

Abstract
Background:
Kearns-Sayre syndrome (KSS) is a mitochondrial DNA (mtDNA) deletion disorder characterized by a triad of onset before 20 years of age, ophthalmoplegia, and pigmentary retinopathy. The heart and central nervous system are commonly involved. We summarized clinical and brain magnetic resonance imaging (MRI) features of a cohort of Chinese KSS patients.
Methods:
Nineteen patients confirmed by muscle biopsy and mtDNA analysis were enrolled. We examined clinical profiles, mainly focusing on changes in electrocardiogram (ECG) and brain MRI. The correlation between genotype and phenotype was statistically analyzed.
Results:
The mean age of onset was 9.6 ± 4.3 years, with all developing the classic triad at the time of diagnosis. Heart conduction block was detected in 63.2%, with four initially presenting as bundle branch block and developing into complete atrioventricular block over 3–72 months. Brain MRI showed symmetric high-T2 signals in 100% of cerebral and cerebellar white matter, as well as brainstem, 46.7% of basal ganglia, and 53.3% of thalamus. There were two patterns of cerebral white matter involvements, one with selective subcortical U-fibers and the other with periventricular white matter. The size of mtDNA deletion did not significantly correlate with age of onset or percentage of ragged blue fibers on muscle pathology.
Conclusions:
The clinical features of KSS evolve dynamically, affecting the cardiac conduction system predominantly, highlighting the significance of ECG monitoring. Brain MRI showed changes involving both the white matter and deep gray nuclei. Clinical presentation or severity of muscle pathological changes is not related to the size of mtDNA deletions.
Keywords: Brain Magnetic Resonance Imaging, Heart Conduction Block, Kearns-Sayre Syndrome
INTRODUCTION
Kearns-Sayre syndrome (KSS) is a rare sporadic mitochondrial cytopathy caused by a single large-scale mitochondrial DNA (mtDNA) deletion.[1] Classically, KSS has a triad of features including onset before 20 years of age, progressive external ophthalmoplegia (PEO), and pigmentary retinopathy.[2,3] In addition, heart blocks, cerebellar ataxia, short stature, and sensory hearing loss are frequently present in these patients.[4] Muscle pathology plays a vital role in differentiating KSS with other diseases which present as ophthalmoplegia,[5] since ragged-red fibers (RRFs), ragged blue fibers (RBFs), and cytochrome oxidase (COX) negative fibers are histopathological hallmarks of mitochondrial disease.[6] The most commonly reported brain magnetic resonance imaging (MRI) findings in KSS are cerebral and cerebellar atrophy with bilateral high-T2 signals in subcortical white matter, thalamus, basal ganglia, and brainstem.[7] Until now, reports on KSS patients in mainland of China were uncommon,[8] making it necessary to clarify the natural history and characteristics of brain MRI changes in more KSS patients. Herein, we report clinical profiles of 19 Chinese patients with KSS, focusing on the dynamic changes in electrocardiogram (ECG), clinical follow-up, brain MRI feature as well as possible relationships between phenotype and genotype.
METHODS
Patients
Nineteen unrelated KSS patients who visited Peking University First Hospital from January 2004 to October 2015 were enrolled in this study [Table 1]. All patients conformed to the diagnostic criteria of KSS, i.e., the triad of PEO, pigmentary retinopathy, and onset before 20 years of age, plus at least one of the following: heart block, cerebellar symptoms, or cerebrospinal fluid (CSF) protein levels above 1000 mg/L.[9] Their clinical records were collected and analyzed, as well as the results of auxiliary examinations, including fundus photography, ECG, and brain MRI. All patients were interviewed and examined by at least two experienced neurologists. MRIs were evaluated by two neuro-radiologists. In addition, 13 of the patients were followed up 6 months to 8 years after their confirmed diagnosis.
Table 1.
