

The crystal structures of three N-(arylsulfonyl)-4-fluorobenzamides, namely 4-fluoro-N-(2-methylphenylsulfonyl)benzamide, (I), N-(2-chlorophenylsulfonyl)-4-fluorobenzamide, (II), and N-(4-chlorophenylsulfonyl)-4-fluorobenzamide monohydrate, (III), are described and compared with related structures. The conformation of the three molecules is very similar with the aromatic rings being inclined to one another by 82.83 (11) and 85.01 (10)° in the two independent molecules of (I), 89.91 (10)° in (II) and 81.82 (11)° in (III).
Keywords: crystal structure, N-(arylsulfonyl)arylamides, N—H⋯O hydrogen bonds, O—H⋯O hydrogen bonds, C—H⋯O interactions
Abstract
The crystal structures of three N-arylsulfonyl-4-fluorobenzamides, namely 4-fluoro-N-(2-methylphenylsulfonyl)benzamide, C14H12FNO3S, (I), N-(2-chlorophenylsulfonyl)-4-fluorobenzamide, C13H9ClFNO3S, (II), and N-(4-chlorophenylsulfonyl)-4-fluorobenzamide monohydrate, C13H9ClFNO3S·H2O, (III), are described and compared with related structures. The asymmetric unit of (I) contains two independent molecules (A and B), while that of (II) contains just one molecule, and that of (III) contains a molecule of water in addition to one main molecule. The dihedral angle between the benzene rings is 82.83 (11)° in molecule A and 85.01 (10)° in molecule B of (I), compared to 89.91 (10)° in (II) and 81.82 (11)° in (III). The crystal structure of (I) features strong N—H⋯O hydrogen bonds between the A and B molecules, resulting in an R 4 4(16) tetrameric unit. These tetrameric units are connected into sheets in the bc plane by various C—H⋯O interactions, and adjacent sheets are further interlinked via C—H⋯πaryl interactions, forming a three-dimensional architecture. The crystal structure is further stabilized by πaryl–πaryl and S=O⋯πaryl interactions. In the crystal of (II), molecules are connected into R 2 2(8) and R 2 2(14) dimers via N—H⋯O hydrogen bonds and C—H⋯O interactions, respectively; the dimers are further interconnected via a weak C=O⋯πaryl interaction, leading to the formation of chains along [1-10]. In the crystal of (III), N—H⋯O and O—H⋯O hydrogen bonds involving both the main molecule and the solvent water molecule results in the formation of sheets parallel to the bc plane. The sheets are further connected by C—H⋯O interactions and weak C—Cl⋯πaryl, C—F⋯πaryl and S=O⋯πaryl interactions, forming a three-dimensional architecture.
Chemical context
Sulfonamide and amide moieties play a very significant role as key constituents in a number of biologically active molecules (Mohan et al., 2013 ▸; Manojkumar et al., 2013 ▸; Hamad & Abed, 2014 ▸). In recent years, N-(arylsulfonyl)arylamides have received much attention as they constitute an important class of drugs for Alzheimer’s disease (Hasegawa & Yamamoto, 2000 ▸), as well as antibacterial inhibitors of tRNA synthetases (Banwell et al., 2000 ▸), antagonists for angiotensin II (Chang et al., 1994 ▸) and leukotriene D4-receptors (Musser et al., 1990 ▸). Further, N-(arylsulfonyl)arylamides are known to be potent anti-tumour agents against a broad spectrum of human tumour xenografts (colon, lung, breast, ovary and prostate) in nude mice (Mader et al., 2005 ▸). In view of the importance of N-(arylsulfonyl)arylamides, the title compounds, (I), (II) and (III), were synthesized and we report herein on their crystal structures.
Structural commentary
The asymmetric unit of compound (I) contains two independent molecules (A and B) (Fig. 1 ▸), that differ slightly in their molecular conformations. The asymmetric unit of compound (II) (Fig. 2 ▸) contains one molecule, while compound (III) (Fig. 3 ▸) crystallizes as a water monosolvate. In molecules A and B of (I), the ortho-methyl substituent on the benzenesulfonyl ring is syn to the N—H bond in the central –C–SO2–N–C(O)– segment (Fig. 1 ▸). This is similar to the syn conformation observed for the N—H bond in the central –C–SO2–N–C(O)– segment with respect to the ortho-chloro substitution on the benzenesulfonyl ring of (II). The dihedral angle between the benzene rings is 82.83 (11)° in molecule A and 85.01 (10)° in molecule B of (I), compared to 89.91 (10)° in (II) and 81.82 (11)° in (III). Further, in (I) the dihedral angles between the benzoic acid ring and the central C8–C7(O3)–N1–S1 segment are 28.99 (1) and 23.81 (9)° in molecules A and B, respectively, while it is 10.41 (10)° in (II) and 21.23 (10)° in (III). The dihedral angles between the sulfonamide ring and the C7(O3)–N1–S1–C1 segment are, respectively, 68.67 (12) and 77.31 (10)° in molecules A and B of (I). The corresponding dihedral angle in (II) is 70.77 (11)°, whereas in (III) the value is much less, viz 48.03 (12)°. An intramolecular C14B–-H14B⋯O2B hydrogen bond (Fig.1 and Table 1 ▸) is observed in molecule B of (I), with an S(6) ring motif.
Figure 1.

A view of the molecular structure of the two independent molecules (A and B) of compound (I), with atom labelling. Displacement ellipsoids are drawn at the 50% probability level.
Figure 2.

A view of the molecular structure of compound (II), with atom labelling. Displacement ellipsoids are drawn at the 50% probability level.
