

Abstract
Nitroxyl or azanone (HNO) represents the redox-related (one electron reduced and protonated) relative of the well-known biological signaling molecule nitric oxide (NO). Despite the close structural similarity to NO, defined biological roles and endogenous formation of HNO remain unclear due to the high reactivity of HNO with itself, soft nucleophiles and transition metals. While significant work has been accomplished in terms of the physiology, biology and chemistry of HNO, important and clarifying work regarding HNO detection and formation has occurred within the last 10 years. This review summarizes advances in the areas of HNO detection and donation and their application to normal and pathological biology. Such chemical biological tools allow a deeper understanding of biological HNO formation and the role that HNO plays in a variety of physiological systems.
Keywords: nitroxyl, azanone, nitric oxide, hydrogen sulfide, nitroxyl donors, nitroxyl detection
1. Introduction
The discovery of nitric oxide (NO) as a biological signaling agent helped establish the importance of endogenously produced gaseous small molecules as modulators of numerous physiological processes.1–3 NO-mediated signaling plays widely-recognized roles in blood pressure and flow control, the immune response and neurotransmission and NO’s role in blood pressure control explains the mechanism of action of clinically long-used drugs like nitroglycerin.2,4–6 These initial findings triggered wide-ranging investigations to support and explore NO-based physiological signaling.4,7,8 For example, metallo-heme proteins, particularly soluble guanylate cyclase (sGC) in terms of smooth muscle relaxation, were identified as valid biological targets of NO that ultimately allowed the development of new clinical phosphodiesterase inhibitors that modulate NO’s actions.4,5,9–12 Protein thiols react with NO or its redox forms to yield S-nitrosothiols (RSNO) that control NO transport, preservation and form an NO-based post-translational modification.13,14 Other early work clearly defined the nitric oxide synthase (NOS) catalyzed oxidation of L-arginine to NO and L-citrulline as a pathway of NO biosynthesis and demonstrated the existence of this system in endothelial and neural tissue and macrophages.5,8,15–20 Numerous analytical methods either existed or were developed, including spectroscopic, electrochemical and chemiluminesence techniques, to measure NO levels allowing researchers to track and monitor this reactive signaling agent.21–24 Commercially available nitric oxide gas provides direct access to solutions of NO but many other NO donors, especially the diazenium diolates (NONOates) achieve more controlled NO delivery.25 The contributions of these basic studies in pharmacology, biochemistry, molecular biology, enzymology, chemistry and physics have driven the current understanding of NO signaling and its translation to physiology and medicine.
Nitroxyl or azanone (HNO) differs from nitric oxide (NO) only by the addition of a hydrogen atom (the formal one-electron reduction and protonation product of NO, Figure 1). Given the close structural similarity of HNO to NO, HNO initially gained significant consideration as a possible component of NO’s biological response or intermediate in NO biosynthesis.26–30 Despite the structural similarity, the high chemical reactivity of HNO has hindered the overall development of our understanding of HNO’s basic chemical and biological properties as compared to NO. HNO represents the smallest and simplest nitroso compound (RN= O, R = H for nitroxyl) and exhibits reactivity like other C-nitroso compounds as HNO reacts with itself to form a dimer (k = 8 × 106 M−1 s−1) that dehydrates to ultimately give nitrous oxide (Figure 1, N2O).31 This property alone requires nitroxyl to be introduced to systems by the use of donor molecules with Angeli’s salt (AS, Na2N2O3) and Piloty’s acid (PA, PhSO2NHOH) being the most commonly utilized. HNO’s self-reactivity also limits HNO detection and the identification of any endogenous generating systems. Nitroxyl also reacts with NO (k = 5.8 × 106 M−1 s−1) and oxygen (k = 3 × 103 M−1 s−1).32,33 Nitroxyl avidly reacts with soft nucleophiles, especially thiols, to yield either disulfides or sulfinamides and many of the observed biological effects of HNO appear to be mediated through the modification of protein thiols (Figure 1).34 Similarly, HNO reacts as a Lewis acid/base or redox partner with numerous metallo-proteins (including both ferrous and ferric heme groups, Figure 1) making both thiol and metal-containing proteins valid biological targets for HNO.32,35 While the stability of C-nitroso compounds and nitrous oxide indicates the accessibility of the formal +1 nitrogen oxidation state of HNO and suggests possible biological roles for HNO, the self-reactivity and the lack of HNO specific metabolites or protein modifications limits our overall understanding of its biological chemistry.
Figure 1.

The chemical biology of HNO
Despite these limitations, notable important discoveries and accomplishments regarding HNO’s chemistry and biology have been reported since the initial identification of NO as a gaseous signaling agent. A number of relatively recent excellent reviews summarize the knowledge to date regarding the chemistry, pharmacology, signaling aspects and physiology of HNO.26,27,29,32,33,35,36 Recently, two special issues (Antioxidants and Redox Signaling, 2011, 14 and the Journal of Inorganic Biochemistry, 2013, 118) have been devoted to various aspects of HNO biology and chemistry. Given this recent attention, the current review does not attempt comprehensive coverage but focuses on advances in the chemical biology in the detection and generation of HNO from within the last 10 years. Specifically, this review will address advances in HNO detection followed by advances in the discovery and development of new HNO sources, including both HNO donors and endogenous HNO formation, which relies heavily on sensitive and selective detection methods. Much progress has been made in the area of HNO’s biological chemistry that sets the stage for major advances in understanding HNO’s role in normal and disease state biology.
2. Advances in HNO Detection
The development of new HNO donors and a complete establishment of HNO’s biological profile requires fast and reliable HNO detection methods. Historically, frequently used methods to detect HNO included tracking the dimerization/dehydration product, N2O, by headspace gas chromatography, identification of sulfinamides that arise from HNO trapping of physiological thiols by high pressure liquid-chromatography mass spectrometry (HPLC-MS) and detection of the nitrosyl adducts of ferric heme proteins, such as FeII-NO myoglobin (MbNO) by ultravioletvisible (UV-Vis) or electron paramagnetic resonance (EPR) spectroscopy. These traditional analytical techniques suffer from either low sensitivity or selectivity and have been extensively reviewed.26,29 Recent intensive study has led to the development of several new detection strategies, including a series of fluorescent probes based on CuII complexes, triphenylphosphine derivatives or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and both electrochemical or mass spectrometric techniques.37–41 These new detection methods hold many advantages over the more traditional methods of HNO detection as each demonstrates greater selectivity and specificity for HNO giving much better confidence. The newer methods operate through distinct chemical or physical mechanisms providing a variety of choices depending on the particular situation. Finally, many of these new methods demonstrate the ability to detect HNO in cells or organisms allowing their application for the discovery of endogenous HNO formation or the role of HNO in biological processes.
2.1 Copper-based Fluorescent Probes
HNO reacts with metallo-proteins, especially heme proteins, via reductive nitrosylation forming the basis of HNO detection by the reaction of ferric heme proteins, such as metmyoglobin (metMb) to yield iron nitrosyl myoglobin (MbNO).42,43 Other examples of colorimetric HNO probes based on reductive nitrosylation include a group of MnIII porphyrins capable of distinguishing HNO from NO.44 This reactivity extends to CuII complexes with the first CuII based fluorescent HNO probe being a bithiophene-substituted poly(p-phenyleneethynylene) derivative (CP1) complexed to CuII that displays good emission enhancement upon exposure to both Angeli’s salt and NO (g).45
In 2010, Lippard developed the first small-molecule fluorescent probe selective for HNO, a CuII complex of a BODIPY reporter linked to a tripodal dipicolylamine-appended receptor via a triazole bridge (CuII[BOT1], Figure 2).46 The BODIPY dye has relatively long-wavelength absorption and emission properties suitable for cellular imaging and the metal free organic moiety BOT1 displays typical BODIPY optical properties with a maximum emission at 526 nm.46 Introduction of CuII immediately quenches the fluorescence via photoinduced electron transfer (PET) from the BODIPY singlet excited state to the bound CuII ion.46 Addition of excess AS to CuII[BOT1] under physiological conditions restores emission inducing a 4.3-fold fluorescence increase revealing HNO detection with significant emission turn-on.46 Further analysis suggests that this probe senses HNO in the 0.5 to 5 mM range and displays selectivity over other biological relevant species such as NO, NO3−, NO2−, ONOO−, H2O2 or OCl−.46,47 The probe detects HNO in biological systems as HeLa cells incubated with CuII[BOT1] show only faint intracellular fluorescence but introduction of AS induces an increase of intracellular fluorescence.46 Similar experiments with an NO donor do not enhance fluorescence.46
Figure 2.

Structure and mechanism of the HNO reaction for CuII[BOT1]
A rigid fluorescent moiety (BOT1), which can be quenched with CuII but not with CuI, represent a key design element of this probe, and direct coordination of BOT1 to CuII affords the non-fluorescent HNO probe. In the fluorescence turn-on process, HNO reduces CuII to CuI, restoring the emission of BOT1 and forming NO (Figure 2) and both electrochemical and quantum chemical studies support this mechanism.47,48 As this mechanism relies on the reducing power of HNO, other biological reductants may reduce CuII in these complexes yielding fluorescence and generating false positives. Treatment of CuII[BOT1] with cysteine restores the emission of BOT1 and positive ion electrospray mass spectrometry shows the reduced species as the CuI complex.46 The lack of a substantial fluorescence signal in HeLa cells incubated with CuII[BOT1] suggests normal levels of intracellular cysteine and other thiols do not reduce this complex.46
After the initial report, various improved CuII based fluorescent HNO probes have emerged (Figure 3).46,49–53 Yao reported CuII[COT1], a CuII-coumarin complex capable of HNO detection by both fluorescence and EPR spectroscopy (Figure 3).49 The same group designed CuII[COET] by replacing one pyridine moiety with an ester group to improve cell membrane permeability (Figure 3)50 and extends HNO detection to both living cells and vertebrate organisms. However, these fluorescent probes suffer from high-energy absorption and emission, and relatively short Stokes shifts. To overcome these limitations, Lippard prepared a CuII-benzoresorufin complex (BRNO1–3, Figure 3).51 The first near infrared emitting fluorescent HNO probe, CuII[DHX1] possesses improved selectivity (Figure 3) and does not react with NO and reduced sulfur species (Figure 3). Recently, a solid-phase prepared modular, lysine-based platform allows easy access to more versatile fluorescent moieties for CuII based HNO probes.53
Figure 3.