Clinical, myopathological and genetic features of 19 KSS patients
| Patient number | Sex | Onset age (years) | Onset symptom | Additional clinical features | EEG | RBF% on muscle pathology | Size and junction of mtDNA deletion |
|---|---|---|---|---|---|---|---|
| 1 | Male | 7 | Short stature | EO, PR | AVB + RBBB* | 3.5 | 5557 bp (8346–13,903) |
| 2 | Female | 9 | Short stature | EO, PR, DM, cerebellar ataxia | – | ND | ND |
| 3 | Male | 13 | EO | PR | AVB + RBBB* | 1.4 | 4977 bp (8482–13,459) |
| 4 | Female | 7 | EO | PR, cerebellar ataxia | ST | 0.7 | 4977 bp (8482–13,459) |
| 5 | Female | 13 | EO | PR, hearing loss, cerebellar ataxia | – | ND | ND |
| 6 | Female | 12 | EO | PR, muscle weakness | RBBB | 6.2 | 4977 bp (8482–13,459) |
| 7 | Male | 4 | EO | PR, hearing loss | RBBB | ND | ND |
| 8 | Female | 5 | EO | PR, cerebellar ataxia | AVB | 0.5 | ND |
| 9 | Male | 9 | EO | PR | AVB* | 2.5 | 4977 bp (8482–13,459) |
| 10 | Female | 9 | Short stature | EO, PR | LAFB | 3.8 | ND |
| 11 | Male | 14 | EO | PR, cerebellar ataxia | AVB* | 0.3 | 4977 bp (8482–13,459) |
| 12 | Female | 2 | EO | PR, hearing loss, cerebellar ataxia | – | ND | 7989 bp (7449–15,438) |
| 13 | Male | 10 | EO | PR, hearing loss | – | 0.9 | 4977 bp (8482–13,459) |
| 14 | Male | 16 | EO | PR, cerebellar ataxia | – | 16.0 | 4977 bp (8482–13,459) |
| 15 | Male | 9 | EO | PR, cerebellar ataxia | RBBB | 0.6 | 6321 bp (8277–14,598) |
| 16 | Male | 2 | EO | PR, cerebellar ataxia | – | 1.0 | 4977 bp (8482–13,459) |
| 17 | Male | 13 | EO | PR | RBBB + LAFB | 0.3 | ND |
| 18 | Female | 15 | EO | PR | AVB + RBBB* | 0.3 | ND |
| 19 | Male | 14 | EO | PR, hearing loss, cerebellar ataxia | AVB + RBBB | 2.0 | 4977 bp (8482–13,459) |
*Receiving permanent pacemaker implantation. EO: External ophthalmoplegia; PR: Pigmentary retinopathy; DM: Diabetes mellitus; AVB: Atrioventricular block; RBBB: Right bundle branch block; ST: Sinus tachycardia; LAFB: Left anterior fascicular block; –: Normal; ND: Not done; RRF: Ragged-red fibers; RBF: Ragged-blue fibers; COX: Cytochrome-oxidase; mtDNA: Mitochondrial DNA; KSS: Kearns-Sayre syndrome.
This study was approved by the Ethical Committee of Peking University First Hospital.
Muscle pathology
Muscle biopsies were performed on 15 patients for diagnostic purpose after they gave written informed consent. For histological examination, serial frozen sections (8 μm) were stained by routine histological and histochemical methods. The percentage of RBFs was counted under the microscope by at least two different experts in muscle pathology.
Molecular genetics
Twelve patients underwent molecular genetic analysis with DNA extracted from biopsied muscle samples. The method of detecting large-scale mtDNA deletion was the same as previously described.[10]
Statistical analyses
Statistical analyses were conducted using SPSS version 22.0 (SPSS Inc., Chicago, IL, USA). Spearman correlation coefficient was performed between the age of onset, the percentage of RBFs, and the length of mtDNA deletions. P < 0.05 was considered statistically significant.