Figure 3.

A view of the molecular structure of compound (III), with atom labelling. Displacement ellipsoids are drawn at the 50% probability level.
Table 1. Hydrogen-bond geometry (Å, °) for (I) .
Cg is the centroid of the fluorobenzene ring of molecule B of (I).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1A—H1A⋯O1B | 0.81 (3) | 2.12 (3) | 2.918 (2) | 167 (2) |
| N1B—H1B⋯O1A i | 0.83 (3) | 2.02 (3) | 2.828 (3) | 162 (3) |
| C6A—H6A⋯O3B | 0.93 | 2.57 | 3.313 (3) | 137 |
| C10B—H10B⋯O1B ii | 0.93 | 2.59 | 3.376 (2) | 143 |
| C10B—H10B⋯O3B ii | 0.93 | 2.46 | 3.215 (2) | 139 |
| C4B—H4B⋯Cg iii | 0.93 | 2.72 | 3.646 (2) | 173 |
| C14B—H14B⋯O2B | 0.96 | 2.45 | 3.058 (3) | 121 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii) 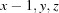 .
.
Supramolecular features
The crystal structure of (I), features two strong N—H⋯O hydrogen bonds, namely, N1A—H1A⋯O1B and N1B—H1B⋯O1A hydrogen bonds (Table 1 ▸) between the A and B molecules, resulting in a tetrameric unit (Fig. 4 ▸). The unitary level graph-set notation for each hydrogen bond is D(2), while in the second level the tetrameric unit has a graph-set motif of R 4 4(16). Adjacent tetramers are connected into sheets in the bc plane (Fig. 4 ▸) via C6A—H6A⋯O3B, C10B—H10B⋯O1B and C10B—H10B⋯O3B interactions (Table 1 ▸). Adjacent sheets are further interconnected via C4B—H4B⋯πaryl interactions (involving the centroid of the fluorobenzoyl ring of molecule B) (Fig. 5 ▸ and Table 1 ▸) to form chains along the a axis, so forming a three-dimensional architecture. The crystal structure of (I), is further stabilized by πaryl–πaryl interactions (Fig. 6 ▸) [Cg1⋯Cg2 = 3.7413 (12) Å; Cg1 and Cg2 are the centroids of the fluorobenzoyl rings of molecules A and B, respectively] and also by weak S1A=O2A⋯πaryl interactions [O⋯Cg3 = 3.7991 (19) Å; Cg3 is the centroid of the benzenesulfonyl ring of molecule B].
Figure 4.

Crystal packing of (I), displaying N—H⋯O hydrogen bonds and C—H⋯O intermolecular interactions (dashed lines), which result in the formation of sheets parallel to the bc plane.
Figure 5.

C—H⋯πaryl interactions (dashed lines) displayed in (I).
Figure 6.

π–π interactions (dashed lines) displayed in (I).
In the crystal of (II), molecules are connected into 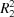 (8) dimers via N1—H1⋯O2 hydrogen bonds (Fig. 7 ▸
a and Table 2 ▸), and the dimers are further interconnected via C13—H13⋯O2 interactions (Fig. 7 ▸
a and Table 2 ▸) with an (14) graph-set motif. Weak C7=O3⋯πaryl interactions [O⋯Cg = 3.9157 (19) Å; Cg is the centroid of the fluorobenzoyl ring] connect these dimers, thus forming a one-dimensional architecture (Fig. 7 ▸
b).
(8) dimers via N1—H1⋯O2 hydrogen bonds (Fig. 7 ▸
a and Table 2 ▸), and the dimers are further interconnected via C13—H13⋯O2 interactions (Fig. 7 ▸
a and Table 2 ▸) with an (14) graph-set motif. Weak C7=O3⋯πaryl interactions [O⋯Cg = 3.9157 (19) Å; Cg is the centroid of the fluorobenzoyl ring] connect these dimers, thus forming a one-dimensional architecture (Fig. 7 ▸
b).
Figure 7.

Crystal packing of (II): (a)display of (8) and (14) dimers formed via N—H⋯O hydrogen bonds and C—H⋯O interactions, both shown as dashed lines; (b) formation of one-dimensional architecture.
Table 2. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O2i | 0.82 (3) | 2.16 (3) | 2.968 (2) | 172 (2) |
| C13—H13⋯O2i | 0.93 | 2.40 | 3.194 (2) | 144 |
Symmetry code: (i) 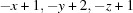 .
.
In the crystal of (III), molecules are connected via bridging water molecules, through strong N1—H1⋯O4, O4—H1O4⋯O1, O4—H2O4⋯O2 and O4—H2O4⋯O3 hydrogen bonds (Table 3 ▸), resulting in the formation of sheets parallel to the bc plane (Figs. 8 ▸ and 9 ▸). The sheets are further connected by C5—H5⋯O1 interactions, forming C6 chains (Table 3 ▸) running parallel to the c axis (Fig. 10 ▸). The crystal structure is also stabilized by several weak C—H⋯π interactions, C4—Cl1⋯Cg1 [Cl⋯Cg1 = 3.7513 (11) Å], C11—F1⋯Cg2 [F1⋯Cg2 = 3.8674 (17) Å] and S1=O2⋯Cg1 interactions [O2⋯Cg1 = 3.2039 (17) Å] (Cg1 and Cg2 are the centroids of the benzenesulfonyl ring and fluorobenzoyl rings, respectively), forming a complex three-dimensional architecture (Fig. 11 ▸).
