

Abstract
The anti-protozoal drug pentamidine is active against opportunistic Pneumocystis pneumonia, but in addition has several other biological targets, including the NMDA receptor (NR). Here we describe the inhibitory potencies of 76 pentamidine analogs at 2 binding sites of the NR, the channel binding site labeled with [3H]MK-801 and the [3H]ifenprodil binding site. Most analogs acted weaker at the ifenprodil than at the channel site. The spermine-sensitivity of NR inhibition by the majority of the compounds was reminiscent of other long-chain dicationic NR blockers. The potency of the parent compound as NR blocker was increased by modifying the heteroatoms in the bridge connecting the 2 benzamidine moieties and also by integrating the bridge into a seven-membered ring. Docking of the 45 most spermine-sensitive bisbenzamidines to a recently described acidic interface between the N-terminal domains of GluN1 and GluN2B mediating polyamine stimulation of the NR revealed the domain contributed by GluN1 as the most relevant target.
Keywords: Pentamidine, NMDA receptor, Ifenprodil, Polyamine, Bisbenzamidine, Alkylation, Coupling, Radioligand binding, Rat brain, Molecular docking
1. Introduction
Pentamidine (1), a bisbenzamidine with two aromatic nuclei and an unbranched saturated five-carbon chain inserted between 2 phenolic ether oxygens (Chart 1), introduced as a trypanocidal agent,1 gained importance with the HIV pandemic as one of the most effective agents against opportunistic Pneumocystis pneumonia.2 Despite successes of antiretroviral therapy, still 20% of HIV-infected patients suffer from neurocognitive disorders, most likely caused by mediators like quinolinic acid released by invading glia.3,4 By virtue of its dicationic elongated structure, 1 is predestined to fit into the DNA minor groove,5,6 an interaction possibly underlying its chemotherapeutic properties. However, 1 has several other biological targets in addition, including the NMDA receptor (NR),7 the imidazoline I2 binding site,8 acid-sensing ion channels,9 and the potassium channel KIR2.1,10 as recently reviewed.11 The NR is one of the most common excitatory neurotransmitter receptors,12 activated by glutamic acid; pathologically increased interstitial quinolinic acid might lead to neurotoxicity by acting as agonist at this receptor.13 Pentamidine has been shown to block the NR at low micromolar concentration, but barely reaches the brain after systemic application.14 The conformationally restricted pentamidine analog N,N′-bis(p-amidinophenyl)homopiperazine (24; Chart 1) inhibits more potently than pentamidine the growth of Pneumocystis carinii in culture15 and the NR on rat brain membranes.16 Inhibition of the NR by 1 and 24 is sensitive to polyamines,7,17 suggesting the involvement of the polyamine regulatory site of the NR.18 The endogenous polyamines spermidine and spermine increase the opening frequency of the NR-associated ion channel by allowing the closure of an interface in the extracellular N-terminal domain between subunits GluN1 and GluN2B; this is brought about by neutralization of negative surface charges, which in the absence of polyamines repel each other and keep the interface open.18 Here, we present inhibitory potencies of several pentamidine analogs at the NR on rat brain membranes, with particular reference to the partial reversal of this inhibition by the polyamine spermine (polyamine sensitivity). While antimicrobial activities of these compounds have been evaluated in vitro, most of them have never been tested at the NR. We test these compounds as inhibitors of specific binding of the channel ligand [3H]MK-801,19 in absence and in presence of 30 µM spermine, and several of them also as inhibitors of [3H]ifenprodil binding.20 Also ifenprodil binds to the GluN1/2B interface, but somewhat more externally.21 The interaction is mainly mediated by H-bonds and hydrophobic interactions, not by charge neutralization. MK-801 is an open-channel blocker that, upon opening of the NR-associated channel, gains access to a site deeply located close to the narrow restriction of the passage way; specific binding of non-saturating concentrations of [3H]MK-801 can be taken as indicator for the accessibility and opening frequency of the channel.19 Finally, we present first attempts to dock most of these compounds to a recently described binding pocket shared by the NR subunits GluN1 and GluN2B.22
Chart 1.

2. Syntheses
All structures are shown in Tables 1–4. The syntheses of several pentamidine analogs with heteroatom variation in the bridge have been described23–28 (see Table S1 in the Supporting information for assignment). The syntheses of 9 and 11 and of their precursors 9a and 11a are described here for the first time (using established procedures). In short, 2 equiv of the appropriate 4-fluorobenzonitrile were subjected to nucleophilic displacement by 1 equiv of a diamino spacer (Scheme 1). The spacers were 1,5-diaminopentane and bis(2-aminoethyl) ether, respectively, and yielded the terminal nitrogens for the bridge. The resulting bisbenzonitriles 9a and 11a were converted in a sealed flask with ethanolic HCl to the iminoesters and thereafter, again in a sealed flask, under alkaline conditions to the respective bisbenzamidines 9 and 11 (Procedure A).23
Table 1.
