive agents N-acetylcysteine (NAC), budesonide, or phenethyl isothiocyanate (PEITC). Treatment with the chemopreventive agents started either after weaning or after discontinuation of exposure to MCS, thus mimicking the situation either in current smokers or in ex-smokers, and lasted until the end of the experiment (210 days). Statistical analysis: 1p < 0.1 (borderline to significance), 2p < 0.05, 3p < 0.01, and 4p < 0.001, as compared with the corresponding MCS.
NAC, budesonide, and PEITC considerably decreased both incidence and multiplicity of lung tumors, when these agents started to be administered to mice after weanling in order to mimic the situation in current smokers. When budesonide and PEITC were administered after having discontinued exposure to MCS, in order to mimic the situation in ex-smokers, budesonide retained its cancer chemopreventive efficacy, which was comparable to that observed in current smokers. In contrast, PEITC lost a part of its protective activity, although in most cases the decreases in lung tumor yield were still statistically significant or borderline to statistical significance as compared with MCS-exposed mice in the absence of chemopreventive agents. It is noteworthy that the chemopreventive agents were able to inhibit all types of lung tumors, also including carcinomas. The protective effects were of the same order of magnitude in the two genders, although males were somewhat more sensitive than females to either NAC or PEITC administered after weaning, with intergender differences that were borderline to statistical significance (p = 0.07 for both NAC and PEITC).
Other histopathological alterations
In addition to lung tumors, exposure to MCS resulted in a significant increase of three other types of lesions. The first one was emphysema, consisting of rupture of alveolar walls, either focal or diffuse, with formation of spaces including at least 5 alveoli. Emphysema was absent in sham-exposed mice and present in the 16.4% of MCS-exposed mice (p < 0.01, as compared with sham). Of the chemopreventive agents tested, budesonide and PEITC decreased the incidence of emphysema, but not to a significant extent (data not shown).
The second lesion was hyperplasia of the alveolar epithelium, consisting of focal or diffuse thickening of alveolar epithelium with formation of at least 3 layers. This lesion affected the 3.8% of sham-exposed mice and the 29.1% of MCS-exposed mice (p < 0.01, as compared with sham). The incidence of this MCS-related lesion was decreased only by treatment with NAC, which lowered incidence to 13.3% (p = 0.06, as compared with sham).
The third lesion was hyperplasia of the urinary bladder epithelium, consisting of macroscopically appreciable thickening of the urothelium, confirmed microscopically as an increased proliferation of transitional epithelium, with formation of at least 10 layers of epithelial cells, sometimes with signs of dedifferentiation such as basophilic cytoplasm and hyperchromic nuclei. This kind of lesion, which was absent in sham-exposed mice, was detected in the 18.2% of MCS-exposed mice (p < 0.01, as compared with sham). Occurrence of this preneoplastic lesion was decreased to a significant extent in NAC-treated mice (4.4%, p < 0.05) and budesonide-treated mice, either current smokers (4.6%, p < 0.01) or ex-smokers (6.6%, p < 0.05), while the decrease observed in PEITC-treated mice, either current smokers (10.0%) or ex-smokers (14.7%), was not statistically significant.
A variety of other histopathological alterations that were detected in mice, including hyperplasia of the bronchial epithelium, pneumonia and abscesses in lung, leukemia, thymoma, and hyperplasia of thymus, were unrelated to exposure of mice to MCS, irrespective of treatment with the chemopreventive agents. The only exception was an increase of incidence of parenchymatous degeneration of the liver observed in MCS-exposed mice treated with both budesonide, either in current smokers (12.6%, p < 0.01 as compared with MCS only) or in ex-smokers (10.5%, p = 0.01), and PEITC, either in current smokers (16.0%, p < 0.01) or in ex-smokers (8.8%, p < 0.05). This lesion was not detectable in any MCS-exposed mouse, in the absence of chemopreventive agents. Furthermore, the only case of hepatocellular carcinoma among the 433 mice studied was detected in an MCS-exposed mouse treated with budesonide.