Table 3. Hydrogen-bond geometry (Å, °) for (III) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O4 | 0.83 (3) | 1.91 (3) | 2.733 (3) | 171 (2) |
| O4—H1O4⋯O1i | 0.79 (3) | 2.25 (3) | 2.884 (2) | 138 (3) |
| O4—H2O4⋯O2ii | 0.82 (2) | 2.29 (3) | 2.955 (2) | 139 (3) |
| O4—H2O4⋯O3ii | 0.82 (2) | 2.16 (3) | 2.841 (2) | 141 (3) |
| C5—H5⋯O1iii | 0.93 | 2.54 | 3.124 (3) | 121 |
Symmetry codes: (i)  ; (ii)
; (ii) 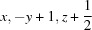 ; (iii)
; (iii) 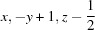 .
.
Figure 8.

Crystal packing of (III), displaying an infinite two-dimensional sheet parallel to the bc plane formed via N—H⋯O and various O—H⋯O hydrogen bonds (dashed lines).
Figure 9.

Crystal packing of (III) when viewed along the b axis; adjacent two-dimensional sheets are seen.
Figure 10.

Display of C5—H5⋯O1 C(6) chains (dashed lines) running parallel to the c axis in (III).
Figure 11.

Display of various weak interactions (dashed lines) in the crystal structure of (III).
Database survey
A search of the Cambridge Structural Database (CSD, Version 5.37, last update February 2016; Groom & Allen, 2014 ▸) for similar compounds viz N-(arylsulfonyl)-4-(substituted)benzamides, gave 14 hits. These fourteen compounds along with the three title compounds, (I)–(III), are grouped into three series; series 1: N-(2-methylphenylsulfonyl)benzamide, N-(2-methylphenylsulfonyl)-4-(chloro/methyl/nitro/methoxy)benzamides and (I), series 2: N-(2-chlorophenylsulfonyl)benzamide, N-(2-chlorophenylsulfonyl)-4-(chloro/methyl/nitro/methoxy)benzamides and (II), and series 3: N-(4-chlorophenylsulfonyl)benzamide, N-(4-chlorophenylsulfonyl)-4-(chloro/methyl/nitro)benzamides and (III).
Series 1: In series 1 (Table 4 ▸), the asymmetric units of three compounds, namely, N-(2-methylphenylsulfonyl)benzamide (Suchetan et al., 2010d ▸), N-(2-methylphenylsulfonyl)-4-nitrobenzamide (Suchetan et al., 2011b ▸) and N-(2-methylphenylsulfonyl)-4-methoxybenzamide (Sreenivasa et al., 2014a ▸) contain one molecule, while those of N-(2-methylphenylsulfonyl)-4-chlorobenzamide (Suchetan et al., 2010e ▸), N-(2-methylphenylsulfonyl)-4-methylbenzamide (Gowda et al., 2010a ▸) and N-(2-methylphenylsulfonyl)-4-fluorobenzamide (I) contain two molecules. In all of the compounds of series 1, the conformation of the ortho-methyl group on the benzenesulfonyl ring is syn to the N—H bond in the central –C–SO2--N–C(O)– segment. The values of the dihedral angle between the two aromatic rings in the molecules of series 1 fall in the range 73.9 (1)– 89.4 (1)°, the smallest dihedral angle being in N-(2-methylphenylsulfonyl)benzamide and the largest in N-(2-methylphenylsulfonyl)-4-chlorobenzamide (Table 4 ▸). Comparison of the intermolecular interactions displayed in the crystal structures of compounds in this series reveals that, except for the methoxy- and fluoro-substituted compounds, the crystal structures all display N—H⋯O(S) hydrogen bonds, while the methoxy- and fluoro-substituted compounds display other weak interactions of the type C—H⋯O, C—H⋯πaryl, πaryl–πaryl in addition to the N—H⋯O(S) hydrogen bonds. However, except for compound (I), all the compounds display one-dimensional supramolecular chains, whereas in (I), the supramolecular architecture is three-dimensional.
Table 4. Comparison of various parameters (°) in the crystal structures of series 1: N-(2-methylphenylsulfonyl)-para-substituted-arylamides.
| Parameters | H | Cl | CH3 | NO2 | OCH3 | F |
|---|---|---|---|---|---|---|
| Crystal System | Orthorhombic | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Z′ | 1 | 2 | 2 | 1 | 1 | 2 |
| Orientation of 2-CH3 group to the N—H bond | syn | syn, syn | syn, syn | syn | syn | syn, syn |
| Angle between aromatic rings | 73.9 (1) | 89.4 (1), 82.4 (1) | 88.1 (1), 83.5 (1) | 83.8 (2) | 80.81 (1) | 82.83 (11), 85.01 (10) |
| Intermolecular interactions | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S), C—H⋯O(S), π–π | N—H⋯O(S), C—H⋯O(S), C—H⋯π, π–π, S=O⋯π |
| Supramolecular architecture | 0D chains | 0D | 0D chains | 0D chains | 1D chains | 3D |
Series 2: The asymmetric units of all of the compounds in series 2 (Table 5 ▸) contain one molecule and the conformation of the ortho-chloro substituent on the benzenesulfonyl ring is syn to the N—H bond in the central –C–SO2–N–C(O)– segment. The values of the dihedral angle between the two aromatic rings in the molecules fall in the range 73.3 (1)–89.91 (10)°, which is almost the same as in series 1, the smallest being in N-(2-chlorophenylsulfonyl)benzamide (Gowda et al., 2010b
▸) and the largest in N-(2-chlorophenylsulfonyl)-4-fluorobenzamide (II) (Table 5 ▸). The crystal structures of N-(2-chlorophenylsulfonyl)-benzamide, N-(2-chlorophenylsulfonyl)-4-chlorobenzamide (Suchetan et al., 2011c
▸) and N-(2-chlorophenylsulfonyl)-4-methylbenzamide (Gowda et al., 2010c
▸) display zero-dimensional architectures featuring inversion-related (8) dimers formed via N—H⋯O(S) hydrogen bonds, while strong N—H⋯O(S) hydrogen bonds in N-(2-chlorophenylsulfonyl)-4-nitrobenzamide (Suchetan et al., 2011d
▸) lead to one-dimensional chains. Similar to that observed in series 1, the methoxy- and fluoro-substituted compounds in series 2 show diversity in their intermolecular interactions. N-(2-chlorophenylsulfonyl)-4-methoxybenzamide (Sreenivasa et al., 2014b
▸) features structure-directing N—H⋯O(S) and C—H⋯O(S) hydrogen bonds and weak πaryl–πaryl interactions, resulting in a two-dimensional structure. However, in N-(2-chlorophenylsulfonyl)-4-fluorobenzamide (II), N—H⋯O(S) and C—H⋯O(S) hydrogen bonds (with no structure-directing characteristics) between molecules form inversion-related dimers, and these dimers are interconnected via C=O⋯πaryl interactions, forming a one-dimensional architecture.