Inhibition of [3H]MK-801 and [3H]ifenprodil binding to rat cortical membranes by pentamidine analogs with heteroatom variations in the bridge
 | |||||||
|---|---|---|---|---|---|---|---|
| X | YRY | RP | [3H]MK-801 | [3H]ifenprodil | |||
| IC50 ± SD (n) (µM) | Ratio | IC50 ± SD (n) | |||||
| No spm | 30 µM spm | No spm | |||||
| Arcaine | — | — | — | 4.60 ± 1.71 (4) | 196 (2) | 33.6 (2) | 24.4 (2) |
| N12N | — | — | — | 7.05 ± 2.38 (11) | 245 ± 30 (5) | 32.9 ± 6.3 (5) | 960 (2) |
| N4T8N | — | — | — | 0.52 ± 0.32 (46) | 6.10 ± 1.43 (24) | 15.1 ± 7.1 (24) | 49.3 (2) |
| 1 | O | CH2 | — | 2.68 ± 0.65 (9) | 24.9 ± 5.7 (7) | 9.1 ± 2.5 (7) | 32.5 (2) |
| 2 | O | O | — | 3.41 ± 0.52 (4) | 148 ± 22 (3) | 40.6 ± 4.2 (3) | 63.8 (2) |
| 3 | O | O | OMe | 10.6 ± 3.9 (4) | 343 ± 27 (3) | 33.1 ± 11.7 (3) | 43.7 (2) |
| 4 | O | S | — | 2.20 ± 0.32 (3) | 29.1 (2) | 12.3 (2) | 19.2 (2) |
| 5 | S | O | — | 1.22 ± 0.23 (3) | 23.8 ± 2.7 (3) | 20.2 ± 6.1 (3) | n.d. |
| 6 | O | O | — | 317 ± 20 (3) | 785 (2) | 2.59 (2) | n.d. |
| 7 | NH | CH2 | — | 3.10 ± 0.11 (3) | 42.3 ± 1.9 (3) | 13.7 ± 0.6 (3) | 20.6 ± 5.4 (3) |
| 8 | NMe | CH2 | — | 0.56 ± 0.08 (4) | 5.62 ± 2.88 (4) | 9.2 ± 3.5 (4) | 16.6 (2) |
| 9 | NH | CH2 | OMe | 2.88 (2) | 20.7 (2) | 7.2 (2) | 4.54 ± 1.43 (3) |
| 10 | NH | O | — | 0.85 ± 0.23 (7) | 38.5 ± 10.4 (6) | 50.6 ± 19.7 (6) | 11.4 ± 3.7 (3) |
| 11 | NMe | O | — | 0.76 ± 0.15 (4) | 12.5 ± 1.8 (3) | 15.2 ± 2.2 (3) | 21.9 ± 5.2 (3) |
| 12 | O | NH+Me | — | 21.2 ± 5.1 (3) | 143 (2) | 7.65 (2) | n.d. |
| 13 | O | N-Bs | — | 0.23 ± 0.06 (4) | 2.13 ± 0.17 (3) | 10.4 ± 1.56 (3) | 1.47 ± 0.55 (3) |
| 14 | O | NTs | — | 0.24 ± 0.08 (5) | 1.32 ± 0.23 (4) | 6.28 ± 1.18 (4) | 0.41 ± 0.13 (3) |
| 15 | O | NTs | OMe | 3.77 ± 1.37 (4) | 28.7 ± 4.4 (3) | 8.11 ± 2.91 (3) | 0.84 ± 0.48 (4) |
Number of experiments (n) in parentheses; if n = 2, no SD is indicated (see Supporting information for single values); spm, spermine; n.d., not determined. The column ‘ratio’ lists the mean ratios IC50 (30 µM spm)/IC50 (no spm). N12N, diaminododecane; N4T8N, 5-(4-aminobutyl)-2-thiopheneoctanamine;41 Bs, benzenesulfonyl.
Table 4.