New CuII based fluorescent probes
2.2 Phosphine-based Fluorescent Probes
Nitroso compounds are electrophilic and react with various nucleophiles at the polar N=O bond. For example, triphenylphosphine reduces α-nitroso-β-napthol to the phosphine oxide and an azaylide.54 In addition to C-nitroso compounds, triphenylphosphine reacts with trityl S-nitrosothiol in a similar pattern, generating an S-azaylide and triphenylphosphine oxide.55 By viewing HNO as the simplest nitroso compound and given its reactivity with soft nucleophiles, our group reported the reaction of HNO with organic phosphines.56 Triphenylphosphine reacts with HNO generated from Angeli’s salt (2:1 ratio) to give equal amounts of triphenylphosphine oxide and an azaylide, a Staudinger reduction intermediate that quickly hydrolyzes to phosphine oxide.56 A similar reaction of a water-soluble phosphine, tris(4,6-dimethyl-3-sulfonatophenyl)phosphine trisodium salt hydrate (TXPTS), with HNO generates a stable azaylide product, serving as a useful HNO detection method.56 Nitroso compounds may react with phosphines via a hetero three-membered ring intermediate followed by another phosphine to produce the corresponding phosphine oxide and azaylide in a 1:1 ratio (Figure 4).56,57
Figure 4.

Trapping nitroso compounds with phosphine
Azaylides produced from these reactions are reactive nucleophilic intermediates that can undergo Staudinger ligation forming the basis of a new HNO detection strategy. An electrophilic ester adjacent to the phosphine allows the intramolecular nucleophilic attack from the azaylide, producing a stable and unique HNO-derived amide that can be tracked for HNO identification and quantification. Figure 4 depicts a triaryl phosphine trapping HNO through reductive Staudinger ligation with an appropriately positioned electrophilic group.56 Reaction of the phosphine with HNO first generates an equal amount of phosphine oxide and azaylide, which will be trapped by the adjacent electrophilic ester group forming a tetrahedral intermediate. The decomposition of this species yields a free alcohol and a phosphonium ion that hydrolyzes to benzamide phosphine oxide as the final product (Figure 4). The benzamide is a unique product from the process and represents a stable and chemically distinct HNO marker. The alcohol release provides a new perspective for the further design and development of colorimetric and fluorescent HNO probes. Based on this strategy, methyl 2-(diphenylphosphino)benzoate and its derivatives have been designed for successful detection of HNO with 31P NMR, UV-Vis spectroscopy or HPLC.56,58
Figure 5 depicts recently developed phosphine based fluorescent probes.40,59–67 These probes share structural similarities (with the exception of the replacement of the methyl ester with a fluorophore-derived ester) and the same basic chemical concept as described in Figure 4. Unlike the CuII based probes, phosphine probes are metal free, reductant resistant and in general demonstrate a greater fluorescence response. Nakagawa reported the first fluorescent probe (P-Rhod, Figure 5) using this strategy based on rhodol, a synthetic hybrid of fluorescein and rhodamine with excellent photophysical properties.40 Incubation of P-Rhod with excess AS in buffer leads to a significant fluorescence increase and parallel experiments demonstrate that P-Rhod possesses excellent selectivity for HNO over other biological species including physiological oxidants such as ONOO−, NO2−, NO3−, and H2O2 as well as biological reductants like H2S and ascorbic acid. P-Rhod detects HNO derived from various systems and demonstrates feasibility for cellular applications as HeLa cells loaded with P-Rhod immediately fluoresce upon introduction of HNO.
Figure 5.

Structure and mechanism of phosphorus based HNO probes
Following this report, several other phosphorus-based fluorescent HNO probes using the same strategy have been described. The coumarin-based fluorescent probe (P-CM, Figure 5) is much easier to prepare and has an improved sensitivity and linear response toward low HNO concentrations.61 The 1,8-naphthalimide-derived probe (P-Nap, Figure 5) shifts fluorescence from blue (418 nm) to green (546 nm) upon introduction of AS, allowing the ratiometric detection of HNO.60 A lysosomal-directed near-infrared probe (Lyso-JN, Figure 5) permits organelle specific HNO detection.59 Our group developed a modified fluorescein-based fluorescent probe suitable for detection of HNO in biological systems.65 This strategy has been extended to prepare several other probes including a FRET (coumarin/fluorescein pair) based ratiometric fluorescent probe (CF), a two-photon fluorescence turn-on probe (GCTPOC-1), a near-infrared fluorescent probe (Cyto-JN) and an excited state intramolecular proton transfer (ESIPT) based ratiometric fluorescent probe (HNO-HBT).62–64,66,67
Earlier reports indicate that phosphines react similarly with S-nitrosothiols leading to the concern that phosphorus-based probes may give false positives when used in more complicated biological systems in the presence of biological S-nitrosothiols.57,68 Experimental data demonstrates that the existing phosphine probes possess relatively good selectivity for HNO over GSNO.40,65 S-Azaylides derived from biologically-relevant S-nitrosothiols do not appear to effectively participate in the reductive ligations required for fluorescence turn-on.69,70 The S-azaylides derived from biological RSNOs all contain free amine and carboxylic acid groups and primarily yield phosphine oxides assuring a selective fluorescence response for HNO.70
2.3 TEMPOL-based fluorescent probes
HNO reacts with 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) through a hydrogen abstraction from HNO by the nitroxide radical (Figure 6).71 Based on this reaction, Toscano designed a nitroxide based prefluorescent probe 4-((9-acridinecarbonyl)amino)-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO-9-AC, Figure 6) that demonstrates weak fluorescence.38 Introduction of HNO from various sources transforms TEMPO-9-AC to the highly fluorescent TEMPO-9-AC-H and this probe displays selectivity for HNO over NO in aqueous solution.38 The use of TEMPO-9-AC may be complicated by intermolecular fluorescence quenching and competitive HNO trapping by the nascent NO (a potential drawback in the CuII probes as well) limiting its use in complicated biological environments.
Figure 6.

The reaction of HNO with TEMPOL and TEMPO-9-AC
2.4 Non-Fluorescent Approaches to HNO Detection
Membrane inlet (or introduction) mass spectrometry (MIMS), a technique for gas detection since 1963, also detects HNO. In 2011, Toscano re-engineered and modified the membrane inlet following the original design of Silverman.39 This specific analytical technique has been successfully applied to monitor HNO generated from AS and 2-bromo-N-hydroxybenzenesulfonamide (2-bromo-Piloty’s acid) in aqueous solution by detecting NO+, HNO+ and N2O+, the respective ions of the ions of NO, HNO and N2O.39 Careful control experiments distinguish these species and allow for HNO real-time HNO detection.39
Electrochemical HNO detection represents another area of rapid and extensive research and progress in the identification of HNO. In 2010, Doctorovich developed a gold electrode with a cobalt porphyrin covalently attached through Au-S bonds.37 The CoII 5,10,15,20-tetrakis[3-(p-acetylthiopropoxy) phenyl]-porphyrin modified electrodes (Co(P), Figure 7) were thoroughly characterized and electrochemical methods demonstrate its ability to discriminate HNO from NO.37,72 The cycle starts with the reaction of HNO with the electrode to yield CoIII(P)-NO− (Figure 7).37,72 Under these conditions, CoIII(P)-NO− is oxidized to CoIII(P)NO, which releases the NO ligand and returns to CoIII(P), allowing the start of a new cycle.37,72 This method allows time-resolved electrochemical quantification of HNO with a linear response in transient HNO concentration as low as 1 to 1000 nM.72 Control experiments indicate that several common biological species do not affect electrode performance, although the presence of O2 leads to a notable decrease in HNO concentration.72
Figure 7.