Table 5. Comparison of various parameters (°) in the crystal structures of series 2: N-(2-chlorophenylsulfonyl)-para-substituted-arylamides.
| Parameters | H | Cl | CH3 | NO2 | OCH3 | F |
|---|---|---|---|---|---|---|
| Crystal System | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Z′ | 1 | 1 | 1 | 1 | 1 | 1 |
| Orientation of 2-Cl group to the N—H bond | syn | syn | syn | syn | syn | syn |
| Angle between aromatic rings | 73.3 (1) | 85.7 (1) | 89.1 (2) | 85.4 (1) | 82.07 (1) | 89.9 (1) |
| Intermolecular interactions | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(S), C—H⋯O(S), π–p | N—H⋯O(S), C—H⋯O(S), C=O⋯π |
| Supramolecular architecture | 0D (ring motifs) | 0D (ring motifs) | 0D (ring motifs) | 1D chains | 2D | 1D |
Series 3: In series 3, the parent compound N-(4-chlorophenylsulfonyl)benzamide (Suchetan et al., 2010a
▸) crystallizes with two molecules in the asymmetric unit, while N-(4-chlorophenylsulfonyl)-4-chlorobenzamide (Suchetan et al., 2010b
▸), N-(4-chlorophenylsulfonyl)-4-methylbenzamide (Suchetan et al., 2010c
▸) and N-(4-chlorophenylsulfonyl)-4-nitrobenzamide (Suchetan et al., 2011a
▸) crystallize with one molecule, and N-(4-chlorophenylsulfonyl)-4-fluorobenzamide (III) crystallizes with one molecule and a molecule of water in the asymmetric unit. The values of the dihedral angle between the two aromatic rings in the molecules are in the range 62.8 (1)–89.5 (1)°, the smallest value being for N-(4-chlorophenylsulfonyl)benzamide and the largest for N-(4-chlorophenylsulfonyl)-4-methylbenzamide (Table 6 ▸). Except for compound (III), the crystals of all of the compounds feature N—H⋯O(S) hydrogen bonds, either forming (8) inversion dimers (zero-dimensional structure) or one-dimensional chains. Once again, the fluoro-substituted compound (III) displays a variety of hydrogen bonds and weak interactions (Tables 3 ▸ and 6 ▸), leading to a three-dimensional architecture.
Table 6. Comparison of various parameters (°) in the crystal structures of series 3: N-(4-chlorophenylsulfonyl)-para-substituted-arylamides.
| Parameters | H | Cl | CH3 | NO2 | F |
|---|---|---|---|---|---|
| Crystal System | Triclinic | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| Z′ | 2 | 1 | 1 | 1 | 1, H2O |
| Angle between aromatic rings | 62.8 (1), 78.6 (1) | 85.6 (1) | 89.5 (1) | 87.8 (1) | 81.82 (11) |
| Intermolecular interactions | N—H⋯O(S) | N—H⋯O(S) | N—H⋯O(C) | N—H⋯O(S) | N—H⋯O(W), O(W)—H⋯O(S), O(W)—H⋯O(C), C—H⋯O(S), C—Cl⋯π, C—F⋯π, S=O⋯π |
| Supramolecular architecture | 0D (ring motifs) | 0D chains | 0D (ring motifs) | D chains | 3D |
Synthesis and crystallization
Compounds (I)–(III) were prepared by refluxing a mixture of 4-fluorobenzoic acid, the corresponding substituted benzenesulfonamides and phosphorousoxychloride for 3 h on a water bath. The resultant mixtures were cooled and poured into ice-cold water. The solids obtained were filtered, washed thoroughly with water and then dissolved in sodium bicarbonate solutions. The compounds were later re-precipitated by acidifying the filtered solutions with dilute HCl. They were filtered, dried and recrystallized. [Melting point (m.p.) of (I) = 410 K, (II) = 428 K and (III) = 456 K]. Prism-like, colourless single crystals of all three of the compounds were obtained from slow evaporation of the respective solutions of the compounds in methanol (with few drops of water).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 7 ▸. The H atoms of the NH groups in (I)–(III) were located in difference Fourier maps and freely refined. The H atoms of the water molecule in (III) were located in a difference Fourier map and were refined with the bond length restraint O—H = 0.83 (3) Å. The other H atoms were positioned with idealized geometry using a riding model: C—H = 0.93–0.96 Å, with U
iso = 1.5U
eq(C-methyl) and 1.2U
eq(C) for other H atoms. In the final cycles of refinement, reflections (0 1 1), (0 0 2) and ( 0 20) in (I), (0 0 2) in (II) and (2 0 0) in (III) were omitted due to large differences in F
2
obs and F
2
calc.