Discussion
The findings of the present study provide convincing evidence that it is possible to prevent lung cancer induced by CS in an animal model by means of dietary and pharmacological agents. The results obtained were clear-cut both in terms of carcinogenicity of MCS in neonatal mice and of cancer protective effects of the investigated chemopreventive agents. In particular, NAC, budesonide, and PEITC strikingly decreased the yield of MCS–induced lung tumors, both benign and malignant, in an experimental setting that mimicked an intervention in current smokers. When given after withdrawal of exposure to ECS, budesonide maintained its protective capacity, while PEITC lost part of its cancer chemopreventive activity. These findings are compatible with the known mechanisms of action of these agents. In fact, PEITC and other isothiocyanates modify the metabolism of carcinogens, such as NNK,29 and affect multigene expression in the lung of smoke–exposed rats.30 PEITC is also an inducer of apoptosis.31 Budesonide is a potent anti-inflammatory agent and is therefore expected to work also in advanced carcinogenesis stages, as it was previously demonstrated in A/J mice treated with B(a)P, in which this glucocorticoid inhibited all stages of tumor progression, from hyperplasia to cancer.32 Budesonide was also found to decrease the size of lung tumors, and reversed DNA hypomethylation and gene expression in lung tumors induced by vinyl carbamate in A/J mice.33 NAC has a variety of protective mechanisms in carcinogenesis, the primary one being its ability to scavenge reactive oxygen species (ROS) and other free radicals.21 Interestingly, NAC inhibited the carcinogenicity of MCS in neonatal mice also when it was administered only during pregnancy.34 In parallel, prenatal NAC inhibited genomic and post-genomic alterations occurring spontaneously at birth in mouse lung as a consequence of birth–related oxidative stress.35
This report shows that chemopreventive agents can determine a profound reduction in incidence and multiplicity of CS–induced lung cancer. Previous chemoprevention studies used mice exposed to ECS. Our recent study in Swiss albino mice exposed to ECS, starting at birth and continuing for 9 months, showed that a variety of early molecular and cytogenetical alterations are detectable immediately after weanling. In addition, 11 months after birth, a number of histopathological changes were detected in the lung, with a modest but significant increase of lung tumors. Both PEITC and budesonide, administered daily with the diet after weanling, protected the lungs from ECS–related histopathological alterations, including tumors.36 However, the bioassay using ECS in neonatal mice is much less effective than the one using MCS. In another experiment, using Swiss albino mice exposed to ECS throughout pregnancy, we found that administration of NAC decreased both incidence (3.7–fold) and multiplicity (4.5–fold) of lung tumors, but these differences did not reach the statistical significance threshold due to the small numbers of mice.16 A variety of chemopreventive agents were evaluated by Witschi and co–workers in A/J mice exposed to ECS for 5 months, followed by recovery in filtered air for an additional 4 months. However, the tumorigenic effect in this model is rather weak, and the narrow positivity window betweem sham–exposed and ECS–exposed mice renders evaluation of chemopreventive agents rather difficult. Using this bioassay, only the combination of myoinositol with the glucocorticoid dexamethasone significantly reduced the ECS–related lung tumor multiplicity.37 Interestingly, this protective effect was also detected when the compounds were administered after cessation of exposure to ECS.38 On the other hand, no protective effect could be detected with several other chemopreventive agents, including NAC, acetylsalicyclic acid, 1, 4–phenylenebis(methylene)selenocyanate, d–limonene, PEITC, either alone or in combination with benzyl isothiocyanate, Bowman–Birk protease inhibitor, green tea, and an aerosol of its major component epigallocatechin gallate.37,39,40 It is intriguing, however, that NAC and β–carotene were able to inhibit the tumorigenicity of the ECS gas phase,41 which is responsible for the carcinogenicity of the whole mixture.10
The mouse model used in the present study appears to be suitable not only to evaluate the efficacy of cancer chemopreventive agents but also to detect their possible toxicity. In fact, although the tested doses of budesonide and PEITC had preliminarily been shown to produce no alteration in body weight gain and general appearance for up to 42 days in smoke–free mice,36 the histopathological analyses performed after 210 days in MCS–exposed mice revealed some signs of hepatotoxicity of PEITC and especially of budesonide. Furthermore, treatment of MCS–exposed mice with PEITC and budesonide tended to decrease their body weights. Note that PEITC has been shown to induce ROS–mediated genotoxicity in vitro,42 while glucocorticoids are known to produce side effects in humans.20 At toxic doses, budesonide even induced liver tumors in rats.43
In conclusion, the proposed bioassay using mice exposed to MCS since birth, which in just 7 months leads to a high yield of both benign and malignant lung tumors, proved to be quite suitable for evaluating both efficacy and side effects of cancer chemopreventive agents of either dietary or pharmacological nature.