Inhibition of [3H]MK-801 binding to rat cortical membranes by pentamidine analogs with additional aromatic rings
| IC50 ± SD (n) (µM) | Ratio | ||
|---|---|---|---|
| no spm | 30 µM spm | ||
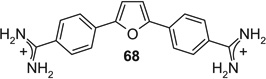 |
4.23 ± 2.47 (4) |
7.48 ± 2.51 (4) |
2.37 ± 1.69 (4) |
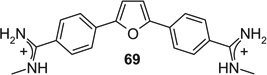 |
6.97 ± 2.15 (3) |
24.0 ± 19.2 (3) |
3.47 ± 2.20 (3) |
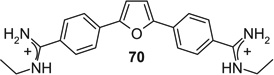 |
12.2 (2) |
30.7 (2) |
2.56 (2) |
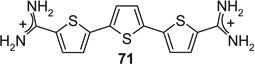 |
5.34 (2) |
6.76 (2) |
1.50 (2) |
 |
6.68 ± 1.25 (4) |
17.7 ± 6.3 (4) |
2.61 ± 0.61 (4) |
 |
9.9 ± 4.4 (3) |
34.2 (2) |
4.63 (2) |
 |
3.21 (2) |
13.4 (2) |
4.14 (2) |
 |
5.01 ± 3.28 (3) |
11.7 (2) |
4.26 (2) |
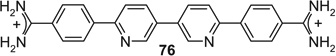 |
26.8 ± 13.1 (3) |
30.0 ± 6.7 (3) |
1.24 ± 0.38 (3) |
Definitions as in caption to Table 1.
Scheme 1.

Preparation of the dinitrile precursors of 9 and 11. Atom numbers in accordance with NMR data.
For the synthesis of 15a, the nitrile precursor of 15, the terminal heteroatoms in the bridge were (as in pentamidine) oxygens; this time, the leaving halogens were on the spacer N,N-bis(2-chloroethyl)-p-toluenesulfonamide, and the bridge oxygens were introduced with the benzonitrile (Scheme 2).
Scheme 2.

Preparation of the dinitrile precursor of 15. Atom numbers in accordance with NMR data.
The piperazine and homopiperazine derivatives are known compounds;15,29,30 also several of the open chain analogs have been described;31 40, 43 and 44 are described here for the first time and were prepared via the bisbenzonitriles in 3 steps. The first one was equivalent to step one in Scheme 1 (with DMF as the dry solvent). As difunctional spacers we used N,N′-dimethylpropylenediamine, 1,7-diaminoheptane and 1,8-diaminooctane, respectively. The obtained bisbenzonitriles were converted into the corresponding bisbenzamidines via the bisbenzamidoximes (Procedure B).31
Most of the remaining compounds are known32–38 (see Table S1). The dinitrile precursor 71a for the terthiophene derivative 71 was prepared by Pd-catalyzed coupling of 2 equiv 5-bromo-2-cyanothiophene to 1 equiv 2,5-bis(trimethylstannyl)thiophene (Scheme 3) as described for the addition of 2 phenyl residues to a central thiophene.39 The 3,3′-bipyridine bisbenzonitrile 76a was prepared in 2 steps, starting with Pd-catalyzed coupling of 2,5-dibromopyridine to 4-cyanophenylboronic acid (Scheme 4) as described for the synthesis of para-terphenyl.36 The resulting bromo nitrile 76b was dimerized (once more under Pd catalysis) to the bisbenzonitrile 76a. Both bisbenzonitriles 71a and 76a were finally converted to the respective bisbenzamidines 71 and 76 by LHMDS in THF (Procedure C).36
Scheme 3.

Preparation of the dinitrile precursor of 71.
Scheme 4.

Preparation of the dinitrile precursor of 76.
3. Results and discussion
3.1. Variation of bridge heteroatoms
The polyamine-sensitivity of the NR is one of the main physiological parameters evaluated in this study. We define this sensitivity as the ratio of two IC50s; the IC50 of a polyamine-sensitive inhibitor of [3H]MK-801 binding will be higher (weaker) in the presence of the polyamine (here 30 µM spermine) than in its absence. Replacing in the connecting chain of 1 the middle methylene group by oxygen (2; sometimes called ‘γ-oxa-pentamidine’), but not by sulfur (4), converted a moderately spermine-sensitive inhibitor into a highly spermine-sensitive one (in the absence of added spermine the potencies of 1, 2 and 4 were similar). Changing the 2 distal oxygens in the highly spermine-sensitive 2 to sulfurs yielded with 5 a compound more potent and more spermine-sensitive than pentamidine (although not reaching to the spermine-sensitivity of 2). Introducing nitrogens instead of O or S had even more pronounced consequences. A nitrogen atom in the center of the pentamidine bridge introduces a positive charge at physiological pH, while at the distal positions nitrogen has no such consequences (due to the neighboring aromatic nuclei). Indeed, 12 with central nitrogen acted much weaker than pentamidine, while at the two distal positions (7) N did not change at all the properties of pentamidine (inhibition of the NR by this compound has already been described40). Again, also in this molecule an oxygen in the center (10) conferred very high sensitivity to spermine (as in 2), but this time also the absolute potency was increased (by a factor 4.0). Remarkably, the potency of 7 (so to say ‘diaza-pentamidine’) was increased considerably (by a factor 5.5) by substituting the Ns with methyl groups (giving 8; without changing sensitivity to spermine). The high potency of 10, with O in the middle and Ns vicinal to the aromatic rings, was not further increased by N-methylation (11), and the extreme spermine sensitivity of 10 was reset to ‘normal’ by this modification.