0 20) in (I), (0 0 2) in (II) and (2 0 0) in (III) were omitted due to large differences in F
2
obs and F
2
calc.
Table 7. Experimental details.
| (I) | (II) | (III) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C14H12FNO3S | C13H9ClFNO3S | C13H9ClFNO3S·H2O |
| M r | 293.31 | 313.72 | 331.74 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, C2/c |
| Temperature (K) | 173 | 173 | 173 |
| a, b, c (Å) | 10.0259 (3), 12.4289 (3), 21.6241 (6) | 7.9009 (2), 9.0775 (3), 18.4216 (5) | 45.5989 (11), 4.8853 (1), 12.6517 (3) |
| β (°) | 92.443 (1) | 99.801 (1) | 94.481 (1) |
| V (Å3) | 2692.15 (13) | 1301.92 (6) | 2809.73 (11) |
| Z | 8 | 4 | 8 |
| Radiation type | Cu Kα | Cu Kα | Cu Kα |
| μ (mm−1) | 2.32 | 4.29 | 4.06 |
| Crystal size (mm) | 0.28 × 0.26 × 0.21 | 0.30 × 0.27 × 0.23 | 0.28 × 0.25 × 0.23 |
| Data collection | |||
| Diffractometer | Bruker APEXII | Bruker APEXII | Bruker APEXII |
| Absorption correction | Multi-scan (SADABS; Bruker, 2009 ▸) | Multi-scan (SADABS; Bruker, 2009 ▸) | Multi-scan (SADABS; Bruker, 2009 ▸) |
| T min, T max | 0.548, 0.614 | 0.317, 0.373 | 0.369, 0.393 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 26667, 4404, 4165 | 10032, 2124, 2074 | 11176, 2320, 2030 |
| R int | 0.042 | 0.042 | 0.054 |
| (sin θ/λ)max (Å−1) | 0.587 | 0.585 | 0.584 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.040, 0.121, 1.00 | 0.041, 0.124, 0.97 | 0.039, 0.118, 0.94 |
| No. of reflections | 4404 | 2124 | 2320 |
| No. of parameters | 371 | 185 | 205 |
| No. of restraints | 0 | 0 | 2 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.36, −0.46 | 0.37, −0.50 | 0.34, −0.35 |
Supplementary Material
Crystal structure: contains datablock(s) I, II, III, global. DOI: 10.1107/S2056989016005089/su5287sup1.cif
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors are thankful to the Institution of Excellence, Vijnana Bhavana, University of Mysore, Mysore, for providing the single-crystal X-ray diffraction data. GMS thanks the Vision Group on Science and Technology (VGST), Karnataka, India, for financial support under its SPiCE project scheme.
supplementary crystallographic information
Crystal data
| C13H9ClFNO3S·H2O | Prism |
| Mr = 331.74 | Dx = 1.568 Mg m−3 |
| Monoclinic, C2/c | Melting point: 456 K |
| Hall symbol: -C 2yc | Cu Kα radiation, λ = 1.54178 Å |
| a = 45.5989 (11) Å | Cell parameters from 163 reflections |
| b = 4.8853 (1) Å | θ = 5.8–64.3° |
| c = 12.6517 (3) Å | µ = 4.06 mm−1 |
| β = 94.481 (1)° | T = 173 K |
| V = 2809.73 (11) Å3 | Prism, colourless |
| Z = 8 | 0.28 × 0.25 × 0.23 mm |
| F(000) = 1360 |
Data collection
| Bruker APEXII diffractometer | 2030 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.054 |
| Graphite monochromator | θmax = 64.3°, θmin = 5.8° |
| phi and φ scans | h = −52→51 |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | k = −5→5 |
| Tmin = 0.369, Tmax = 0.393 | l = −14→14 |
| 11176 measured reflections | 1 standard reflections every 1 reflections |
| 2320 independent reflections | intensity decay: 0.1% |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.118 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.94 | w = 1/[σ2(Fo2) + (0.0912P)2 + 2.087P] where P = (Fo2 + 2Fc2)/3 |
| 2320 reflections | (Δ/σ)max = 0.001 |
| 205 parameters | Δρmax = 0.34 e Å−3 |
| 2 restraints | Δρmin = −0.35 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.356285 (11) | 0.64551 (10) | 0.55127 (4) | 0.0147 (2) | |
| Cl1 | 0.261887 (12) | −0.14924 (12) | 0.34233 (4) | 0.0273 (2) | |