Acknowledgments
We thank Dr. Ilaria Righi for skillful assistance in preparation of this manuscript.
Grant sponsor: The Bulgarian Ministry of Education and Science. Grant sponsor: The U.S. National Cancer Institute; Grant number: N01-CN53301.
References
- 1.International Agency for Research on Cancer. Tobacco smoke and involuntary smoking IARC monographs on the evaluation of the carcinogenic risks to humans. Vol. 83. Lyon: IARC; 2004. [PMC free article] [PubMed] [Google Scholar]
- 2.Blot WJ, Fraumeni JF., Jr . Cancers of the lung and pleura. In: Schottenfeld D, Fraumeni JF Jr, editors. Cancer Epidemiology and Prevention. New York: Oxford University Press; 1996. pp. 637–65. [Google Scholar]
- 3.Winterhalder RC, Hirsch FR, Kotantoulas GK, Franklin WA, Bunn PA., Jr Chemoprevention of lung cancer – from biology to clinical reality. Ann Oncol. 2004;15:185–96. doi: 10.1093/annonc/mdh051. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society. Cancer Facts & Figures 2001. Atalanta: 2001. Tobacco use; pp. 4–5. [Google Scholar]
- 5.De Flora S, Quaglia A, Bennicelli C, Vercelli M. The epidemiological revolution of the 20th Century. FASEB J. 2005;19:892–7. doi: 10.1096/fj.04-3541rev. [DOI] [PubMed] [Google Scholar]
- 6.Hirsch FR, Lippman SM. Advances in the biology of lung cancer chemoprevention. J Clin Oncol. 2005;23:3186–97. doi: 10.1200/JCO.2005.14.209. [DOI] [PubMed] [Google Scholar]
- 7.van Zandwijk N. Chemoprevention in lung carcinogenesis--an overview. Eur J Cancer. 2005;41:1990–2002. doi: 10.1016/j.ejca.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Omenn GS. Chemoprevention of lung cancers: lessons from CARET, the beta-carotene and retinol efficacy trial, and prospects for the future. Eur J Cancer Prev. 2007;16:184–91. doi: 10.1097/01.cej.0000215612.98132.18. [DOI] [PubMed] [Google Scholar]
- 9.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–9. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 10.Witschi H. The complexities of an apparently simple lung tumor model: The A/J mouse. Exp Toxicol Pathol. 2005;57(1):171–81. doi: 10.1016/j.etp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Mertens VE. Zigarettenrauch, eine Ursache des Lungenkrebses. Z Krebsforsch. 1930;32:82. [Google Scholar]
- 12.Lorenz E, Stewart HL, Daniel JH, Warren S. The effects of breathing tobacco smoke on strain A mice. Cancer Res. 1943;3:123. [Google Scholar]
- 13.Hecht SS. Carcinogenicity studies of inhaled cigarette smoke in laboratory animals: old and new. Carcinogenesis. 2005;26:1488–92. doi: 10.1093/carcin/bgi148. [DOI] [PubMed] [Google Scholar]
- 14.Coggins CR. A minireview of chronic animal inhalation studies with mainstream cigarette smoke. Inhal Toxicol. 2002;14:991–1002. doi: 10.1080/08958370290084746. [DOI] [PubMed] [Google Scholar]
- 15.Witschi H. A/J mouse as a model for lung tumorigenesis caused by tobacco smoke: strengths and weaknesses. Exp Lung Res. 2005;31:3–18. doi: 10.1080/01902140490494959. [DOI] [PubMed] [Google Scholar]
- 16.De Flora S, D'Agostini F, Balansky R, Camoirano A, Bennicelli C, Bagnasco M, Cartiglia C, Tampa E, Longobardi M, Lubet RA, Izzotti A. Modulation of cigarette smoke-related end-points in mutagenesis and carcinogenesis. Mutat Res. 2003:523–524. 237–52. doi: 10.1016/s0027-5107(02)00340-8. [DOI] [PubMed] [Google Scholar]