Already these first observations demonstrate that absolute potencies and spermine-sensitivities of pentamidine congeners follow separate SARs. Substitution with methoxy at the aryl nuclei had minor or unfavorable influence (3 weaker than 2; 9 similar to 7; 15 weaker than 14), but aromatic substituents at a central N were always favorable, most likely because they take away the central positive charge. This latter strategy resulted in the most potent compounds of this series (13, 14), with spermine-sensitivities equivalent to or slightly weaker than that of pentamidine. Since the loss in potency after central introduction of N was not only equalized, but even reversed to a considerable gain, the aromatic residue may find a partner for favorable interaction in this middle part of the pharmacophore, as already suggested by previous studies.41–43 Imidazoline instead of amidine substituents (as in 6) were not tolerated at this target, as already shown for other pentamidine derivatives.44
3.2. Piperazine and homopiperazine as bridge
N,N′-Bis(p-amidinophenyl)homopiperazine (24)15 may be seen as related to the diaza-analogs 7 and 8 described above, constraining the bridge between the 2 Ns into a strictly defined ring. All homopiperazine- and piperazine-bridged bisbenzamidines inhibited the NR with similar spermine-sensitivities (most of them slightly above the sensitivity of 1), but the prototypic 24 clearly stands out from the rest by its absolute potency: none of the 17 analogs in Table 2 (16–23, 25–33) came close to this value (not even by a factor 10). All modifications, be it by switching from a seven- to a six-membered ring, be it by adding alkyl substituents to the terminal amidino groups or by joining these groups to fiveor six-membered rings, resulted in a clear loss of biological activity. Remarkably, the advantage of the prototypic homopiperazine 24 (IC50 0.25 µM) over the prototypic piperazine 16 (IC50 3.08 µM) disappeared upon amidine alkylation (IC50s of all compounds around 10 µM). This is in contrast to favorable effects of amidine alkylation in pentamidine analogs with furan- and pyridine dicarboxamide bridges (see below, Table 3, 63–66).
Table 2.
Inhibition of [3H]MK-801 and [3H]ifenprodil binding to rat cortical membranes by pentamidine analogs with piperazine and homopiperazine bridge (including open-chain isosters)
 | ||||||
|---|---|---|---|---|---|---|
| R | R′ | [3H]MK-801 | [3H]ifenprodil | |||
| IC50 ± SD (n) (µM) | Ratio | IC50 ± SD (n) | ||||
| No spm | 30 µM spm | No spm | ||||
| 16 | H | — | 3.08 ± 0.59 (4) | 70.7 (2) | 26.2 (2) | 17.0 (2) |
| 17 | i-Pr | — | 21.4 ± 5.2 (4) | 544 ± 34 (3) | 32.9 ± 5.8 (3) | n.d. |
| 18 | c-Pr | — | 19.4 ± 7.4 (4) | 466 (2) | 21.8 (2) | n.d. |
| 19 | n-Pr | — | 15.9 ± 4.9 (4) | 371 (2) | 21.8 (2) | n.d. |
| 20 | n-Bu | — | 10.3 ± 3.6 (5) | 139 ± 32 (4) | 16.0 ± 3.2 (4) | n.d. |
| 21 | c-Pent | — | 9.25 ± 3.85 (5) | 159 ± 59 (4) | 19.3 ± 6.4 (4) | n.d. |
| 22 | (CH2)2 | — | 6.43 ± 1.64 (4) | 106 ± 20 (3) | 15.1 ± 4.1 (3) | n.d. |