| O2 | 0.36322 (3) | 0.8548 (3) | 0.47847 (11) | 0.0176 (4) | |
| O1 | 0.34790 (3) | 0.7192 (3) | 0.65445 (11) | 0.0195 (4) | |
| F1 | 0.49577 (3) | −0.3420 (3) | 0.62191 (12) | 0.0384 (4) | |
| O3 | 0.39675 (3) | 0.4091 (3) | 0.40288 (11) | 0.0207 (4) | |
| O4 | 0.38058 (4) | 0.1618 (4) | 0.75835 (13) | 0.0290 (4) | |
| N1 | 0.38535 (4) | 0.4484 (4) | 0.57495 (14) | 0.0161 (4) | |
| C7 | 0.40210 (5) | 0.3508 (4) | 0.49595 (16) | 0.0165 (5) | |
| C8 | 0.42713 (5) | 0.1710 (4) | 0.53341 (17) | 0.0174 (5) | |
| C3 | 0.28843 (5) | 0.1252 (5) | 0.50950 (18) | 0.0216 (5) | |
| H3 | 0.2753 | 0.0371 | 0.5507 | 0.026* | |
| C13 | 0.43853 (5) | −0.0018 (5) | 0.45932 (17) | 0.0218 (5) | |
| H13 | 0.4305 | −0.0004 | 0.3895 | 0.026* | |
| C2 | 0.30931 (5) | 0.3027 (5) | 0.55508 (17) | 0.0206 (5) | |
| H2 | 0.3105 | 0.3346 | 0.6278 | 0.025* | |
| C1 | 0.32848 (4) | 0.4331 (4) | 0.49134 (16) | 0.0155 (5) | |
| C6 | 0.32692 (5) | 0.3910 (5) | 0.38239 (16) | 0.0174 (5) | |
| H6 | 0.3397 | 0.4821 | 0.3406 | 0.021* | |
| C5 | 0.30619 (5) | 0.2126 (5) | 0.33712 (16) | 0.0189 (5) | |
| H5 | 0.3049 | 0.1814 | 0.2644 | 0.023* | |
| C4 | 0.28742 (5) | 0.0806 (5) | 0.40070 (17) | 0.0190 (5) | |
| C9 | 0.43954 (5) | 0.1702 (5) | 0.63795 (18) | 0.0241 (5) | |
| H9 | 0.4321 | 0.2858 | 0.6878 | 0.029* | |
| C10 | 0.46276 (5) | −0.0009 (6) | 0.66765 (19) | 0.0295 (6) | |
| H10 | 0.4712 | −0.0013 | 0.7369 | 0.035* | |
| C12 | 0.46171 (5) | −0.1755 (5) | 0.48843 (19) | 0.0265 (6) | |
| H12 | 0.4694 | −0.2917 | 0.4393 | 0.032* | |
| C11 | 0.47306 (5) | −0.1710 (5) | 0.5923 (2) | 0.0264 (6) | |
| H1O4 | 0.3743 (8) | 0.011 (6) | 0.761 (3) | 0.067 (12)* | |
| H1 | 0.3854 (6) | 0.373 (6) | 0.634 (2) | 0.037 (8)* | |
| H2O4 | 0.3812 (7) | 0.229 (6) | 0.8178 (19) | 0.039 (8)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0164 (3) | 0.0130 (3) | 0.0148 (3) | 0.0011 (2) | 0.0022 (2) | −0.00059 (18) |
| Cl1 | 0.0245 (4) | 0.0239 (4) | 0.0326 (4) | −0.0091 (2) | −0.0040 (3) | 0.0024 (2) |
| O2 | 0.0191 (8) | 0.0126 (8) | 0.0213 (8) | 0.0000 (6) | 0.0024 (6) | 0.0004 (6) |
| O1 | 0.0221 (8) | 0.0194 (8) | 0.0173 (7) | 0.0012 (7) | 0.0040 (6) | −0.0034 (6) |
| F1 | 0.0277 (8) | 0.0398 (10) | 0.0466 (9) | 0.0179 (7) | −0.0037 (7) | 0.0018 (7) |
| O3 | 0.0240 (8) | 0.0209 (8) | 0.0173 (8) | 0.0031 (7) | 0.0034 (6) | 0.0016 (6) |
| O4 | 0.0504 (12) | 0.0206 (11) | 0.0165 (8) | −0.0034 (8) | 0.0067 (8) | −0.0021 (7) |
| N1 | 0.0180 (10) | 0.0154 (10) | 0.0149 (9) | 0.0003 (8) | 0.0004 (7) | 0.0021 (7) |
| C7 | 0.0165 (11) | 0.0135 (11) | 0.0195 (11) | −0.0041 (8) | 0.0010 (8) | −0.0018 (8) |
| C8 | 0.0145 (11) | 0.0155 (11) | 0.0225 (11) | −0.0025 (8) | 0.0030 (8) | 0.0011 (8) |
| C3 | 0.0181 (11) | 0.0233 (13) | 0.0240 (12) | −0.0016 (9) | 0.0048 (9) | 0.0060 (9) |
| C13 | 0.0210 (11) | 0.0217 (13) | 0.0227 (11) | 0.0006 (10) | 0.0017 (9) | −0.0018 (9) |
| C2 | 0.0211 (12) | 0.0232 (13) | 0.0175 (11) | 0.0012 (10) | 0.0022 (9) | 0.0013 (9) |
| C1 | 0.0156 (11) | 0.0114 (11) | 0.0195 (10) | 0.0029 (9) | 0.0014 (8) | 0.0003 (8) |
| C6 | 0.0159 (11) | 0.0176 (12) | 0.0191 (10) | 0.0016 (9) | 0.0031 (8) | 0.0031 (9) |
| C5 | 0.0183 (11) | 0.0213 (12) | 0.0168 (10) | 0.0010 (9) | 0.0001 (8) | −0.0011 (9) |
| C4 | 0.0158 (11) | 0.0133 (11) | 0.0273 (11) | −0.0003 (9) | −0.0024 (8) | 0.0010 (9) |
| C9 | 0.0224 (12) | 0.0268 (14) | 0.0230 (11) | 0.0033 (10) | 0.0005 (9) | −0.0032 (9) |
| C10 | 0.0225 (12) | 0.0397 (16) | 0.0255 (12) | 0.0043 (11) | −0.0040 (9) | 0.0016 (11) |