Reaction cycle in the amperometrical detection of HNO by Co (P)
3. Advances in HNO Sources
Despite the recent described interest in HNO biochemistry, pharmacology and physiology, endogenous production of HNO remains questioned and poorly understood. Compounds that release HNO demonstrate unique biological properties compared to NO donors, especially in the area of cardiac muscle contractility and show promise as new therapeutics for congestive heart failure. A need exists to both develop improved HNO donors for basic studies and therapeutics and to better understand endogenous HNO production.
3.1 HNO Donors
Currently, two popular strategies exist for HNO release: disproportionation/oxidation of hydroxylamine derivatives with good leaving groups attached to the nitrogen atom (type a, Figure 8) and decomposition of nitroso compounds (type b, Figure 8).73 For hydroxylamine derivatives (type a), the nitrogen atom possesses a formal −1 oxidation state and disproportionates to HNO (+1 oxidation state). Transformation of nitroso compounds (type b) to HNO usually occurs through hydrolysis or some type of displacement reaction. Traditional donors such as Angeli’s Salt, isopropylamine diazeniumdiolate (IPA/NO) and its analogs, Piloty’s acid and cyanamide are type a donors while acyl and acyloxy nitroso compounds release HNO via a type b mechanism. Improvements in HNO detection and a better understanding of both basic nitrogen and HNO chemistry have boosted the development of HNO generation systems. Inspired by the potential of endogenous HNO production and its promising pharmaceutical application, efforts have been made to generate physiologically useful donors/prodrugs that release HNO in a controlled, sustainable and tunable fashion.
Figure 8.
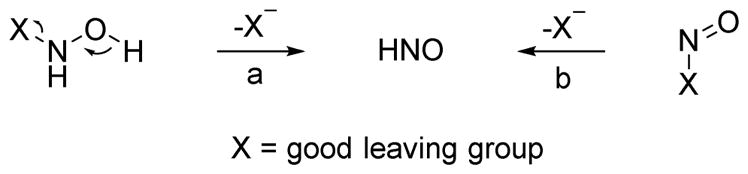
Chemical strategies for HNO generation
3.1.1 Diazeniumdiolate-based donors
Angeli’s Salt still remains the most widely used HNO donor and generates HNO and NO2− under physiological pH with a short half-life and releases NO in acidic media.73 IPA/NO, a primary amine-derived diazeniumdiolate, acts as a dual HNO and NO donor at neutral pH.73 AS is an easily synthesized inorganic salt that has not been successfully structurally modified but the organic character of IPA/NO allows the preparation of derivatives with versatile HNO releasing properties.
To achieve a prolonged HNO release profile, Miranda and Keefer reported a nitroxyl prodrug IPA/NO-AcOM (iPrHN-N(O)=NOCH2OAc, Figure 9), a stable, easily purified derivative from IPA/NO.74 Spontaneous hydrolysis of IPA/NO-AcOM generates HNO with a longer half-life than AS (t1/2 = 41 min, pH 7.4) and a negligible amount of NO.74 In the absence of esterase, IPA/NO-AcOM hydrolyzes and rearranges to produce HNO via an acyl nitroso intermediate.74 The extended release rate enhances the amount of trappable HNO due to limited self-dimerization.74 In the presence of an esterase, IPA/NO-AcOM directly dissociates to acetate, formaldehyde, and IPA/NO.74 This new donor displays a significantly improved ability to strengthen contraction of beating cardiac myocytes compared to IPA/NO.74 Inspired by IPA/NO-AcOM, a nonsteroidal anti-inflammatory drug, IPA/NO-aspirin, (Figure 9, t1/2=7.5 h, pH 7.4, no esterase) was developed that retains the anti-inflammatory properties of aspirin while reducing gastric toxicity.75 IPA/NO-aspirin exhibits significantly enhanced cytotoxicity compared to both parents toward non-small cell lung carcinoma cells but not normal endothelial cells, suggesting a potential application in cancer treatment.75 Knaus reported a series of O2-sulfonylethyl protected IPA/NO derivatives with half-lives that ranged from 6.6 to 17.1 h under physiological conditions.76 These stable non-explosive derivatives release the parent donor via base-induced β-elimination that subsequently and preferentially releases HNO.76 The 2-methylsulfonylethyl analog exhibits an antihypertensive effect and a significant in vitro inotropic effect by increasing contraction and relaxation without inducing a chronotropic effect (Figure 9).76 Besides primary amine based diazeniumdiolates, a secondary amine analog 1-(7-azabenzobicyclo[2.2.1]heptane)diazen-1-ium-1,2-diolate acts as a promising dual NO/HNO donor (t1/2=17.8 min) with significant in vitro inotropic anti-hypertensive effects has been reported and serves as a novel lead.77 A group of cyclic primary amine-based diazeniumdiolates (for example, AcOM-CPA/NO, Figure 9) release HNO with a wide-range of half-lives and hold promise in cancer cell targeting.78
Figure 9.

New IPA/NO type HNO prodrugs
3.1.2 Hydroxylamine derivatives
Piloty’s acid (PA) is another well-known HNO donor that finds less use in biological experiments due to its relatively low production of HNO at physiological pH. Structural modification of PA can greatly improve HNO release and a library of PA derivatives with various substitutions at different positions or based on different aromatic heterocycles have been reported.39,73,79,80 Some of these derivatives release significant amounts of HNO at neutral pH such as 2-bromo-PA and 2-nitro-PA (Figure 10).39,79,80 2-Bromo-PA is an easily prepared stable organic compound that only produces HNO under physiological conditions, properties that suggest its increased use as an HNO donor.39,79 2-Nitro-PA is a more efficient HNO donor due to electronic and steric effects.80 Besides aromatic substitutions, N-,O-diacylated or alkylated derivatives of PA have been prepared as HNO pro-drugs that first generate acyl nitroso compounds that release HNO through hydrolysis. (Figure 10).81 A library of N, O-bis-acylated N-hydroxysulfonamide derivatives with tunable half-lives (minutes - hours) demonstrate efficient HNO release at physiological pH (Figure 10).82 Knaus prepared ethanesulfohydroxamic acid esters of indomethacin, (S)-naproxen, and ibuprofen as potential clinical prodrugs (Figure 10).83 These compounds display diverse biological activities and show much higher NO release compared to other N-hydroxysulfonamides.
Figure 10.

New hydroxylamine derived HNO donors
Toscano reported a new unique series of highly tunable HNO donors based on N-substituted hydroxylamines with carbon-based leaving groups (Figure 10).84–86 The initial report reveals hydroxylamine-derived HNO donors based on Meldrum’s acid (t1/2=0.9 min), barbituric acid (t1/2=0.7 min) and pyrazolone (t1/2=9.5 min) that release HNO with high efficiency under physiological conditions without any enzymatic requirement.84 Further modification of (hydroxylamino)-barbituric acid (HABA) successfully extended the half-lives of this new group of HNO donors from 19 to 107 min.86 An extensive investigation of the (hydroxylamino)- pyrazolone (HAPY) series shows these donors quantitatively produce HNO with half-lives spanning from minutes to days under physiological conditions.86 In addition, this work shows the pyrazolone by-product reacts reversibly with HNO via an HNO-aldol reaction.85 These new structurally diverse HNO donors with extended half-lives hold promise as pharmacological tools and therapeutics.
3.1.3 Nitroso Compounds
Currently two type of nitroso compounds find use as HNO precursors, the acyl nitroso and the acyloxy nitroso compounds. Acyl nitroso compounds are highly reactive species and no stable acyl nitroso compounds have been successfully isolated.73 Traditional strategies for acyl nitroso compound formation include oxidation of hydroxamic acids or hydroxyureas by either chemical oxidants or enzymatic systems, such as catalase or horseradish peroxidase (HRP).87,88 Thermal decomposition of Diels-Alder adducts of acyl nitroso compounds and 9, 10-dimethylanthracene (9, 10-DMA) presents another popular pathway to prepare acyl nitroso compounds.89–92 Inspired by this approach, Nakagawa developed several photo cleavable HNO donors based on the retro-Diels-Alder reaction capable of controlled HNO release (Figure 11).93–95 The introduction of conjugated nitroaromatic groups to known hetero-Diels–Alder cycloadducts greatly enhance photoinduced HNO formation, which was confirmed by EPR and GC-MS analysis.93 Besides these 9,10-DMA hetero cycloadducts, nitrodiazo compounds, nitronates with α-leaving groups and 1,2,4-oxadiazole-4-oxides demonstrate acyl nitroso compound formation upon irradiation making these compounds potential HNO donors (Figure 11).96 Both HPLC and GC–MS analysis reveal photo degradation of nitrodiazo compounds yields quantitative HNO in acetonitrile/water mixtures while nitronates with α-leaving groups in methanol also release HNO upon photolysis.
Figure 11.

Photo controllable acyl nitroso sources
Acyloxy nitroso compounds represent another type of recently described HNO donor. Acid or base catalyzed cleavage of the ester group leads to an unstable nitroso alcohol intermediate that quickly decomposes to HNO and ketone (Figure 12).97 Gas chromatographic headspace analysis of 1-nitrosocyclohexyl acetate (NCA) in a mixture of methanol and neutral phosphate buffer indicates the formation of HNO.97 NCA is relatively stable under buffered conditions but quickly hydrolyzes under basic conditions.97 Interestingly, smaller ring-derived acyloxy nitroso compounds (cyclopentane or cyclobutane) do not hydrolyze to produce HNO but rearrange to cyclic hydroxamic acids.98,99 Acyloxy nitroso compounds can be easily obtained from direct oxidation of the corresponding oxime with lead tetra-acetate and the addition of an excess amount of the appropriate acid generates various esters.100,101 This approach allows structural modification to control HNO release as a function of the ease of hydrolysis of the ester group of the acyloxy nitroso compound.97 King engineered several cyclohexanone-derived acyloxy nitroso donors with half-lives ranging from milliseconds to days.97,102–104 Replacement of a methylene group with an oxygen atom in these compounds improves their water solubility allowing for more biological applications.103,104 Chemical analysis reveals acyloxy nitroso compounds also directly react with both small molecule and protein thiols to yield disulfides, suflinamides and sulfinic acids indicating that hydrolytically stable acyloxy nitroso compounds retain the ability to act as thiol modifying agents. 102–105
Figure 12.