| 23 | (CH2)3 | — | 23.8 ± 8.9 (5) | 264 ± 26 (3) | 13.6 ± 5.2 (3) | n.d. |
| 24 | H | — | 0.25 ± 0.07 (14) | 5.47 ± 1.42 (6) | 20.4 ± 7.3 (6) | 19.1 (2) |
| 25 | i-Pr | — | 17.7 ± 4.8 (4) | 389 ± 85 (3) | 25.3 ± 10.5 (3) | n.d. |
| 26 | c-Pr | — | 11.2 ± 2.9 (4) | 212 ± 32 (4) | 19.9 ± 5.7 (4) | n.d. |
| 27 | t-Bu | — | 34.6 ± 4.1 (3) | 562 (2) | 16.8 (2) | n.d. |
| 28 | n-Bu | — | 9.94 ± 3.72 (3) | 103 ± 3 (3) | 11.2 ± 3.3 (3) | n.d. |
| 29 | c-Pent | — | 8.13 ± 0.29 (3) | 85.7 ± 7.8 (3) | 10.6 ± 1.2 (3) | n.d. |
| 30 | 2-HO-Et | — | 9.44 ± 2.70 (6) | 324 ± 34 (5) | 35.7 ± 6.9 (5) | n.d. |
| 31 | 3-HO-Pr | — | 41.2 ± 13.2 (4) | 427 (2) | 9.72 (2) | n.d. |
| 32 | (CH2)2 | — | 3.03 ± 0.21 (3) | 92.2 (2) | 30.1 (2) | n.d. |
| 33 | (CH2)3 | — | 12.8 ± 4.3 (3) | 143 ± 15 (3) | 11.7 ± 2.5 (3) | n.d. |
| 34 | H | H | 2.01 ± 0.76 (6) | 50.7 ± 13.8 (5) | 30.2 ± 10.4 (5) | 0.76 ± 0.05 (3) |
| 35 | Me | H | 3.18 ± 0.60 (3) | 85.3 (2) | 30.2 (2) | 7.14 (2) |
| 36 | Et | H | 2.61 ± 0.55 (4) | 48.1 ± 6.5 (3) | 19.3 ± 7.8 (3) | 13.0 (2) |
| 37 | Me | Me | 2.71 ± 0.41 (3) | 54.5 (2) | 20.9 (2) | 16.9 (2) |
| 38 | Et | Et | 1.14 ± 0.19 (3) | 13.8 (2) | 11.8 (2) | 9.16 (2) |
| 39 | H | H | 10.0 ± 2.3 (3) | 148 (2) | 17.0 (2) | 77.7 (2) |
| 40 | Me | Me | 1.84 ± 0.65 (4) | 24.7 ± 0.8 (3) | 14.7 ± 5.3 (3) | 31.2 (2) |
Definitions as in caption to Table 1;
c-pent, cyclopentyl.
Table 3.
Inhibition of [3H]MK-801 and [3H]ifenprodil binding to rat cortical membranes by pentamidine analogs with aliphatic linkers of increasing length, and with aromatic linkers
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| X | Y | n | R | Ar | [3H]MK-801 | [3H]ifenprodil | |||
| IC50 ± SD (n) (µM) | Ratio | IC50 ± SD (n) | |||||||
| No spm | 30 µM spm | No spm | |||||||
| 34 | NH | CH2 | 0 | — | — | 2.01 ± 0.76 (6) | 50.7 ± 13.8 (5) | 30.2 ± 10.4 (5) | 0.76 ± 0.05 (3) |
| 39 | NH | CH2 | 1 | — | — | 10.0 ± 2.3 (3) | 148 (2) | 17.0 (2) | 77.7 (2) |
| 41 | NH | CH2 | 2 | — | — | 4.55 ± 1.74 (3) | 84.2 (2) | 17.2 (2) | n.d. |
| 7 | NH | CH2 | 3 | — | — | 3.10 ± 0.11 (3) | 42.3 ± 1.9 (3) | 13.7 ± 0.6 (3) | 20.6 ± 5.4 (3) |
| 42 | NH | CH2 | 4 | — | — | 4.37 ± 0.32 (3) | 23.9 (2) | 5.26 (2) | n.d. |
| 43 | NH | CH2 | 5 | — | — | 4.67 ± 0.38 (3) | 25.4 (2) | 5.37 (2) | n.d. |
| 44 | NH | CH2 | 6 | — | — | 7.26 (2) | 31.1 (2) | 4.37 (2) | n.d. |
| 45 | NH | CO | 3 | — | — | 230 ± 97 (4) | 675 ± 131 (3) | 2.81 ± 0.76 (3) | 54.5, 65.6 (2) |
| 46 | NH | CO | 4 | — | — | 165 ± 68 (3) | 437 (2) | 3.46 (2) | n.d. |
| 47 | NH | CO | 5 | — | — | >1000 | >1000 | n.d. | n.d. |
| 48 | NH | CO | 6 | — | — | 22.9 ± 6.7 (4) | 170 ± 15 (3) | 7.25 ± 1.95 (3) | n.d. |
| 49 | NH | CO | 7 | — | — | 26.5 ± 2.6 (3) | 111 ± 13 (3) | 4.25 ± 0.94 (3) | n.d. |
| 50 | NH | CO | 8 | — | — | 15.7 ± 2.2 (3) | 62.6 ± 4.8 (3) | 4.07 ± 0.96 (3) | n.d. |
| 51 | NH | CO | 10 | — | — | 9.63 ± 1.45 (3) | 27.7 ± 6.9 (3) | 2.98 ± 0.58 (3) | n.d. |
| 52 | CO | NH | 3 | — | — | >1000 | >1000 | n.d. | n.d. |