- 17.Mauderly JL, Gigliotti AP, Barr EB, Bechtold WE, Belinsky SA, Hahn FF, Hobbs CA, March TH, Seilkop SK, Finch GL. Chronic inhalation exposure to mainstream cigarette smoke increases lung and nasal tumor incidence in rats. Toxicol Sci. 2004;81:280–92. doi: 10.1093/toxsci/kfh203. [DOI] [PubMed] [Google Scholar]
- 18.Hutt JA, Vuillemenot BR, Barr EB, Grimes MJ, Hahn FF, Hobbs CH, March TH, Gigliotti AP, Seilkop SK, Finc GL, Mauderly JL, Belinsky SA. Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways. Carcinogenesis. 2005;26:1999–2009. doi: 10.1093/carcin/bgi150. [DOI] [PubMed] [Google Scholar]
- 19.Balansky RM, Ganchev G, Iltcheva M, Steele VE, D'Agostini F, De Flora S. Potent carcinogenicity of cigarette smoke in mice exposed early in life. Carcinogenesis. 2007;28:2236–43. doi: 10.1093/carcin/bgm122. [DOI] [PubMed] [Google Scholar]
- 20.Wattenberg LW, Wiedmann TS, Estensen RD, Zimmerman CL, Galbraith AR, Steele VE, Kelloff GJ. Chemoprevention of pulmonary carcinogenesis by brief exposures to aerosolized budesonide or beclomethasone dipropionate and by the combination of aerosolized budesonide and dietary myo-inositol. Carcinogenesis. 2000;21:179–82. doi: 10.1093/carcin/21.2.179. [DOI] [PubMed] [Google Scholar]
- 21.De Flora S, Izzotti A, D'Agostini F, Balansky RM. Mechanisms of N–acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking–related end–points. Carcinogenesis. 2001;22:999–1013. doi: 10.1093/carcin/22.7.999. [DOI] [PubMed] [Google Scholar]
- 22.Balansky R, Blagoeva PM, Mircheva ZI. Investigation of the mutagenic activity of tobacco smoke. Mutat Res. 1987;188:13–9. doi: 10.1016/0165-1218(87)90109-1. [DOI] [PubMed] [Google Scholar]
- 23.Balansky R, D'Agostini F, Zanacchi P, De Flora S. Protection by N-acetylcysteine of the histopathological and cytogenetical damage produced by exposure of rats to cigarette smoke. Cancer Lett. 1992;64:123–31. doi: 10.1016/0304-3835(92)90072-4. [DOI] [PubMed] [Google Scholar]
- 24.Balansky R, D'Agostini F, De Flora S. Induction, persistence and modulation of cytogenetic alterations in cells of smoke-exposed mice. Carcinogenesis. 1999;20:1491–7. doi: 10.1093/carcin/20.8.1491. [DOI] [PubMed] [Google Scholar]
- 25.Bagnasco M, Bennicelli C, Camoirano A, Balansky R, De Flora S. Metabolic alterations produced by cigarette smoke in rat lung and liver, and their modulation by oral N-acetylcysteine. Mutagenesis. 1992;7:295–301. doi: 10.1093/mutage/7.4.295. [DOI] [PubMed] [Google Scholar]
- 26.Balansky R, Izzotti A, Scatolini L, D'Agostini F, De Flora S. Induction by carcinogens and chemoprevention by N-acetylcysteine of adducts to mitochondrial DNA in rat organs. Cancer Res. 1996;56:1642–7. [PubMed] [Google Scholar]
- 27.D'Agostini F, Balansky R, Izzotti A, Lubet RA, Kelloff GJ, De Flora S. Modulation of apoptosis by cigarette smoke and cancer chemopreventive agents in the respiratory tract of rats. Carcinogenesis. 2001;22:375–80. doi: 10.1093/carcin/22.3.375. [DOI] [PubMed] [Google Scholar]
- 28.D'Agostini F, Mastracci L, Izzotti A, Balansky R, Pennisi TM, Steele VE, De Flora S. Modulation by phenethyl isothiocyanate and budesonide of molecular and histopathological alterations induced by environmental cigarette smoke in mice. Cancer Prev Res. 2009;2:546–56. doi: 10.1158/1940-6207.CAPR-08-0235. [DOI] [PubMed] [Google Scholar]
- 29.Hecht SS. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J Nutr. 1999;129:768S–74S. doi: 10.1093/jn/129.3.768S. [DOI] [PubMed] [Google Scholar]