Generation of HNO by acyloxy nitroso compounds
Acyloxy nitroso compounds also demonstrate a number of biological actions including vasodilation, enhanced cardiac contractility and the inhibition of platelet aggregation.26,102,104 Acyloxy nitroso compounds relax pre-constricted rat aorta with the rapid donors following a profile similar to AS and acyloxy nitroso compounds also activate soluble guanylate cyclase.97,102,106 Acyloxy nitroso compounds increase rat cardiac muscle contractility by modifying cardiac myofilament proteins by increasing both maximum force and calcium sensitivity in both intact and skinned muscle.107 A modified biotin switch procedure and modern mass spectrometry defined specific HNO-modified thiols with disulfides formed between Cys257 in actin and Cys190 in tropomyosin and Cys81 in myosin light chain and Cys37 in myosin heavy chain.107 These results provide an initial glimpse into how HNO-based structural modification alters cardiac contractility. Acyloxy nitroso compounds also reverse the isoflurane-induced depression of myocardial contractility108 and block the leukemia inhibitory factor (LIF) signaling in endothelial and cardiac myocytes indicating the redox sensitivity of the signal transducer and activator of transcription 3 (STAT3) pathway.108,109 Recent work shows these compounds limit left ventricular diastolic dysfunction in a mouse model of diabetes suggesting the possibility of long-term use of HNO donors in chronic diseases.110
3.1.4 Metal nitrosyl (MNO) complexes
Metallo-proteins play important roles in HNO chemistry and MNO complexes represent an alternative type of HNO-releasing agents. Harrop reported the first potentially useful MNO complex for HNO delivery.111 The {CoNO}8 complex [Co(LN4PhCl)(NO)] is relatively stable at physiological pH but addition of stoichiometric amounts of acid releases HNO that is trapped by [Fe(TPP)Cl] or Ph3P to afford the {FeNO}7 complex or Ph3P=O/Ph3P=NH, providing evidence for HNO release.111 A ruthenium complex has also been identified as a NO/HNO-donor in aqueous media.112 Trans-[Ru(NO)(NH3)P(O−)(OEt)2] (PF6)2, is stable in aqueous media over pH 3.0–7.5 but electrochemical activation of this complex at potentials of −0.50 and −0.80 V versus SCE releases NO/HNO with a half-life of 1.5 h (pH 7.5 and 25 °C) and such metal-based HNO donors comprise a structurally and mechanistically unique group.112
3.2 Potential Endogenous HNO Sources
Due to the previous limitations in HNO detection, only proposed pathways of endogenous HNO formation exist including: NOS-catalyzed reactions, oxidation of hydroxylamine-derivatives, reductions of NO, and the direct reaction of S-nitrosothiols with nucleophiles.113–115 The development of new methods of HNO detection in the last few years greatly aids the identification of endogenous HNO formation. Recently, the reaction of NO and hydrogen sulfide (H2S) has drawn attention as a possible pathway for HNO formation. Similar to NO, H2S acts as an endogenously produced gaseous biological signaling agent.3,116–118 Like NO, H2S demonstrates toxicity at higher concentrations but mediates numerous physiological processes associated with the cardiovascular, nervous and immune (inflammatory response) systems.116–120 Hydrogen sulfide forms during normal L-cysteine metabolism from the pyridoxal-5′-phosphate (PLP)-dependent cystathione β-synthetase (CBS) and cystathione γ-lyase (CSE) catalyzed reactions of cysteine, homocysteine, and cystathione or the action of 3-mercaptopyruvate sulfur transferase (MST) on 3-mercaptopyruvate.119,121,122 Hydrogen sulfide formation occurs in numerous tissues and excellent reviews regarding H2S chemistry and biology exist.119–125
Generally, H2S/HS− acts as a nucleophile and reducing agent and should form addition complexes with electrophilic organic compounds or nitrogen oxides and reduce NO or its oxidized forms (nitrite, nitrate, peroxynitrite or S-nitrosothiols) to lower oxidation state nitrogen oxides including HNO, thus altering the biological properties of the system.123,126 Given that H2S exists as the diamagnetic acid/base pair of H2S/HS− at physiological pH and the paramagnetic nature of NO, the direct reaction of H2S and NO appears unlikely.127–129 However, the reaction of thiols with NO yields radical complexes that decompose to various products depending on reaction conditions and gives a possible mechanism for HNO formation that parallels the recently reported proton assisted reduction of NO by alcohols (Figure 13).130 Calculations provide a bond dissociation energy of H2S = 90 kcal/mol making H atom abstraction by NO to form HNO (H-N bond dissociation energy 47 kcal/mol) thermodynamically unfavorable.123,131 Direct reaction of NO with H2S to give an S-nitrosothiol requires oxidation of either H2S to the HS• radical followed by coupling with NO or oxidation of NO to the nitrosonium ion (NO+) followed by reaction with −SH, reactions that both yield the simplest S-nitrosothiol, HSNO (thionitrous acid, Figure 13), similar to thiol nitrosation to generate S-nitrosothiols.127,128,132,133
Figure 13.

Potential reactions of H2S and NO
3.2.1 Reactions of NO with H2S
Early work by Moore shows the addition of H2S (as NaSH) to different NO donors scavenges released NO or blocks NO release and the addition of H2S to NO donors changes the expected NO-based biological function.134 Treatment of these reaction mixtures with HgCl2 or CuCl2 produces nitrite or NO and restores the expected biological function suggesting S-nitrosothiol formation.134 While not explicitly defined, a likely candidate for this species would be HSNO.134
Bian similarly reports that administration of NO donors and H2S (as NaSH) to cardiac myocytes elicits distinct responses compared to either the NO donor or NaSH alone.135 Specifically, NaSH does not affect myocyte contractility and three mechanistically distinct NO donors including sodium nitroprusside (SNP, 2Na+[Fe(CN)5NO]2−) decrease contractility (negative inotropic effect).135 Addition of both NaSH and the NO donors however yields an increase in myocyte contractility (positive inotropic effect).135 The SNP + NaSH system also increases the resting calcium level in the cell, which depends on intracellular calcium stores, suggesting increased calcium cycling within these cells.135 These results mimic the effects of HNO on cardiac myocytes leading to the suggestion that the reaction of NO and H2S generates HNO.34,135 Experiments using AS show an identical response to the mixture of SNP and H2S.135 The addition of various thiols blocks both the AS and the SNP + NaSH mixture response suggesting HNO intermediacy.135,136 Further experiments show that the effects of SNP and NaSH do not involve cGMP or cyclic adenylate monophosphate (cAMP)-mediated pathways.135 Given the previous evidence of S-nitrosothiol formation from the NO and H2S reaction, direct displacement of HSNO by H2S (a reduction) would in principle generate HNO and HSSH (Figure 14).114 These biological results strongly support HNO generation from the reaction of SNP and NaSH and provide the basis for the further chemical exploration of these speculative H2S-mediated pathways to HNO.
Figure 14.

Reactions of hydrogen sulfide with SNP
Hydrogen sulfide actively adds to SNP to form a red violet complex, the basis of the “Gmelin” test of sulfide.137 A re-examination of the H2S/HS− reaction by Olabe with the metal coordinated NO of SNP reveals initial addition of HS− to SNP produces a nitrosyl addition product with a pKa of 10.5.138 This product/anion pair decomposes to HS22− and [Fe(CN)5NO]3− radicals or further reacts with H2S to give addition products that ultimately yield NH3, N2O and HS2−, which may indicate HNO generation.138 By using a combination of various spectroscopic and chemical methods as well as pharmacological measures, Filipovic further defined HNO release from SNP upon reaction with H2S.139 Using a copper-based fluorescence HNO probe, these experiments show HNO generation in human umbilical vein endothelial cells (HUVECs) treated with both SNP and H2S.139 Control experiments do not show HNO formation in the presence of only SNP or H2S suggesting HNO formation arises from their reaction.139 Similarly, only the combination of SNP and H2S result in the release of calcitonin gene related peptide (CGRP), a potent vasodilator and known HNO marker in an isolated mouse heart model.139 Mechanistically, H2S adds to the coordinated nitrosonium ion of SNP to give a formally coordinated HNSO adduct that yields HNO and HSSH upon reduction by a second molecule of H2S (Figure 14).139 Overall, this group of studies indicates that addition of H2S to NO donors (especially SNP) changes both the chemical and biological outcome and clearly results in HNO formation in the case of SNP. Given the varied structures of available NO donors and potential variability of different biological systems, the generality of these reactions to produce HNO remains to be determined but this work clearly demonstrates the possibility of HNO formation through these pathways.
Using the recently developed HNO electrode for detection as well as following the disappearance of NO and H2S electrochemically, Filipovic recently showed the reaction of H2S and NO directly produces HNO.140 While the chemical mechanism for this conversion remains unclear, H2S may add to NO to form a radical complex that decomposes to HNO and a thiyl radical, similar to the proton coupled nucleophilic attack (Figure 13 and Section 3.2.3).130 Both the pKa of H2S and the ability of sulfur to stabilize radicals would support this pathway. Subsequent experiments indicate HNO formation in dorsal root ganglion (DRG) neurons using a copper-based HNO fluorescent probe from enzymatically generated NO and H2S.140 A basal level of fluorescence possibly suggests endogenous HNO formation in these cells that can produce both NO and H2S.140 Fluorescence decreases upon addition of inhibitors of either NOS or CBS inhibitors or by depleting the substrates L-arginine or L-cysteine revealing the requirement of both NO and H2S for HNO formation.140 The combination of NO and H2S also causes CGRP release in this system and these results provide some of the strongest evidence for endogenous HNO formation arising from the reaction of enzymatically generated NO and H2S.140 As hydrogen sulfide reduces the copper-based fluorescence probe yielding fluorescence in vitro, the use of such probes under situations that require H2S for HNO generation should warrant caution and perhaps a combination of HNO detection methods. The HNO generated in the DRG neurons reacts with the transient receptor potential channel A1 (TRPA1) to form specific disulfide bonds near the N-terminus of the protein that were identified biotin-switch technology and mass spectrometry.140 This protein modification results in an increase in calcium flux that leads to CGRP release and subsequent local and systemic vasodilation.140 This work provides some of the strongest evidence for endogenous HNO formation arising from the reaction of enzymatically generated NO and H2S and clearly defines an HNO-TRPA1-CGRP pathway that describes HNO’s biological effects.140 This work nicely shows the application of new HNO detection methods both for the discovery of endogenous HNO formation and elucidation of HNO biology.
3.2.2 Reactions of S-Nitrosothiols with H2S
Similar to the production of HNO from the reaction of NO and H2S, the reactions of S-nitrosothiols with H2S have garnered increasing attention as a pathway to HNO formation. The addition of H2S to any S-nitrosothiol should yield an initial N-hydroxysulfenamide adduct that could undergo trans-nitrosation to yield RSH and HSNO or decompose to RSSH and reduced nitrogen products through radical intermediates as previously described (Figure 15).141 Alternatively, direct displacement by the reaction of H2S with any S-nitrosothiol would yield HNO and RSSH (Figure 15).114 Trans-nitrosation provides a mechanism for the formation of HSNO whose basic chemistry and reactivity in terms of biology remains relatively unexplored.
Figure 15.