| 53 | CO | NH | 4 | — | — | >1000 | >1000 | n.d. | n.d. |
| 54 | CO | NH | 5 | — | — | 688 (2) | >1000 | n.d. | n.d. |
| 55 | CO | NH | 6 | — | — | 266 (2) | 1208 (2) | 4.59 (2) | n.d. |
| 56 | CH2 | O | — | H | 1,2-Ph | 2.19 ± 0.85 (5) | 18.4 ± 3.2 (4) | 10.1 ± 3.2 (4) | 7.29 ± 2.85 (4) |
| 57 | O | CH2 | — | H | 1,2-Ph | 1.46 ± 0.60 (4) | 5.54 ± 1.23 (3) | 4.00 ± 0.93 (3) | 1.42, 2.05 (2) |
| 58 | O | CH2 | — | H | 1,3-Ph | 0.79 ± 0.32 (4) | 6.70 ± 0.89 (4) | 9.04 ± 1.93 (4) | 3.97 ± 1.59 (3) |
| 59 | O | CH2 | — | H | 1,4-Ph | 5.24 ± 1.12 (4) | 25.7 ± 0.9 (3) | 5.25 ± 1.29 (3) | 10.8, 16.8 (2) |
| 60 | O | CO | — | H | 1,4-Ph | 10.8 ± 1.7 (3) | 46.6 (2) | 3.98 (2) | 38.6, 27.0 (2) |
| 61 | NH | CO | — | H | 1,3-Ph | 42.3 ± 6.8 (4) | 36.7 (2) | 0.97 (2) | n.d. |
| 62 | NH | CO | — | H | 1,4-Ph | 53.9 ± 2.4 (3) | 52.9 (2) | 0.96 (2) | n.d. |
| 63 | NH | CO | — | H | 2,5-Fur | 152 ± 10 (3) | 153 ± 7 (3) | 1.01 ± 0.02 (3) | n.d. |
| 64 | NH | CO | — | i-Pr | 2,5-Fur | 10.6 ± 1.3 (3) | 174 ± 45 (3) | 16.9 ± 6.0 (3) | n.d. |
| 65 | NH | CO | — | H | 2,6-Pyr | 58.2 (2) | 70.9 (2) | 1.22 (2) | >100 |
| 66 | NH | CO | — | i-Pr | 2,6-Pyr | 6.94 ± 1.22 (3) | 74.9 ± 13.5 (3) | 10.8 ± 1.2 (3) | 50.1, 49.4 (2) |
| 67 | CO | NH | — | H | 2,6-Pyr | 125 (2) | 233 (2) | 1.85 (2) | n.d. |
Definitions as in caption to Table 1;
Fur, furandiyl; Pyr, pyridinediyl.
Breaking up the successful homopiperazine ring sets back the high potency of 24 to the ‘normal’ level of pentamidine (34 with short –CH2CH2– bridge) or even below (39 with long –CH2CH2CH2– bridge), while keeping the somewhat increased sensitivity to spermine. This is surprising, since formally the longer propyl bridge is closer to the pentyl bridge of pentamidine than the shorter ethyl bridge. Apparently, the 5 carbons chain in pentamidine forms a rather sharp loop, bringing the 2 benzamidine moieties as close as in 34 and closer than in 39. The potency of 34 was not increased by providing methyl or ethyl substituents to the ethyl bridge: 35–37 exhibited all pentamidine-like potencies (only the diethyl derivative 38 with IC50 1.14 µM exceeded somewhat this potency). On the other hand, the rather poor performance of the propyl bridge (in 39 with IC50 10 µM) was raised to the pentamidine level by methyl substituents (in 40 with IC50 1.84 µM). These results suggest a double role of the bridge, tuning the extension and orientation of the molecule and providing itself favorable attachment points. It might be worth to explore larger substituents, given the success of central aromatic substituents in the first series (13 and 14).
3.3. Bridges of increasing length
The shorter chain homologs of pentamidine (butamidine and propamidine) have been tested as inhibitors of [3H]MK-801 binding to rat brain membranes,40 returning the IC50s 2.59, 17.7 and 5.54 µM for 5, 4 and 3 methylene groups between the oxygens, respectively. Also in our diaza-analogs we observed two optima, one with an ethyl and another with a pentyl bridge (34 and 7; Table 3).