RESULTS
General clinical features
All 19 patients were sporadic and included 11 males and 8 females. The mean age of onset was 9.6 ± 4.3 years (2–16 years) and the mean age at diagnosis was 16.6 ± 6.1 years (8–31 years). The symptoms at onset were ptosis and external ophthalmoplegia in 16 cases (84.2%) and short stature in three cases (15.8%). All patients developed ptosis, external ophthalmoplegia, and pigmentary retinopathy with disease progression. In addition, ten cases (52.6%) showed cerebellar ataxia, mainly presenting as unstable gait and cerebellar tremor. Five cases (26.3%) developed different degrees of hearing loss. One case (5.3%) was diagnosed with diabetes mellitus and one case (5.3%) complained of limb weakness but could still walk independently. At the early stage of the disease, patients usually presented with extraocular muscle symptoms, and during the course of the disease gradually developed other clinical features related to cerebellum, audition, and heart conduction [Figure 1]. Four patients who underwent lumbar puncture showed a significant increase in CSF protein levels (>1000 mg/L). In the follow-up studies of 13 patients, eight patients showed no significant deterioration in condition. Walking difficulties were seen in three due to cerebellar ataxia, and loss of ambulation in two patients due to heart failure.
Figure 1.
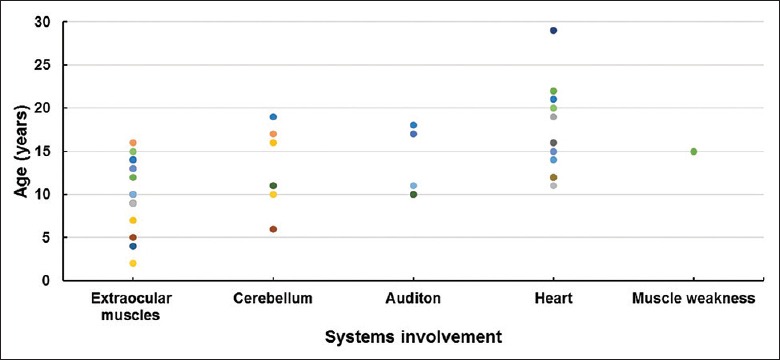
The developing age of different symptoms in 19 KSS patients. Different spot colors represent different individuals. KSS: Kearns-Sayre syndrome.
Cardiac manifestations
All patients had ECG examinations. Twelve cases (63.2%) showed conduction blocks, including right bundle branch block (RBBB) in three cases, complete atrioventricular block (AVB) in three cases, complete AVB with RBBB in three cases, left anterior fascicular block (LAFB) in one case, RBBB with LAFB in one case, and Mobitz type II 2nd degree AVB with RBBB in one case. One case (5.3%) showed sinus tachycardia, and the remaining six cases (31.6%) had no abnormalities of ECG at the time of their last visit. It is noteworthy that although RBBB was detected in four patients on their first abnormal ECG, all of them developed RBBB combined with LAFB, and finally deteriorated to complete AVB within 3–72 months during the following monitoring of ECGs. During the follow-up, five patients with complete AVB showed relatively stable situations after receiving permanent pacemaker implantations.
Brain magnetic resonance imaging findings
Abnormalities were found in all 15 patients who had brain MRI examinations [Table 2 and Figure 2]. All cases showed symmetric high-T2 signals in cerebral and cerebellar white matter, as well as brainstem with various extent. In addition, symmetric high-T2 signals were found in basal ganglia (mainly globus pallidus) in seven cases (46.7%), and in thalamus in eight cases (53.3%). According to the distribution of cerebral white matter lesions, these patients can be divided into two groups. Group 1 included six patients who, on brain MRIs, showed subcortical U-fiber involvement with sparing of the periventricular white matter. In this group, abnormal high-T2 signals were detected in the corpus callosum (mainly in the splenium), basal ganglia, and dorsal brain stem in all six patients, in the thalamus of five patients (83.3%), and in the cerebral peduncle of five patients (83.3%). The remaining nine patients belonged to Group 2 who had mainly periventricular white matter involvement on brain MRIs. In this group, abnormal high-T2 signals were seen in the corpus callosum of four patients (44.4%), basal ganglia in 2 (22.2%), thalamus in 3 (33.3%), cerebral peduncle in 8 (88.8%), and dorsal brain stem in six patients (66.6%). Patients in both Group 1 and Group 2 showed involvement of bilateral internal capsule, especially the posterior limb, and brachium pontis. There were three cases (20.0%) with abnormal high-T2 signals in cerebellar dentate nuclei. Cerebellar atrophy was observed in five cases (33.3%), while obvious cerebral atrophy was not found in any of the 15 patients. Moreover, of the eight patients who received diffusion-weighted imaging (DWI) sequence scanning, 6 (75.0%) showed restricted diffusion on DWI in white matter and basal ganglia.