- 30.Izzotti A, Bagnasco M, Cartiglia C, Longobardi M, Camoirano A, Tampa E, Lubet RA, De Flora S. Modulation of multigene expression and proteome profiles by chemopreventive agents. Mutat Res. 2005;591:212–23. doi: 10.1016/j.mrfmmm.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 31.D'Agostini F, Izzotti A, Balansky RM, Bennicelli C, De Flora S. Modulation of apoptosis by chemopreventive agents. Mutat Res. 2005;591:173–86. doi: 10.1016/j.mrfmmm.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 32.Estensen RD, Jordan MM, Wiedmann TS, Galbraith AR, Steele VE, Wattenberg LW. Effect of chemopreventive agents on separate stages of progression of benzo[alpha]pyrene induced lung tumors in A/J mice. Carcinogenesis. 2004;25:197–201. doi: 10.1093/carcin/bgg196. [DOI] [PubMed] [Google Scholar]
- 33.Pereira M, Tao L, Liu Y, Li L, Steele VE, Lubet RA. Modulation by budesonide of DNA methylation and mRNA expression in mouse lung tumors. Int J Cancer. 2007;120:1150–3. doi: 10.1002/ijc.22468. [DOI] [PubMed] [Google Scholar]
- 34.Balansky R, Ganchev G, Iltcheva M, Steele VE, De Flora S. Prenatal N-acetylcysteine prevents cigarette smoke-induced lung cancer in neonatal mice. Carcinogenesis. 2009;30:1398–401. doi: 10.1093/carcin/bgp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izzotti A, Balansky RM, Camoirano A, Cartiglia C, Longobardi M, Tampa E, De Flora S. Birth related genomic and transcriptional changes in mouse lung. Modulation by transplacental N–acetylcysteine. Mutat Res Rev. 2003;544:441–9. doi: 10.1016/j.mrrev.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 36.D'Agostini F, Mastracci L, Izzotti A, Balansky R, Pennisi TM, Steele VE, De Flora S. Modulation by phenethyl isothiocyanate and budesonide of molecular and histopathological alterations induced by environmental cigarette smoke in mice. Cancer Prev Res. 2009;2:546–56. doi: 10.1158/1940-6207.CAPR-08-0235. [DOI] [PubMed] [Google Scholar]
- 37.Witschi H. Successful and not so successful chemoprevention of tobacco smoke-induced lung tumors. Exp Lung Res. 2000;26:743–55. doi: 10.1080/01902140150216792. [DOI] [PubMed] [Google Scholar]
- 38.Witschi H, Uyemiami D, Moran D, Espiritu I. Chemoprevention of tabacco smoke lung carcinogenesis in mice after cessation of smoke exposure. Carcinogenesis. 2000;21:977–82. doi: 10.1093/carcin/21.5.977. [DOI] [PubMed] [Google Scholar]
- 39.Witschi H, Espiritu I. Development of tobacco smoke-induced lung tumors in mice fed Bowman-Birk protease inhibitor concentrate (BBIC) Cancer Lett. 2002;183:141–6. doi: 10.1016/s0304-3835(02)00156-8. [DOI] [PubMed] [Google Scholar]
- 40.Witschi H, Espiritu I, Ly M, Uyeminami D, Morin D, Raabe OG. Chemoprevention of tobacco smoke-induced lung tumors by inhalation of an epigallocatechin gallate (EGCG) aerosol: a pilot study. Inhal Toxicol. 2004;16:763–70. doi: 10.1080/08958370490490400. [DOI] [PubMed] [Google Scholar]
- 41.Witschi H. Carcinogenic activity of cigarette smoke gas phase and its modulation by beta-carotene and N-acetylcysteine. Toxicol Sci. 2005;84:81–7. doi: 10.1093/toxsci/kfi043. [DOI] [PubMed] [Google Scholar]
- 42.Kassie F, Knasmüller S. Genotoxic effects of allyl isothiocyanate and phenethyl isothiocyanate (PEITC) Chem Biol Interact. 2000;127:163–80. doi: 10.1016/s0009-2797(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 43.Ryrfeldt A, Squire RA, Ekman L. Liver tumors in male rats following treatment with glucocorticoids. Toxicol Pathol. 1992;20:115–7. doi: 10.1177/019262339202000114. [DOI] [PubMed] [Google Scholar]