| C12 | 0.0227 (12) | 0.0236 (14) | 0.0335 (13) | 0.0022 (10) | 0.0045 (10) | −0.0069 (10) |
| C11 | 0.0163 (12) | 0.0251 (14) | 0.0374 (13) | 0.0061 (10) | −0.0016 (10) | 0.0050 (10) |
Geometric parameters (Å, º)
| S1—O2 | 1.4279 (15) | C3—H3 | 0.9300 |
| S1—O1 | 1.4346 (14) | C13—C12 | 1.382 (3) |
| S1—N1 | 1.6466 (18) | C13—H13 | 0.9300 |
| S1—C1 | 1.763 (2) | C2—C1 | 1.389 (3) |
| Cl1—C4 | 1.740 (2) | C2—H2 | 0.9300 |
| F1—C11 | 1.360 (3) | C1—C6 | 1.390 (3) |
| O3—C7 | 1.217 (3) | C6—C5 | 1.377 (3) |
| O4—H1O4 | 0.79 (3) | C6—H6 | 0.9300 |
| O4—H2O4 | 0.82 (2) | C5—C4 | 1.380 (3) |
| N1—C7 | 1.389 (3) | C5—H5 | 0.9300 |
| N1—H1 | 0.84 (3) | C9—C10 | 1.378 (3) |
| C7—C8 | 1.488 (3) | C9—H9 | 0.9300 |
| C8—C13 | 1.392 (3) | C10—C11 | 1.375 (4) |
| C8—C9 | 1.397 (3) | C10—H10 | 0.9300 |
| C3—C2 | 1.380 (3) | C12—C11 | 1.374 (3) |
| C3—C4 | 1.391 (3) | C12—H12 | 0.9300 |
| O2—S1—O1 | 119.70 (9) | C2—C1—C6 | 121.5 (2) |
| O2—S1—N1 | 108.70 (9) | C2—C1—S1 | 118.95 (16) |
| O1—S1—N1 | 104.43 (9) | C6—C1—S1 | 119.53 (16) |
| O2—S1—C1 | 109.39 (9) | C5—C6—C1 | 119.1 (2) |
| O1—S1—C1 | 107.77 (9) | C5—C6—H6 | 120.4 |
| N1—S1—C1 | 105.99 (10) | C1—C6—H6 | 120.4 |
| H1O4—O4—H2O4 | 109 (3) | C6—C5—C4 | 119.3 (2) |
| C7—N1—S1 | 123.36 (15) | C6—C5—H5 | 120.3 |
| C7—N1—H1 | 122 (2) | C4—C5—H5 | 120.3 |
| S1—N1—H1 | 112 (2) | C5—C4—C3 | 122.0 (2) |
| O3—C7—N1 | 122.4 (2) | C5—C4—Cl1 | 118.63 (17) |
| O3—C7—C8 | 122.48 (19) | C3—C4—Cl1 | 119.37 (17) |
| N1—C7—C8 | 115.13 (18) | C10—C9—C8 | 120.4 (2) |
| C13—C8—C9 | 119.4 (2) | C10—C9—H9 | 119.8 |
| C13—C8—C7 | 117.45 (19) | C8—C9—H9 | 119.8 |
| C9—C8—C7 | 123.2 (2) | C9—C10—C11 | 118.3 (2) |
| C2—C3—C4 | 118.7 (2) | C9—C10—H10 | 120.8 |
| C2—C3—H3 | 120.6 | C11—C10—H10 | 120.8 |
| C4—C3—H3 | 120.6 | C11—C12—C13 | 117.9 (2) |
| C12—C13—C8 | 120.7 (2) | C11—C12—H12 | 121.0 |
| C12—C13—H13 | 119.6 | C13—C12—H12 | 121.0 |
| C8—C13—H13 | 119.6 | F1—C11—C12 | 118.4 (2) |
| C3—C2—C1 | 119.4 (2) | F1—C11—C10 | 118.3 (2) |
| C3—C2—H2 | 120.3 | C12—C11—C10 | 123.3 (2) |
| C1—C2—H2 | 120.3 | ||
| O2—S1—N1—C7 | −45.29 (19) | O1—S1—C1—C6 | 163.03 (17) |
| O1—S1—N1—C7 | −174.12 (17) | N1—S1—C1—C6 | −85.63 (19) |
| C1—S1—N1—C7 | 72.20 (19) | C2—C1—C6—C5 | −1.0 (3) |
| S1—N1—C7—O3 | 1.5 (3) | S1—C1—C6—C5 | 176.33 (16) |
| S1—N1—C7—C8 | −178.80 (14) | C1—C6—C5—C4 | 0.2 (3) |
| O3—C7—C8—C13 | −20.8 (3) | C6—C5—C4—C3 | 1.0 (3) |
| N1—C7—C8—C13 | 159.45 (19) | C6—C5—C4—Cl1 | −178.71 (16) |
| O3—C7—C8—C9 | 158.7 (2) | C2—C3—C4—C5 | −1.4 (3) |
| N1—C7—C8—C9 | −21.0 (3) | C2—C3—C4—Cl1 | 178.32 (18) |
| C9—C8—C13—C12 | 0.7 (3) | C13—C8—C9—C10 | −0.3 (3) |
| C7—C8—C13—C12 | −179.7 (2) | C7—C8—C9—C10 | −179.8 (2) |
| C4—C3—C2—C1 | 0.5 (3) | C8—C9—C10—C11 | −0.6 (4) |
| C3—C2—C1—C6 | 0.7 (3) | C8—C13—C12—C11 | −0.2 (3) |
| C3—C2—C1—S1 | −176.74 (17) | C13—C12—C11—F1 | 179.7 (2) |
| O2—S1—C1—C2 | −151.16 (17) | C13—C12—C11—C10 | −0.8 (4) |
| O1—S1—C1—C2 | −19.5 (2) | C9—C10—C11—F1 | −179.3 (2) |
| N1—S1—C1—C2 | 91.82 (19) | C9—C10—C11—C12 | 1.2 (4) |
| O2—S1—C1—C6 | 31.4 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···O4 | 0.83 (3) | 1.91 (3) | 2.733 (3) | 171 (2) |
| O4—H1O4···O1i | 0.79 (3) | 2.25 (3) | 2.884 (2) | 138 (3) |
| O4—H2O4···O2ii | 0.82 (2) | 2.29 (3) | 2.955 (2) | 139 (3) |
| O4—H2O4···O3ii | 0.82 (2) | 2.16 (3) | 2.841 (2) | 141 (3) |
| C5—H5···O1iii | 0.93 | 2.54 | 3.124 (3) | 121 |
Symmetry codes: (i) x, y−1, z; (ii) x, −y+1, z+1/2; (iii) x, −y+1, z−1/2.