Reactions of H2S with S-nitrosothiols
In 2012, Filipovic reported the generation of HSNO via pulse radiolysis, treatment of H2S with acidified nitrite and the trans-nitrosation of glutathione S-nitrosothiol (GSNO) with H2S.142 The product of the last reaction was characterized by high resolution MS, IR and 15N NMR spectroscopy to provide evidence of HSNO formation.142 Further experiments show that HSNO decomposes, to yield NO, likely through S-N bond homolysis, and HNO as judged by methemoglobin trapping and identification of nitrous oxide and hydroxylamine.142 Treatment of HUVECs with GSNO and H2S yields HNO within the cells as judged by a copper-based fluorescent probe.142 Other experiments show that HSNO capably trans-nitrosates thiols of bovine serum albumin and hemoglobin within red blood cells identifying HSNO as a diffusible nitrosonium carrier.142 Filipovic extended this work showing that nitrite reacts with H2S to form HSNO and HNO in the presence of water soluble ferric iron porphyrin complexes.143 The combination of nitrite and H2S applied to HUVECs generates more NO (than nitrite alone) and HNO localized to the mitochondria.143 While HNO could arise from an oxygen atom transfer to H2S mechanism, the authors propose initial reduction of the ferric iron by H2S, binding and reduction of nitrite to give an FeIINO+ species that reacts with H2S to yield an HSNO adduct that subsequently decomposes to NO and HNO.143,144
The trans-nitrosation of any small molecule or protein S-nitrosothiol with H2S will yield an equilibrium mixture of HSNO, an extremely interesting species given the recent information regarding its ability to release NO, HNO and shuttle the nitrosonium group.142 HSNO represents the simplest S-nitrosothiol and the sulfur analog of nitrous acid and would be expected to be more acidic than HNO2 (pKa = 3.5) indicating the likely formation of thionitrite (−SNO) at physiological pH. Unlike nitrite, HSNO could exist as a tautomeric pair with HONS and both cis and trans stereoisomers of both HSNO and HONS theoretically exist (Figure 16).145–147 Both HSNO and HONS appear extremely unstable and have only been isolated by low temperature photolysis reactions of HNSO in an argon matrix and examined by infrared spectroscopy and theoretical calculations.145–147 Further theoretical calculations of HSNO indicate an S-N bond dissociation energy of 29.2 kcal/mol indicating the weakness of this bond and suggesting homolytic S-N bond cleavage to NO.148 These calculations also consider the structure as a combination of covalent non-charged, zwitterionic and ion pair species to predict S-nitrosothiol structure.148 Recent calculations indicate the formation of the tautomers HONS and SN(H)O from HSNO appear thermodynamically feasible and that a solvent assisted reaction of SN(H)O with H2S provides a theoretical pathway to HNO.149 The chemical reactivity of HSNO or SNOremains poorly described but many reactions appear possible including sulfur-nitrogen bond homolysis to yield NO, reactions with nucleophiles to yield HNO, hydrolysis to nitrite and various dimerization and disproportionation pathways to other products (Figure 16). These highly unstable species have not been clearly identified in biological systems but based on this chemistry may form during the reaction of H2S with S-nitrosothiols or other oxidized nitrogen oxides. Whether HSNO/NSO− can be generated or detected under biological conditions as well as its chemistry remains to be answered. New work shows the reaction of H2S with peroxynitrite generates sulfinyl nitrite [HS(O)NO], that was characterized by spectroscopic and computation methods and may act as a biological NO donor.150
Figure 16.

Reactivity of HSNO and its tautomers
Feelisch has also examined the chemistry and biology of the reaction of NO or S-nitrosothiols with H2S and found that similar to previous reports the mixture of NO and H2S elicits modified biological effects compared to either NO or H2S.151 The addition of H2S and NO to rat fibroblastoid like (RFL-6) cells alters sGC activity and cGMP formation in a concentration-dependent manner.151 At low ratios of H2S:NO, sGC activity is inhibited but enhanced at equimolar amounts of H2S to NO.151 Ultraviolet-visible spectroscopy suggests rapid formation of HSNO and in the presence of excess H2S, a yellow compound forms with an absorption at 412 nm that is attributed to nitrosopersulfide (−SSNO).151 Compared to HSNO, −SSNO displays relative stability and slowly decomposes to NO and polysulfides resulting in sustained sGC activation.151 Reasonable mechanisms for both HSNO and SSNO− formation are suggested that demonstrate the complexity of the intersection of nitrogen and sulfur chemistry. Feelisch and collaborators followed this initial report and further characterized the chemical products of the reaction from NO (generated from NO donors or S-nitrosothiols) and H2S by Uv-vis spectroscopy, mass spectrometry and various chemical methods and show that the three major bioactive products of the reaction include the above mentioned nitrosopersulfide (−SSNO), polysulfides (HSn−) and dintrososulfite (or N-nitrosohydroxylamine N-sulfonate), (SULFI/NO), a known sulfite-derived adduct of the NO dimer.152 Each of these products has specific biological attributes and further releases other bioactive species with SSNO− acting as a long-term NO donor capable of sGC activation leading to vasorelaxation.152 −SSNO also releases polysulfides but the exact biological effects of these compounds remain unclear.152 SULFI/NO acts as both an NO and HNO donor that does not significantly effect sGC activation/vasorelaxation but increases cardiac output, stroke volume and peak blood flow velocity, all indications of increased cardiac function and indicators of HNO release.152 Of interest in these studies is the inability to provide evidence of HSNO formation (by mass spectrometry) although they suggest HSNO as an intermediate in −SSNO formation.152
The recent intense work in this area indicates 1) that H2S modulates the biological effects of NO and 2) that reactions of NO or RSNOs and H2S generates HNO, explaining a portion of the modified biology. Clearly chemical and biological “crosstalk” between NO and H2S exists that likely includes nitroxyl but questions remain regarding the chemistry (Figure 17). The Filipovic group proposes the biological activity arises from NO and HNO formation from HSNO while the Feelisch group proposes that the biological activity arises from the formation of −SSNO (an NO donor) and SULFI/NO (an NO/HNO donor) suggesting that HSNO may only be an intermediate towards their formation (Figure 17). The biological redox chemistry of both nitrogen and sulfur is complex and their combination only further complicates these chemistries. HSNO appears a likely and logical candidate intermediate in these reactions but its history of only being isolated in a frozen matrix argues against its long-term stability and the lack of a pure standard decreases confidence in its characterization and identification. One study identifies HSNO by mass spectrometry while another fails to reveal this species highlighting the difficulty in working with such unstable compounds. Further work on HNO formation through these H2S-mediated pathways will be required to elucidate these pathways.
Figure 17.

Cross talk between H2S and S-nitrosothiol signaling
Alternatively, Filipovic recently synthesized an organic salt of −SSNO and confirmed its structure by X-ray crystallography, UV-vis, IR and 15N NMR spectroscopy in an organic solvent.153 A verified SSNO− sample allows a number of experiments to investigate its properties and reactivity.153 Particularly, −SSNO appears unstable in aqueous solutions and decomposes to disulfide ion and HNO.153 Contrary to other reports, SSNO− reacts with H2S and actually forms HSNO by trans-nitrosation.151–153 Taken together, these results cast doubt on the role and importance of −SSNO in the biological chemistry of these reactions. This work provides insight into the biological chemistry of −SSNO and further work will be required to completely clarify interaction of nitrogen and sulfur biochemistry and its potential for HNO generation. However, these reactions clearly generate HNO and the known biochemical generation of NO and H2S establish the basis of endogenous HNO production.
3.2.3 Proton Coupled Nitric Oxide Reduction
New exciting work shows the ability of biologically relevant alcohols to reduce NO to HNO through a process termed proton coupled nucleophilic attack (PCNA).130 Addition of various alcohols including ascorbic acid, tyrosine and hydroquinone to a solution of NO results in HNO formation as determined by electrochemical methods.130 Both nitrous oxide and nitrite form during these reactions presumably from HNO dimerization or the reaction of HNO with NO.130 EPR spectroscopy indicates the generation of the corresponding alkoxy radicals, especially for ascorbic acid.130 Aliphatic alcohols including methanol, D-mannitol and malic acid do not facilitate this transformation.130 Figure 18 depicts a proposed mechanism of HNO formation that involves a proton coupled nucleophilic attack of the alcohol to NO to yield an intermediate radical species that decomposes to the alkoxy radical, which can be trapped by excess NO, and HNO.130 Treatment of endothelial and macrophage cells that produce NO with excess ascorbic acid shows evidence of HNO formation by fluorescence and electrochemical detection, respectively.130 Extended work shows the ability of other biologically relevant phenols including vitamin E (tocopherol) and the drugs acetaminophen and salicylic acid facilitate the conversion of NO to HNO.154 Overall, these results provide a unique potential endogenous pathway for the conversion of NO to HNO in the presence of these “reducing” alcohols and theoretically make any NO source a source of HNO. Given the numerous systems capable of reducing nitrite (NO2−) to NO and the known conversion of dietary nitrate to nitrite by oral bacteria in humans and the subsequent chemical or enzymatic reduction of nitrite to NO, a large number of potential endogenous HNO generating systems appear possible.155
Figure 18.