We also tested the influence of chain length with two other types of bridges. In the first one, the carbons vicinal to the nitrogens were carbonyl groups, changing the chain geometry at this position from sp3 (tetraedric) to sp2 (planar), with dramatic consequences. Although the dianilide 45 was equivalent in total bridge length to pentamidine, it turned out 100-fold weaker, with practically no sensitivity to spermine; the same was true for 46, with butyl between the two amide groups (–NH–CO–butyl–CO–NH–). The next homolog 47 was still weaker (IC50 >1 mM). Surprisingly, activity returned in the higher homologs 48–51, even with some indication for spermine sensitivity (48). Potencies were very weak, if the amide groups were inverted (–CO–NH–butyl–NH–CO–, 53), and also here the weakest compounds were those of middle size. We conclude that the chain linking the two benzamidine moieties should either be short or should exhibit enough degrees of freedom for folding to adopt the critical short distance between the end charges. With rigid segments like amide bonds, such a distance appears difficult to achieve.
3.4. Aryl inserted into the bridge
Finally, we explored the influence of aromatic nuclei inserted into the bridge linking the two benzamidine moieties. This kind of modification was not only well tolerated, but even increased the potency, if the geometry of pentamidine was maintained as in 58 (IC50 0.79 µM). In this compound, still a continuous chain of 5 carbon atoms separated the 2 oxygens. Also a chain of 4 carbon atoms as in 57 was compatible with high potency (IC50 1.46 µM), but only with quite low spermine sensitivity. Interestingly, this sensitivity was restored simply by switching atom order from –O–C–(1,2-Ph)–C–O– (57) to –C–O–(1,2-Ph)–O–C– (56), with no loss in potency. Inserting the phenyl nucleus in 1,4 orientation (59) was less favorable.
Still weaker compounds resulted from insertions of an aromatic group between two amide bonds, nevertheless returning some surprising SARs. While two ester bonds on both sides of a central phenyl nucleus (with the alcoholic oxygens at the outer positions; 60) were tolerated almost as well as the ether bonds in 59, amide bonds (with the nitrogens at the outer positions; 61 and 62) led to a strong drop in potency and to the complete loss of spermine sensitivity. Replacing the phenyl nucleus between these amide bonds by a furan nucleus (63) lowered these figures even more, but to our surprise potency returned (IC50 10.6 µM) upon alkylation of the distal amidine groups (64), with parallel restoration of spermine sensitivity. An analogous phenomenon was observed after insertion of a pyridine nucleus: weak potency without (65), but appreciable potency with amidine alkylation (66; IC50 6.94 µM), also here with remarkable restoration of sensitivity to spermine. It should be taken into consideration, however, that these alkylations achieved medium potencies starting from a rather weak level (similar potencies had been achieved by alkylations of 16 and 24—Table 2—starting from a much higher level).
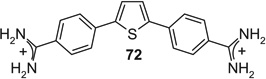
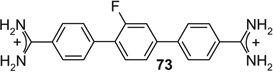
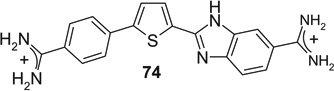
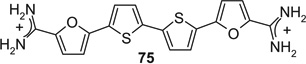
If aryl nuclei were inserted without interspersed flexible linkers, compounds were obtained with absolute potencies (Table 4) either similar to pentamidine (68, 71, 74, 75) or slightly weaker (69, 70, 72, 73, 76). In all these compounds, however, sensitivity to spermine was practically absent. In the bioisosteric sequence 71, 72 and 73, potencies seem to weaken with increasing distance between the distal charges (IC50s 5.3 < 6.7 < 9.9 µM). The low potency of the tetracyclic 76 may be due to the still longer distance between them. Although 75 was also a tetracyclic compound, here the chain of five-membered rings would allow for some adjustment of length, possibly explaining the higher potency of this compound. Such flexibility cannot explain the relatively high potency of the benzimidazole-containing 74, one of the more extended molecules in this series; here, the particular imidazole signature may have contributed favorably to receptor interaction.
3.5. Inhibition of [3H]ifenprodil binding
Polyamines activate the NR at an interface between the subunits GluN1 and GluN2B.18,21 This site is formed by acidic amino acid residues contributed by both extra-cellular N-terminal domains; their neutralization by polyamines is hypothesized to result in the close apposition of these domains, facilitating the activation of the NR by agonists. A second site located more externally mediates inhibition of the NR by ifenprodil.21 No high affinity ligands have been described binding directly to the first site; however, the outer site can be labeled with [3H]ifenprodil. Compounds can usually be tested directly for their affinity to this site via their potency to displace this radioligand from neuronal membranes, with the precaution that indirect influences of polyamines on [3H]ifenprodil binding have been demonstrated.45
Some of our test compounds were quite potent displacers of [3H]ifenprodil (last column in Tables 1–3). The most potent was 14, a pentamidine with tosylated N in the middle of the bridge (IC50 0.41 µM), that was also a potent displacer of [3H]MK-801 (0.24 µM). Quite potent too were 15 (the methoxy derivative of 14; 0.84 µM), 13 (also a congener of 14; 1.47 µM), 34 (the diaza analog with the shortest bridge; 0.76 µM), and 57 (with phenyl in the bridge; 1.74 µM). Of all 31 compounds tested at both sites, only 3 were more potent at the ‘IF-site’, the other 28 more potent at the ‘MK-site’ (9 of them even more than 10-fold). Affinities of 31 compounds tested at both sites (including 3 prototypic polyamine inverse agonists) were significantly correlated to each other (Fig. 1A). The correlation line had no unity slope; high affinity compounds were more similar at both sites, but low affinity compounds more dissimilar. On the other hand, affinity at the IF-site was no predictor of spermine-sensitivity at the MK-site (Fig. 1B).