Table 2.
Distribution of high-T2 signals on brain MRIs of 15 KSS patients*
| Patient number | Cerebral white matter† | Basal ganglia | Thalamus | Brainstem‡ | Cerebellum | ||||
|---|---|---|---|---|---|---|---|---|---|
| Subcortical | Periventricular | Corpus callosum | Cerebral peduncles | Dorsal brain stem | White matter | Dentate nuclei | |||
| 1 | + | – | + | + | + | + | + | + | – |
| 2 | + | – | + | + | + | + | + | + | + |
| 3 | – | + | – | – | – | + | – | + | – |
| 4 | + | – | + | + | + | – | + | + | – |
| 5 | – | + | – | – | – | + | – | + | – |
| 6 | – | + | + | – | – | + | + | + | – |
| 7 | – | + | – | + | – | – | – | + | – |
| 8 | + | – | + | + | + | + | + | + | + |
| 10 | – | + | + | + | + | + | + | + | – |
| 11 | – | + | + | – | – | + | + | + | – |
| 12 | – | + | – | – | + | + | + | + | – |
| 13 | – | + | – | – | + | + | + | + | – |
| 15 | + | – | + | + | + | + | + | + | – |
| 17 | + | – | + | + | – | + | + | + | – |
| 19 | – | + | + | – | – | + | + | + | + |
*The rows in gray were cases with subcortical white matter involved, while the rest were cases with prominent periventricular white matter involved; †Internal capsule in this group was affected in all 15 patients; ‡Brachium pontis in this group was affected in all 15 patients. KSS: Kearns-Sayre syndrome; MRI: Magnetic resonance imaging.
Figure 2.

Brain MRIs of two KSS patients. (a-d) Brain MRI in patient 2 showed abnormal high-T2 signals in the subcortical white matter with U-fibers involved (a), splenium of corpus callosum, internal capsule, basal ganglia, and thalamus (b), dorsal brainstem (c), and brachium pontis and cerebellar white matter (d), respectively. (e-h) Brain MRI in patient 6 showed abnormal high-T2 signal signals in the periventricular white matter (e), splenium of corpus callosum, internal capsule (f), dorsal brainstem (g), brachium pontis, and cerebellar white matter (h), respectively. MRI: Magnetic resonance imaging; KSS: Kearns-Sayre syndrome.
Muscle pathology and molecular genetics
Myopathological examination showed RRFs, RBFs, and COX-negative fibers with different ratios in all 15 patients receiving biopsy [Figure 3]. The percentage of RBFs ranged from 0.3% to 16.0%. Single large-scale deletions of mtDNA were detected in all 12 patients’ muscle DNA who underwent mtDNA mutation analysis. Nine patients (75.0%) carried mtDNA 4977 bp deletions (nt8482–nt13,459), three patients carried three other different larger mtDNA deletions.
Figure 3.