References
- Banwell, M. G., Crasto, C. F., Easton, C. J., Forrest, A. K., Karoli, T., March, D. R., Mensah, L., Nairn, M. R., O’Hanlon, P. J., Oldham, M. D. & Yue, W. (2000). Bioorg. Med. Chem. Lett. 10, 2263–2266. [DOI] [PubMed]
- Bruker (2009). APEX2, SADABS, SAINT-Plus and XPREP. Bruker AXS Inc., Madison, Wisconsin, USA.
- Chang, L. L., Ashton, W. T., Flanagan, K. L., Chen, T. B., O’Malley, S. S., Zingaro, G. J., Siegl, P. K. S., Kivlighn, S. D., Lotti, V. J., Chang, R. S. L. & Greenlee, W. J. (1994). J. Med. Chem. 37, 4464–4478. [DOI] [PubMed]
- Gowda, B. T., Foro, S., Suchetan, P. A. & Fuess, H. (2010a). Acta Cryst. E66, o747. [DOI] [PMC free article] [PubMed]
- Gowda, B. T., Foro, S., Suchetan, P. A. & Fuess, H. (2010b). Acta Cryst. E66, o794. [DOI] [PMC free article] [PubMed]
- Gowda, B. T., Foro, S., Suchetan, P. A. & Fuess, H. (2010c). Acta Cryst. E66, o1466. [DOI] [PMC free article] [PubMed]
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Hamad, A. S. & Abed, F. S. (2014). J. Appl. Chem, 3, 56–63.
- Hasegawa, T. & Yamamoto, H. (2000). Bull. Chem. Soc. Jpn, 73, 423–428.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Mader, M., Shih, C., Considine, E., De Dios, A., Grossman, C., Hipskind, P., Lin, H., Lobb, K., Lopez, B., Lopez, J., Cabrejas, L., Richett, M., White, W., Cheung, Y., Huang, Z., Reilly, J. & Dinn, S. (2005). Bioorg. Med. Chem. Lett. 15, 617–620. [DOI] [PubMed]
- Manojkumar, K. E., Sreenivasa, S., Mohan, N. R., Madhu Chakrapani Rao, T. & Harikrishna, T. (2013). J. Appl. Chem, 2, 730–737.
- Mohan, N. R., Sreenivasa, S., Manojkumar, K. E. & Chakrapani Rao, T. M. (2013). J. Appl. Chem. 2, 722–729.
- Musser, J. H., Kreft, A. F., Bender, R. H. W., Kubrak, D. M., Grimes, D., Carlson, R. P., Hand, J. M. & Chang, J. (1990). J. Med. Chem. 33, 240–245. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sreenivasa, S., Palakshamurthy, B. S., Madankumar, S., Lokanath, N. K. & Suchetan, P. A. (2014a). Acta Cryst. E70, o193. [DOI] [PMC free article] [PubMed]
- Sreenivasa, S., Palakshamurthy, B. S., Pampa, K. J., Lokanath, N. K. & Suchetan, P. A. (2014b). Acta Cryst. E70, o199. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Foro, S. & Gowda, B. T. (2011a). Acta Cryst. E67, o904. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Foro, S. & Gowda, B. T. (2011b). Acta Cryst. E67, o929. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Foro, S. & Gowda, B. T. (2011c). Acta Cryst. E67, o146. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Foro, S. & Gowda, B. T. (2011d). Acta Cryst. E67, o930. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Gowda, B. T., Foro, S. & Fuess, H. (2010a). Acta Cryst. E66, o766. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Gowda, B. T., Foro, S. & Fuess, H. (2010b). Acta Cryst. E66, o1253. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Gowda, B. T., Foro, S. & Fuess, H. (2010c). Acta Cryst. E66, o1501. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Gowda, B. T., Foro, S. & Fuess, H. (2010d). Acta Cryst. E66, o1024. [DOI] [PMC free article] [PubMed]
- Suchetan, P. A., Gowda, B. T., Foro, S. & Fuess, H. (2010e). Acta Cryst. E66, o1997. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, II, III, global. DOI: 10.1107/S2056989016005089/su5287sup1.cif
Additional supporting information: crystallographic information; 3D view; checkCIF report