HNO formation from proton coupled NO reduction
CONCLUSION
Nitroxyl (HNO) continues to emerge as a small molecule signaling agent with a distinct chemistry and biology from other recognized gaseous transmitters (NO, CO and H2S). This review describes recent chemical biological advances in both HNO detection and HNO donation/formation that provide a basis of our expanding understanding of HNO. The development of numerous mechanistically distinct HNO detection systems permits greatly improved specific and selective HNO detection and these methods are now routinely being used in various cell types and systems to define HNO-mediated pathways. The use of electrochemical and copper-based fluorescence detection methods point to at least one endogenous source of HNO formation, the reaction of enzymatically produced NO and H2S and suggest another in the reduction of NO by biologically relevant alcohols.130,140 These detection methods also verify HNO production from a group of new synthetic HNO donors allowing their confident use in a variety of experiments to discover new therapeutic agents. On-going studies outside the scope of this review continue to explore HNO’s role in basic cardiac physiology or define HNO’s reactivity with oxygen or new targets, such as cobalt-containing proteins or manganese quercetin dioxygenase.110,156–158 Reactions of HNO with the proteins that control hydrogen peroxide degradation reveals a “cross-talk” between HNO and hydrogen peroxide that may define new redox based signaling or gene expression pathways.159 The combination of the advances in HNO detection and donation with these emerging areas highlights the enormous potential of these new chemical biology tools to understanding HNO’s actions in biology, physiology and medicine.
Highlights.
New fluorescent, mass spectrometric and electrochemical HNO detection
New structurally varied HNO donors
Endogenous HNO formation from NO and H2S “cross-talk”
Better defined roles for HNO in specific biological processes
Acknowledgments
Work performed in the author’s laboratories was supported by the National Institutes of Health (HL62198) and Wake Forest University.
Abbreviations
- AS
Angeli’s Salt
- cAMP
cyclic adenylate monophosphate
- cGMP
cyclic guanylate monophosphate
- CBS
cystathione β-synthetase
- CSE
cystathione γ-lyase
- EPR
electron paramagnetic resonance
- GSH
glutathione
- GSNO
glutathione S-nitrosothiol
- GSSG
oxidized glutathione
- GSSH
glutathione persulfide
- LPS
lipo-polysaccharide
- MST
3-mercaptopyruvate sulfur transferase
- NO
nitric oxide
- NOS
nitric oxide synthase
- PLP
pyridoxal-5′-phosphate
- sGC
soluble guanylate cyclase
- SNP
sodium nitroprusside
- SCE
saturated calomel electrode
- NCA
1-nitrosocyclohexyl acetate
- TEMPO
2,2,6,6-tetramethyl-1-piperidinyloxy
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl
- MIMS
membrane inlet mass spectrometry
- Mb
myoglobin
- PA
Piloty’s acid
- HABA
(hydroxylamino)barbituric acid
- HAPY
(hydroxylamino)-pyrazolone
- DMA
dimethylanthracene
- TXPTS
tris(4,6-dimethyl-3-sulfonatophenyl)phosphine trisodium salt hydrate
- ESIPT
excited state intramolecular proton transfer
- HUVEC
human umbilical vein endothelial cells
- CGRP
calcitonin gene related peptide
- DRG
dorsal root ganglion
- TRPA1
transient receptor potential channel A1
- RFL
rat fibroblastoid like
- SULFI/NO
N-nitrosohydroxylamine N-sulfonate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palmer RM, Ferrige AG, Moncada S. Nature. 1987;327:524. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 2.Moncada S, Palmer RMJ, Higgs EA. Pharmacol Rev. 1991;43:109. [PubMed] [Google Scholar]
- 3.Li L, Hsu A, Moore PK. Pharmacol Ther. 2009;123:386. doi: 10.1016/j.pharmthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA. Antioxid Redox Sign. 2009;11:2717. doi: 10.1089/ars.2009.2721. [DOI] [PubMed] [Google Scholar]
- 5.Kerwin JF, Lancaster JR, Feldman PL. J Med Chem. 1995;38:4343. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 6.Murad F. Biosci Rep. 2004;24:452. doi: 10.1007/s10540-005-2741-8. [DOI] [PubMed] [Google Scholar]
- 7.Wink DA, Mitchell JB. Free Radical Biol Med. 1998;25:434. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 8.Bogdan C. Nat Immunol. 2001;2:907. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 9.Zamora R, Grzesiok A, Weber H, Feelisch M. Biochem J. 1995;312:333. doi: 10.1042/bj3120333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dierks EA, Burstyn JN. Biochem Pharmacol. 1996;51:1593. doi: 10.1016/0006-2952(96)00078-0. [DOI] [PubMed] [Google Scholar]
- 11.Cooper CE. Biochim Biophys Acta. 1999;1411:290. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 12.Ignarro LJ, Cirino G, Casini A, Napoli C. J Cardiovasc Pharmacol. 1999;34:879. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 13.Arnelle DR, Stamler JS. Arch Biochem Biophys. 1995;318:279. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 14.Hogg N. Ann Rev Pharmacol Toxicol. 2002;42:585. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 15.Ignarro LJ. Pharm Res. 1989;6:651. doi: 10.1023/a:1015926119947. [DOI] [PubMed] [Google Scholar]
- 16.Yoo J, Fukuto JM. Biochem Pharmacol. 1995;50:1995. doi: 10.1016/0006-2952(95)02098-5. [DOI] [PubMed] [Google Scholar]
- 17.Wigand R, Meyer J, Busse R, Hecker M. Ann Rheum Dis. 1997;56:330. doi: 10.1136/ard.56.5.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannon RO. Clin Chem. 1998;44:1809. [PubMed] [Google Scholar]
- 19.Stuehr DJ. Biochim Biophys Acta. 1999;1411:217. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 20.Hibbs JB. Res Immunol. 1991;142:565. doi: 10.1016/0923-2494(91)90103-p. [DOI] [PubMed] [Google Scholar]
- 21.Bryan NS, Grisham MB. Free Rad Biol Med. 2007;43:645. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujii S, Yoshimura T. Antioxid Redox Sign. 2000;2:879. doi: 10.1089/ars.2000.2.4-879. [DOI] [PubMed] [Google Scholar]
- 23.Jiang S, Cheng R, Wang X, Xue T, Liu Y, Nel A, Huang Y, Duan XF. Nat Commun. 2013;4 doi: 10.1038/ncomms3225. [DOI] [PubMed] [Google Scholar]
- 24.Hetrick EM, Schoenfisch MH. Annu Rev Anal Chem. 2009;2:409. doi: 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 26.Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. Pharmacol Ther. 2007;113:442. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Trends Pharmacol Sci. 2008;29:601. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Fukuto JM, Chiang K, Hszieh R, Wong P, Chaudhuri G. J Pharmacol Exp Ther. 1992;263:546. [PubMed] [Google Scholar]
- 29.Fukuto JM, Bartberger MD, Dutton AS, Paolocci N, Wink DA, Houk KN. Chem Res Toxicol. 2005;18:790. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- 30.Pufahl RA, Wishnok JS, Marletta MA. Biochemistry (Mosc) 1995;34:1930. doi: 10.1021/bi00006a014. [DOI] [PubMed] [Google Scholar]
- 31.Shafirovich V, Lymar SV. Proc Natl Acad Sci USA. 2002;99:7340. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miranda KM. Coord Chem Rev. 2005;249:433. [Google Scholar]
- 33.Fukuto JM, Dutton AS, Houk KN. Chem Bio Chem. 2005;6:612. doi: 10.1002/cbic.200400271. [DOI] [PubMed] [Google Scholar]
- 34.Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, Pagliaro P, Gao WD, van Eyk J, Kass DA, Wink DA, Paolocci N. Antioxid Redox Sign. 2011;14:1687. doi: 10.1089/ars.2010.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farmer PJ, Sulc F. J Inorg Biochem. 2005;99:166. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Fukuto JM, Bianco CL, Chavez TA. Free Rad Biol Med. 2009;47:1318. doi: 10.1016/j.freeradbiomed.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 37.Suarez SA, Fonticelli MH, Rubert AA, de la Llave E, Scherlis D, Salvarezza RC, Marti MA, Doctorovich F. Inorg Chem. 2010;49:6955. doi: 10.1021/ic1007022. [DOI] [PubMed] [Google Scholar]
- 38.Cline MR, Toscano JP. J Phys Org Chem. 2011;24:993. [Google Scholar]
- 39.Cline MR, Tu CK, Silverman DN, Toscano JP. Free Radical Biol Med. 2011;50:1274. doi: 10.1016/j.freeradbiomed.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Kawai K, Ieda N, Aizawa K, Suzuki T, Miyata N, Nakagawa H. J Am Chem Soc. 2013;135:12690. doi: 10.1021/ja404757s. [DOI] [PubMed] [Google Scholar]
- 41.Rivera-Fuentes P, Lippard SJ. Acc Chem Res. 2015;48:2927. doi: 10.1021/acs.accounts.5b00388. [DOI] [PubMed] [Google Scholar]
- 42.Bazylinski DA, Hollocher TC. J Am Chem Soc. 1985;107:7982. [Google Scholar]
- 43.Doyle MP, Mahapatro SN, Broene RD, Guy JK. J Am Chem Soc. 1988;110:593. [Google Scholar]