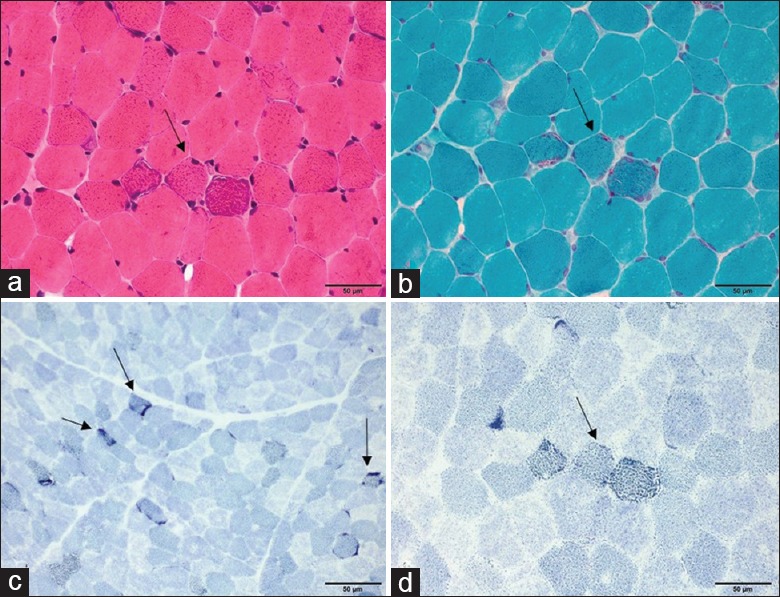
Muscle pathology of patient 6. Basophilic fibers on H & E staining (arrow) (a), RRFs on MGT staining (arrow) (b), and RBFs on SDH staining (arrows) (c and d). RRF: Ragged-red fiber; MGT: Modified Gomori trichrome; RBF: Ragged-blue fiber; SDH: Succinate dehydrogenase; KSS: Kearns-Sayre syndrome. Scale bar=50 μm.
Correlation between phenotype and genotype
Statistical analyses showed no correlations between size of mtDNA deletions and age of onset (P = 0.848) or percentage of RBFs (P = 0.722). Six cases carrying the same type of mtDNA 4977bp deletion had brain MRIs. However, the distribution and severity of brain lesions revealed by MRI findings varied greatly in these patients.
DISCUSSION
In this study, we recruited 19 patients with KSS - the largest cohort in mainland China studied so far. All patients presented with the classic triad of symptoms. The mean age of onset in our patients was 9.6 ± 4.3 years, which was similar to other reports on Chinese KSS patients.[8,11] It has been reported that the age of onset was between 13 and 15 years in Korean KSS patients,[12] and 6–10 years in Japanese KSS patients.[13] Therefore, the age of onset in Asian patients with KSS is a little younger than the US patients with this disease where the age of onset is 17 ± 10 years old.[4] The average time interval from disease onset to diagnosis in our case studies was 7 years, similar to the study in the US patients,[4] suggesting that early diagnosis of KSS is a challenge for clinicians worldwide.
All patients developed full KSS phenotype after a disease duration of 2–15 years. As indicated in Figure 1, in most patients the evolution of disease course showed a similar pattern. Most of them presented with ptosis or ophthalmoplegia at the initial stage of the disease. With disease progression, other systems were affected gradually within several years, including the appearance of neurological dysfunction before 20 years of age and cardiac conduction blockages before the third decade. Cerebellar ataxia was the most common neurological symptom in our cohort (52.6%), which was also frequently described in other reports.[11,14,15] However, it only appeared in one of 35 US patients and was not described in other Japanese or Korean case series.[4,12,13] We previously found 71.0% of 73 patients with different subtypes of mitochondrial encephalomyopathy had a hearing impairment.[16] In the present study, however, only five KSS patients (26.3%) had hearing loss, much lower than the incidence of 64.0% in Japanese patients,[13] suggesting thorough auditory function evaluation is needed in these KSS patients to detect subclinical hearing loss in the future. Proximal muscle weakness was reported in some KSS patients in other studies,[4,17] but it was not common in our patients. Follow-up showed that the greatest impairments affecting KSS patients were cerebellar signs and heart problems.
Our study showed that heart conduction block was very common among Chinese KSS patients, which was consistent with other reports.[18] There were different types of conduction blocks including RBBB, LAFB, and different degrees of AVBs. The most common type of ventricular arrhythmia reported in KSS patients by others was bradycardia-related polymorphic ventricular tachycardia.[19] However, this was neither found in any of our patients nor in other Chinese KSS patients reported so far. We found it was very common that in KSS, conduction defects were aggravating and not constant, as seen in most of our patients who gradually deteriorated from bundle branch blockages to complete AVBs. The progression rate of conduction defects differed widely in patients, ranging from several months to several years. Other reports also stressed that the primary cause of sudden death in KSS patients was high-grade heart block,[4,20] and the incidence of sudden cardiac death in KSS patients was 20% as reported,[18,20] making prompt pacemaker implantation a prerequisite in such conditions. However, most cases in our cohort showed no cardiac symptoms with bundle block detected on ECGs, and when syncope or sudden cardiac death presented, it had usually already progressed to a high-grade heart block. Therefore, we strongly suggest that all patients diagnosed with KSS should have ECG screening at least once a year to detect conduction defects, ventricular arrhythmias, and QT prolongation.[4] For those already having bundle branch heart block, further closer monitoring should be carried out to notice complete AVB, which can lead to sudden cardiac death.[18]
Brain MRIs were abnormal in all of our patients who received MRI examinations, although only half of them were clinically affected, indicating that the severity of clinical manifestations was not correlated with brain MRI changes.[21] Previous studies by other researchers reported that the percentage involvement was 90.9% subcortical white matter, 63.6% basal ganglia, 63.6% brainstem, 54.5% thalamus, and 25.0% cerebellum.[9,22,23] In contrast, we found all patients had involvement of cerebral and cerebellar white matter, as well as brainstem with varying degrees. About 53.3% of our patients presented with high-T2 signals in basal ganglia and thalamus, which was similar to other reports. Saneto et al. reported that the characteristic MRI findings of white matter changes in KSS patients are the involvement of subcortical U-fibers with sparing of the periventricular white matter, differentiating KSS from most lysosomal, and peroxisomal disorders where subcortical regions are only affected late in the course.[7,9] Our study, however, showed there were two patterns of cerebral white matter involvement in KSS: one with selective subcortical U-fibers involvement and the other with prominent periventricular white matter affected, while the latter was also previously reported in an occasional single case.[24,25] The percentages of corpus callosum, basal ganglia, thalamus, and dorsal brain stem involvement were significantly higher in the group affecting subcortical white matter than in the group affecting periventricular white matter, while the involvement of cerebral peduncles showed no difference between the two groups. However, we did not detect the differences in the severity of clinical features between the two groups. In addition, the involvement of the splenium of the corpus callosum and the posterior limb of internal capsule was common in our patients, suggesting another characteristic neuro-radiological feature of KSS patients. The involvement of the dentate nuclei was not correlated to clinical cerebellar ataxia, while the common findings of abnormal signals involving brachium pontis fibers and cerebellar white matter might provide a possible explanation. The findings of restricted diffusion on DWI were not reported previously and might be a characteristic feature of KSS patients. In our study, brain MRIs were carried out at different disease stages; future research should include long-term clinical and MRI follow-up to further determine the evolving state of brain MRIs and its relationship to clinical features.
MtDNA deletions were detected in all 12 patients who were examined by molecular analysis, with 75.0% of them carrying 4977 bp “common deletions”. According to previous studies, the size of the deletion, the deletion heteroplasmy level in skeletal muscle, and the location of the deletion were significantly correlated with age at onset of symptoms and progression of disease burden in KSS and other mtDNA deletion syndromes.[26] However, our study could not find any correlations between the size of the deletion and the age of onset or the percentage of RBFs in our case series. As the percentage of mtDNA deletion was not determined in our study, we assume the deletion heteroplasmy might be the major factor associated with phenotype. In the future, more detailed research with a larger cohort of KSS patients may help determine the actual relationship between mtDNA mutations and MRI changes.