- 44.Marti MA, Bari SE, Estrin DA, Doctorovich F. J Am Chem Soc. 2005;127:4680. doi: 10.1021/ja044632n. [DOI] [PubMed] [Google Scholar]
- 45.Tennyson AG, Do L, Smith RC, Lippard SJ. Polyhedron. 2007;26:4625. [Google Scholar]
- 46.Rosenthal J, Lippard SJ. J Am Chem Soc. 2010;132:5536. doi: 10.1021/ja909148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Royzen M, Wilson JJ, Lippard SJ. J Inorg Biochem. 2013;118:162. doi: 10.1016/j.jinorgbio.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 48.Michael MA, Pizzella G, Yang L, Shi YL, Evangelou T, Burke DT, Zhang Y. J Phys Chem Lett. 2014;5:1022. doi: 10.1021/jz5002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Liu K, Li JY, Fang YA, Zhao TC, Yao C. Org Lett. 2011;13:1290. doi: 10.1021/ol103077q. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y, Yao YW, Li JY, Yao C, Lin BP. Sensor Actuat B-Chem. 2012;174:414. [Google Scholar]
- 51.Apfel UP, Buccella D, Wilson JJ, Lippard SJ. Inorg Chem. 2013;52:3285. doi: 10.1021/ic302793w. [DOI] [PubMed] [Google Scholar]
- 52.Wrobel AT, Johnstone TC, Liang AD, Lippard SJ, Rivera-Fuentes P. J Am Chem Soc. 2014;136:4697. doi: 10.1021/ja500315x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loas A, Radford RJ, Liang AD, Lippard SJ. Chem Sci. 2015;6:4131. doi: 10.1039/c5sc00880h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sidky MM, Soliman FM, Shabana R. Egypt J Chem. 1978;21:29. [Google Scholar]
- 55.Haake M. Tetrahedron Lett. 1972;13:3405. [Google Scholar]
- 56.Reisz JA, Klorig EB, Wright MW, King SB. Org Lett. 2009;11:2719. doi: 10.1021/ol900914s. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Xian M. Angew Chem Int Ed. 2008;47:6598. doi: 10.1002/anie.200801654. [DOI] [PubMed] [Google Scholar]
- 58.Reisz JA, Zink CN, King SB. J Am Chem Soc. 2011;133:11675. doi: 10.1021/ja203652z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jing XT, Yu FB, Chen LX. Chem Commun. 2014;50:14253. doi: 10.1039/c4cc07561g. [DOI] [PubMed] [Google Scholar]
- 60.Liu CY, Wu HF, Wang ZK, Shao CX, Zhu BC, Zhang XL. Chem Commun. 2014;50:6013. doi: 10.1039/c4cc00980k. [DOI] [PubMed] [Google Scholar]
- 61.Mao GJ, Zhang XB, Shi XL, Liu HW, Wu YX, Zhou LY, Tan WH, Yu RQ. Chem Commun. 2014;50:5790. doi: 10.1039/c4cc01440e. [DOI] [PubMed] [Google Scholar]
- 62.Liu CY, Cao ZM, Wang ZH, Jia P, Liu J, Wang ZK, Han BJ, Huang X, Li X, Zhu BC, Zhang XL. Sensor Actuat B-Chem. 2015;220:727. [Google Scholar]
- 63.Liu P, Jing XT, Yu FB, Lv CJ, Chen LX. Analyst. 2015;140:4576. doi: 10.1039/c5an00759c. [DOI] [PubMed] [Google Scholar]
- 64.Lv HM, Chen Y, Lei J, Au CT, Yin SF. Anal Methods. 2015;7:3883. [Google Scholar]
- 65.Miao Z, Reisz JA, Mitroka SM, Pan J, Xian M, King SB. Bioorg Med Chem Lett. 2015;25:16. doi: 10.1016/j.bmcl.2014.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang HT, Liu RC, Tan Y, Xie WH, Lei HP, Cheung HY, Sun HY. ACS Appl Mater Interfaces. 2015;7:5438. doi: 10.1021/am508987v. [DOI] [PubMed] [Google Scholar]
- 67.Zheng KB, Lin WY, Cheng D, Chen H, Liu Y, Liu KY. Chem Commun. 2015;51:5754. doi: 10.1039/c4cc10382c. [DOI] [PubMed] [Google Scholar]
- 68.Zhang D, Chen W, Miao Z, Ye Y, Zhao Y, King SB, Xian M. Chem Commun. 2014;50:4806. doi: 10.1039/c4cc01288g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR. J Am Chem Soc. 2013;135:7693. doi: 10.1021/ja401565w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miao Z, King SB. ChemistryOpen. 2016 doi: 10.1002/open.201500200. [DOI] [Google Scholar]
- 71.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc Natl Acad Sci U S A. 2003;100:9196. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suárez SA, Bikiel DE, Wetzler DE, Martí MA, Doctorovich F. Anal Chem. 2013;85:10262. doi: 10.1021/ac402134b. [DOI] [PubMed] [Google Scholar]
- 73.DuMond JF, King SB. Antioxid Redox Sign. 2011;14:1637. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andrei D, Salmon DJ, Donzelli S, Wahab A, Klose JR, Citro ML, Saavedra JE, Wink DA, Miranda KM, Keefer LK. J Am Chem Soc. 2010;132:16526. doi: 10.1021/ja106552p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basudhar D, Bharadwaj G, Cheng RY, Jain S, Shi S, Heinecke JL, Holland RJ, Ridnour LA, Caceres VM, Spadari-Bratfisch RC, Paolocci N, Velazquez-Martinez CA, Wink DA, Miranda KM. J Med Chem. 2013;56:7804. doi: 10.1021/jm400196q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang ZJ, Kaur J, Bhardwaj A, Alsaleh N, Reisz JA, DuMond JF, King SB, Seubert JM, Zhang YH, Knaus EE. J Med Chem. 2012;55:10262. doi: 10.1021/jm301303p. [DOI] [PubMed] [Google Scholar]
- 77.Bhardwaj A, Huang ZJ, Kaur J, Yang FH, Seubert JM, Knaus EE. Bioorg Med Chem Lett. 2013;23:2769. doi: 10.1016/j.bmcl.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 78.Bharadwaj G, Benini PGZ, Basudhar D, Ramos-Colon CN, Johnson GM, Larriva MM, Keefer LK, Andrei D, Miranda KM. Nitric Oxide. 2014;42:70. doi: 10.1016/j.niox.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toscano JP, Brookfield FA, Cohen AD, Courtney SM, Frost LM, Kalish VJ. 8674132 B2. US Patent. 2007
- 80.Aizawa K, Nakagawa H, Matsuo K, Kawai K, Ieda N, Suzuki T, Miyata N. Bioorg Med Chem Lett. 2013;23:2340. doi: 10.1016/j.bmcl.2013.02.062. [DOI] [PubMed] [Google Scholar]
- 81.Miranda KM, Nagasawa HT, Toscano JP. Curr Top Med Chem. 2005;5:649. doi: 10.2174/1568026054679290. [DOI] [PubMed] [Google Scholar]
- 82.Sutton AD, Williamson M, Weismiller H, Toscano JP. Org Lett. 2012;14:472. doi: 10.1021/ol203016c. [DOI] [PubMed] [Google Scholar]
- 83.Huang ZJ, Velazquez CA, Abdellatif KRA, Chowdhury MA, Reisz JA, DuMond JF, King SB, Knaus EE. J Med Chem. 2011;54:1356. doi: 10.1021/jm101403g. [DOI] [PubMed] [Google Scholar]
- 84.Guthrie DA, Kim NY, Siegler MA, Moore CD, Toscano JP. J Am Chem Soc. 2012;134:1962. doi: 10.1021/ja2103923. [DOI] [PubMed] [Google Scholar]
- 85.Guthrie DA, Ho A, Takahashi CG, Collins A, Morris M, Toscano JP. J Org Chem. 2015;80:1338. doi: 10.1021/jo502330w. [DOI] [PubMed] [Google Scholar]
- 86.Guthrie DA, Nourian S, Takahashi CG, Toscano JP. J Org Chem. 2015;80:1349. doi: 10.1021/jo5023316. [DOI] [PubMed] [Google Scholar]
- 87.Huang JM, Kim-Shapiro DB, King SB. J Med Chem. 2004;47:3495. doi: 10.1021/jm030547z. [DOI] [PubMed] [Google Scholar]
- 88.Huang JM, Sommers EM, Kim-Shapiro DB, King SB. J Am Chem Soc. 2002;124:3473. doi: 10.1021/ja012271v. [DOI] [PubMed] [Google Scholar]
- 89.Christie CC, Kirby GW, Mcguigan H, Mackinnon JWM. J Chem Soc Perk T 1. 1985:2469. [Google Scholar]
- 90.Corrie JET, Kirby GW, Laird AE, Mackinnon LW, Tyler JK. J Chem Soc Chem Comm. 1978:275. [Google Scholar]
- 91.Xu YP, Alavanja MM, Johnson VL, Yasaki G, King SB. Tetrahedron Lett. 2000;41:4265. [Google Scholar]
- 92.Zeng BB, Huang JM, Wright MW, King SB. Bioorg Med Chem Lett. 2004;14:5565. doi: 10.1016/j.bmcl.2004.08.062. [DOI] [PubMed] [Google Scholar]
- 93.Adachi Y, Nakagawa H, Matsuo K, Suzuki T, Miyata N. Chem Commun. 2008:5149. doi: 10.1039/b811985f. [DOI] [PubMed] [Google Scholar]
- 94.Nakagawa H. J Inorg Biochem. 2013;118:187. doi: 10.1016/j.jinorgbio.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 95.Matsuo K, Nakagawa H, Adachi Y, Kameda E, Tsumoto H, Suzuki T, Miyata N. Chem Commun. 2010;46:3788. doi: 10.1039/c001502d. [DOI] [PubMed] [Google Scholar]
- 96.Evans AS, Cohen AD, Gurard-Levin ZA, Kebede N, Celius TC, Miceli AP, Toscano JP. Can J Chem. 2011;89:130. [Google Scholar]
- 97.Sha X, Isbell TS, Patel RP, Day CS, King SB. J Am Chem Soc. 2006;128:9687. doi: 10.1021/ja062365a. [DOI] [PubMed] [Google Scholar]
- 98.Banerjee R, King SB. Org Lett. 2009;11:4580. doi: 10.1021/ol9018198. [DOI] [PubMed] [Google Scholar]