In conclusion, the present study showed that the clinical features of KSS had a tendency to evolve. The progression rate of heart block varied widely among KSS patients, highlighting the significance of close ECG monitoring. Brain MRI often showed pronounced changes involving cerebral and cerebellar white matter and the deep gray nucleus. The group with the dominant involvement of subcortical white matter showed higher percentages of corpus callosum, basal ganglia, thalamus, and dorsal brain stem involvement than the group with the involvement of periventricular white matter. Clinical presentation or the severity of muscle pathological changes is not related to the size of mtDNA deletions.
Financial support and sponsorship
This work was supported by grants from the Ministry of Science and Technology of China (No. 2011ZX09307-001-07) and Beijing Municipal Science and Technology Commission (No. Z151100003915126).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yi Cui
REFERENCES
- 1.Zeviani M, Moraes CT, Dimauro S, Nakase H, Bonilla E, Schon EA, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1998;51:1525–33. doi: 10.1212/wnl.51.6.1525-a. doi: 10.1212/WNL.51.6.1525-a. [DOI] [PubMed] [Google Scholar]
- 2.Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol. 1958;60:280–9. doi: 10.1001/archopht.1958.00940080296016. [PubMed] [Google Scholar]
- 3.Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10:118–26. doi: 10.1007/s11910-010-0096-4. doi: 10.1007/s11910-010-0096-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khambatta S, Nguyen DL, Beckman TJ, Wittich CM. Kearns-Sayre syndrome: A case series of 35 adults and children. Int J Gen Med. 2014;7:325–32. doi: 10.2147/IJGM.S65560. doi: 10.2147/IJGM.S65560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoser BG, Pongratz D. Extraocular mitochondrial myopathies and their differential diagnoses. Strabismus. 2006;14:107–13. doi: 10.1080/09273970600701218. doi: 10.1080/09273970600701218. [DOI] [PubMed] [Google Scholar]
- 6.Filosto M, Tomelleri G, Tonin P, Scarpelli M, Vattemi G, Rizzuto N, et al. Neuropathology of mitochondrial diseases. Biosci Rep. 2007;27:23–30. doi: 10.1007/s10540-007-9034-3. doi: 10.1007/s10540-007-9034-3. [DOI] [PubMed] [Google Scholar]
- 7.Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion. 2008;8:396–413. doi: 10.1016/j.mito.2008.05.003. doi: 10.1016/j.mito.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang F, Ding C, Xiao J, Wang X, Lv J, Ma Y, et al. Clinical characteristics of eight cases with Kearns-Sayre syndrome in children. Chin J Evid Based Pediatr. 2011;6:431–8. doi: 10.3969/j.issn.1673-5501.2011.06.006. [Google Scholar]
- 9.Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293–9. doi: 10.1056/NEJM198905183202001. doi: 10.1056/NEJM198905183202001. [DOI] [PubMed] [Google Scholar]
- 10.Liu Q, Liu J, Leng Y, Zhao J, Lv H, Zhang W, et al. Clinical phenotype and genotype analysis in 61 patients with large scale single deletion in mitochondrial DNA. Chin J Neurol. 2015;48:382–9. doi: 10.3760/cma.j.issn.1006-7876.2015.05.007. [Google Scholar]
- 11.Yau EK, Chan KY, Au KM, Chow TC, Chan YW. A novel mitochondrial DNA deletion in a Chinese girl with Kearns-Sayre syndrome. Hong Kong Med J. 2009;15:374–7. [PubMed] [Google Scholar]
- 12.Park SB, Ma KT, Kook KH, Lee SY. Kearns-Sayre syndrome – 3 case reports and review of clinical feature. Yonsei Med J. 2004;45:727–35. doi: 10.3349/ymj.2004.45.4.727. doi: 10.3349/ymj.2004.45.4.727. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita S, Nishino I, Nonaka I, Goto Y. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J Hum Genet. 2008;53:598–606. doi: 10.1007/s10038-008-0289-8. doi: 10.1007/s10038-008-0289-8. [DOI] [PubMed] [Google Scholar]
- 14.Kamata Y, Mashima Y, Yokoyama M, Tanaka K, Goto Y, Oguchi Y. Patient with Kearns-Sayre syndrome exhibiting abnormal magnetic resonance image of the brain. J Neuroophthalmol. 1998;18:284–8. doi: 10.1097/00041327-199812000-00014. [PubMed] [Google Scholar]
- 15.Kosinski C, Mull M, Lethen H, Töpper R. Evidence for cardioembolic stroke in a case of Kearns-Sayre syndrome. Stroke. 1995;26:1950–2. doi: 10.1161/01.str.26.10.1950. doi: 10.1161/01.STR.26.10.1950. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Xue J, Zhao D, Chen L, Yuan Y, Wang Z. Audiological evaluation in Chinese patients with mitochondrial encephalomyopathies. Chin Med J. 2014;127:2304–9. doi: 10.3760/cma.j.issn.0366-6999.20132799. [PubMed] [Google Scholar]
- 17.Phadke M, Lokeshwar MR, Bhutada S, Tampi C, Saxena R, Kohli S, et al. Kearns Sayre syndrome – Case report with review of literature. Indian J Pediatr. 2012;79:650–4. doi: 10.1007/s12098-011-0618-3. doi: 10.1007/s12098-011-0618-3. [DOI] [PubMed] [Google Scholar]
- 18.van Beynum I, Morava E, Taher M, Rodenburg RJ, Karteszi J, Toth K, et al. Cardiac arrest in kearns-sayre syndrome. JIMD Rep. 2012;2:7–10. doi: 10.1007/8904_2011_32. doi: 10.1007/8904_2011_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabunga P, Lau AK, Phan K, Puranik R, Liang C, Davis RL, et al. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. 2015;181:303–10. doi: 10.1016/j.ijcard.2014.12.038. doi: 10.1016/j.ijcard.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal H, Ekhomu O, Choi HW, Naheed Z. Natural history of conduction abnormalities in a patient with Kearns-Sayre syndrome. Pediatr Cardiol. 2013;34:1044–7. doi: 10.1007/s00246-012-0365-x. doi: 10.1007/s00246-012-0365-x. [DOI] [PubMed] [Google Scholar]
- 21.Lerman-Sagie T, Leshinsky-Silver E, Watemberg N, Luckman Y, Lev D. White matter involvement in mitochondrial diseases. Mol Genet Metab. 2005;84:127–36. doi: 10.1016/j.ymgme.2004.09.008. doi: 10.1016/j.ymgme.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Serrano M, García-Silva MT, Martin-Hernandez E, O’Callaghan Mdel M, Quijada P, Martinez-Aragón A, et al. Kearns-Sayre syndrome: Cerebral folate deficiency, MRI findings and new cerebrospinal fluid biochemical features. Mitochondrion. 2010;10:429–32. doi: 10.1016/j.mito.2010.04.001. doi: 10.1016/j.mito.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Wray SH, Provenzale JM, Johns DR, Thulborn KR. MR of the brain in mitochondrial myopathy. AJNR Am J Neuroradiol. 1995;16:1167–73. [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa E, Hirano S, Yamanouchi H, Goto Y, Nonaka I, Takashima S. Progressive brainstem and white matter lesions in Kearns-Sayre syndrome: A case report. Brain Dev. 1994;16:416–8. doi: 10.1016/0387-7604(94)90133-3. doi: 10.1016/0387-7604(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 25.Sacher M, Fatterpekar GM, Edelstein S, Sansaricq C, Naidich TP. MRI findings in an atypical case of Kearns-Sayre syndrome: A case report. Neuroradiology. 2005;47:241–4. doi: 10.1007/s00234-004-1314-z. doi: 10.1007/s00234-004-1314-z. [DOI] [PubMed] [Google Scholar]
- 26.Grady JP, Campbell G, Ratnaike T, Blakely EL, Falkous G, Nesbitt V, et al. Disease progression in patients with single, large-scale mitochondrial DNA deletions. Brain. 2014;137(Pt 2):323–34. doi: 10.1093/brain/awt321. doi: 10.1093/brain/awt321. [DOI] [PMC free article] [PubMed] [Google Scholar]