- 99.Hadimani MB, Mukherjee R, Banerjee R, Shoman ME, Aly OM, King SB. Tetrahedron Lett. 2015;56:5870. doi: 10.1016/j.tetlet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rehse K, Herpel M. Arch Pharm. 1998;331:111. doi: 10.1002/(sici)1521-4184(199803)331:3<111::aid-ardp111>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 101.Rehse K, Herpel M. Arch Pharm. 1998;331:104. doi: 10.1002/(sici)1521-4184(199803)331:3<104::aid-ardp104>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 102.Shoman ME, DuMond JF, Isbell TS, Crawford JH, Brandon A, Honovar J, Vitturi DA, White CR, Patel RP, King SB. J Med Chem. 2011;54:1059. doi: 10.1021/jm101432z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DuMond JF, Wright MW, King SB. J Inorg Biochem. 2013;118:140. doi: 10.1016/j.jinorgbio.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mohamed HAH, Abdel-Aziz M, Abuo-Rahma GEAA, King SB. Biorg Med Chem. 2015;23:6069. doi: 10.1016/j.bmc.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mitroka S, Shoman ME, DuMond JF, Bellavia L, Aly OM, Abdel-Aziz M, Kim-Shapiro DB, King SB. J Med Chem. 2013;56:6583. doi: 10.1021/jm400057r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miller TW, Cherney MM, Lee AJ, Francoleon NE, Farmer PJ, King SB, Hobbs AJ, Miranda KM, Burstyn JN, Fukuto JM. J Biol Chem. 2009;284:21788. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao WD, Murray CI, Tian Y, Zhong X, DuMond JF, Shen X, Stanley BA, Foster DB, Wink DA, King SB, Van Eyk JE, Paolocci N. Circ Res. 2012;111:1002. doi: 10.1161/CIRCRESAHA.112.270827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ding WG, Li ZT, Shen XX, Martin J, King SB, Sivakumaran V, Paolocci N, Gao WD. J Pharmacol Exp Ther. 2011;339:825. doi: 10.1124/jpet.111.185272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zgheib C, Kurdi M, Zouein FA, Gunter BW, Stanley BA, Zgheib J, Romero DG, King SB, Paolocci N, Booz GW. Plos One. 2012;7 doi: 10.1371/journal.pone.0043313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cao N, Wong YG, Rosli S, Kiriazis H, Huynh K, Qin CX, Du XJ, Kemp-Harper BK, Ritchie RH. Circ Heart Fail. 2015;8:572. doi: 10.1161/CIRCHEARTFAILURE.114.001699. [DOI] [PubMed] [Google Scholar]
- 111.Rhine MA, Rodrigues AV, Bieber Urbauer RJ, Urbauer JL, Stemmler TL, Harrop TC. J Am Chem Soc. 2014;136:12560. doi: 10.1021/ja5064444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Truzzi DR, Franco DW. Inorg Chim Acta. 2014;421:74. [Google Scholar]
- 113.Reisz JA, Bechtold E, King SB. Dalton Trans. 2010;39:5203. doi: 10.1039/c000980f. [DOI] [PubMed] [Google Scholar]
- 114.Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Biochemistry (Mosc) 1998;37:5362. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 115.Adak S, Wang Q, Stuehr DJ. J Biol Chem. 2000;275:33554. doi: 10.1074/jbc.M004337200. [DOI] [PubMed] [Google Scholar]
- 116.Gadalla MM, Snyder SH. J Neurochem. 2010;113:14. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mustafa AK, Gadalla MM, Snyder SH. Sci Signal. 2009;2 doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang GD, Wu LY, Jiang B, Yang W, Qi JS, Cao K, Meng QH, Mustafa AK, Mu WT, Zhang SM, Snyder SH, Wang R. Science. 2008;322:587. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Olson KR. Am J Physiol-Reg I. 2011;301:R297. doi: 10.1152/ajpregu.00045.2011. [DOI] [PubMed] [Google Scholar]
- 120.Olson KR. Antioxid Redox Sign. 2012;17:32. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caliendo G, Cirino G, Santagada V, Wallace JL. J Med Chem. 2010;53:6275. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- 122.Kabil O, Banerjee R. J Biol Chem. 2010;285:21903. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, Chen A, Wink DA. Chem Res Toxicol. 2012;25:769. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang R. Proc Natl Acad Sci U S A. 2012;109:8801. doi: 10.1073/pnas.1206646109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li HY, Shimizu H, Flinspach M, Jamal J, Yang WP, Xian M, Cai TW, Wen EZ, Jia QA, Wang PG, Poulos TL. Biochemistry (Mosc) 2002;41:13868. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 126.Fago A, Jensen FB, Tota B, Feelisch M, Olson KR, Helbo S, Lefevre S, Mancardi D, Palumbo A, Sandvik GK, Skovgaard N. Comp Biochem Phys A. 2012;162:1. doi: 10.1016/j.cbpa.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 127.Singh RJ, Hogg N, Joseph J, Kalyanaraman B. J Biol Chem. 1996;271:18596. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- 128.Keszler A, Zhang YH, Hogg N. Free Rad Bio Med. 2010;48:55. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Demaster EG, Quast BJ, Redfern B, Nagasawa HT. Biochemistry (Mosc) 1995;34:11494. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 130.Suarez SA, Neuman NI, Munoz M, Alvarez L, Bikiel DE, Brondino CD, Ivanovic--Burmazovic I, Miljkovic JL, Filipovic MR, Marti MA, Doctorovich F. J Am Chem Soc. 2015;137:4720. doi: 10.1021/ja512343w. [DOI] [PubMed] [Google Scholar]
- 131.Fukuto JM, Carrington SJ. Antioxid Redox Sign. 2011;14:1649. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- 132.Zhang YH, Hogg N. Free Rad Bio Med. 2004;36:947. doi: 10.1016/j.freeradbiomed.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 133.Keshive M, Singh S, Wishnok JS, Tannenbaum SR, Deen WM. Chem Res Toxicol. 1996;9:988. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- 134.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK. Biochem Biophys Res Commun. 2006;343:303. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 135.Yong QC, Hu LF, Wang SH, Huang DJ, Bian JS. Cardiovasc Res. 2010;88:482. doi: 10.1093/cvr/cvq248. [DOI] [PubMed] [Google Scholar]
- 136.Flores-Santana W, Salmon DJ, Donzelli S, Switzer CH, Basudhar D, Ridnour L, Cheng R, Glynn SA, Paolocci N, Fukuto JM, Miranda KM, Wink DA. Antioxid Redox Sign. 2011;14:1659. doi: 10.1089/ars.2010.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Butler AR, Calsyharrison AM, Glidewell C, Sorensen PE. Polyhedron. 1988;7:1197. [Google Scholar]
- 138.Quiroga SL, Almaraz AE, Amorebieta VT, Perissinotti LL, Olabe JA. Chem Eur J. 2011;17:4145. doi: 10.1002/chem.201002322. [DOI] [PubMed] [Google Scholar]
- 139.Filipovic MR, Eberhardt M, Prokopovic V, Mijuskovic A, Orescanin-Dusic Z, Reeh P, Ivanovic-Burmazovic I. J Med Chem. 2013;56:1499. doi: 10.1021/jm3012036. [DOI] [PubMed] [Google Scholar]
- 140.Deem S, Kim SS, Min JH, Eveland R, Moulding J, Martyr S, Wang XD, Swenson ER, Gladwin MT. Am J Physiol Heart Circ Physiol. 2004;287:H2561. doi: 10.1152/ajpheart.00310.2004. [DOI] [PubMed] [Google Scholar]
- 141.Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. Proc Natl Acad Sci U S A. 1996;93:14428. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Filipovic MR, Miljkovic JL, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanović-Burmazović I. J Am Chem Soc. 2012;134:12016. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miljkovic JL, Kenkel I, Ivanović-Burmazović I, Filipovic MR. Angew Chem Int Ed. 2013;52:12061. doi: 10.1002/anie.201305669. [DOI] [PubMed] [Google Scholar]
- 144.Heinecke JL, Khin C, Pereira JCM, Suárez SA, Iretskii AV, Doctorovich F, Ford PC. J Am Chem Soc. 2013;135:4007. doi: 10.1021/ja312092x. [DOI] [PubMed] [Google Scholar]
- 145.Tchir PO, Spratley RD. Can J Chem. 1975;53:2311. [Google Scholar]
- 146.Tchir PO, Spratley RD. Can J Chem. 1975;53:2318. [Google Scholar]
- 147.Tchir PO, Spratley RD. Can J Chem. 1975;53:2331. [Google Scholar]
- 148.Timerghazin QK, English AM, Peslherbe GH. Chem Phys Lett. 2008;454:24. [Google Scholar]
- 149.Ivanova LV, Anton BJ, Timerghazin QK. PCCP. 2014;16:8476. doi: 10.1039/c4cp00469h. [DOI] [PubMed] [Google Scholar]
- 150.Filipovic MR, Miljkovic J, Allgauer A, Chaurio R, Shubina T, Herrmann M, Ivanovic-Burmazovic I. Biochem J. 2012;441:609. doi: 10.1042/BJ20111389. [DOI] [PubMed] [Google Scholar]
- 151.Cortese-Krott MM, Fernandez BO, Santos JLT, Mergia E, Grman M, Nagy P, Kelm M, Butler A, Feelisch M. Redox Biol. 2014;2:234. doi: 10.1016/j.redox.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cortese-Krott MM, Kuhnle GGC, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, Nagy P, King SB, Saavedra JE, Keefer LK, Singer M, Kelm M, Butler AR, Feelisch M. Proc Natl Acad Sci U S A. 2015;112:E4651. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wedmann R, Zahl A, Shubina TE, Durr M, Heinemann FW, Bugenhagen BEC, Burger P, Ivanovic-Burmazovic I, Filipovic MR. Inorg Chem. 2015;54:9367. doi: 10.1021/acs.inorgchem.5b00831. [DOI] [PubMed] [Google Scholar]
- 154.Hamer M, Suarez SA, Neuman NI, Alvarez L, Munoz M, Marti MA, Doctorovich F. Inorg Chem. 2015;54:9342. doi: 10.1021/acs.inorgchem.5b01347. [DOI] [PubMed] [Google Scholar]
- 155.Lundberg JO, Weitzberg E, Gladwin MT. Nat Rev Drug Discov. 2008;7:156. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 156.Kumar MR, Zapata A, Ramirez AJ, Bowen SK, Francisco WA, Farmer PJ. Proc Natl Acad Sci U S A. 2011;108:18926. doi: 10.1073/pnas.1111488108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Subedi H, Brasch NE. Eur J Inorg Chem. 2015:3825. [Google Scholar]
- 158.Jorolan JH, Buttitta LA, Cheah C, Miranda KM. Nitric Oxide-Biology and Chemistry. 2015;44:39. doi: 10.1016/j.niox.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 159.Jackson MI, Fields HF, Lujan TS, Cantrell MM, Lin J, Fukuto JM. Arch Biochem Biophys. 2013;538:120. doi: 10.1016/j.abb.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]