Figure 1.

Inhibition by 31 dicationic compounds at the ifenprodil site of the NR correlates with inhibition at the channel binding site (A, interrupted line; r2 = 0.22, p<0.01), but not with the sensitivity of this inhibition to 30 µM spermine (B; r2 = 0.08, n.s.); data from Tables 1–3.
3.6. Modeling of potential sites of action
In an attempt to identify the possible molecular target mediating spermine-sensitive inhibition of the NR by bisbenzamidines, we performed docking calculations for the 45 most spermine-sensitive bisbenzamidines (see Supporting information, Fig. S1) at a site proposed to mediate polyamine-stimulation of the NR.18,21,22 The endogenous polyamines spermidine and spermine (and also Mg2+) by virtue of their positive charges neutralize surface charges on the N-terminal domains of subunits GluN1 and GluN2B. In the non-stimulated state (Low Po mode; Po stands for open probability), these negative surface charges keep the GluN1/2B domains apart from each other. Polyamines induce the High Po mode by inserting themselves between the domains, leading them together. Antagonists interfering with this mechanism can be expected to stabilize the Low Po mode, by binding to only one of the 2 surfaces (without leading them together). Although stimulation of the NR by polyamines exhibits parallels to stimulation by increased pH, not only GluN1/2B, but also GluN1/2A and GluN1/2D combinations are subject to proton inhibition.46 Systematic examination of mutated receptors did not result in correlations between spermine and proton sensitivities,18 and also in our own studies,42,47 inhibition by similar dicationic compound families was differently influenced by spermine and pH. Thus, stimulation by polyamines seems to depend on the pattern of specific acidic amino acid residues on the domain surfaces, rather than on the general neutralization of a local acidic milieu. Bisbenzamidines were docked separately to the putative polyamine domains either of GluN1 or of GluN2B, departing from the whole GluN1/N2B complex in the Low Po mode, i.e. with the domains separated from each other. We obtained highly significant (r2 = 0.42; p<0.001) correlation for GluN1 (Fig. 2A), but not for GluN2B (Fig. 2B). No correlation was obtained with the domains close to each other (not shown). We conclude that more than 40% of the variability in the potency of these bisbenzamidines is explained by their interaction with targets on the polyamine domain of GluN1 in the Low Po mode. A still higher correlation may have been obtained at pure GluN1/2B heterodimeric NRs; the majority of native NRs as used by us, however, seem to consist of GluN1/2A/2B heterotrimers,48 with consequences for the GluN1/2B interface.49 The GluN1/2B selective antagonist ifenprodil blocks with high affinity only 23% of NRs in our native membranes prepared from rat frontoparietal cortex and hippocampus.50 Nevertheless, we tried to dock the same compounds also to the ifenprodil site proposed more externally at the GluN1/2B interface.21 The obtained correlation (r2 = 0.18; Fig. S2B), while less pronounced than for the ‘polyamine site’ on GluN1, was significant (p<0.01). Thus, also this site may have contributed to the variability of inhibitory potency; however, for none of these compounds we observed partial inhibition of [3H]MK-801 binding as typical for inhibition of [3H]MK-801 binding by ifenprodil, and in general binding of [3H]MK-801 was more potently inhibited than binding of [3H]ifenprodil (Fig. 1A). To some extent, significant docking of bisbenzamidines also to the IF-site may be explained by mutual influences between spermine and ifenprodil at native NRs.44
Figure 2.

Relationship between goodness of fit to a polyamine site modeled at the GluN1/2B interface and potency to inhibit the binding of [3H]MK-801 to native rat brain membranes. (A) Docking to GluN1, r2 = 0.42. (B) Docking to GluN2B, r2 = 0.06.
Finally, we created an overlay of pentamidine (1) with 8 of the most potent analogs (5, 8, 10, 11, 13, 14, 38, 58), docked to the amino acid residues on GluN1 allegedly mediating stimulation by polyamines (Fig. 3). Bisbenzamidines with flexible linker (as 1 itself) were able to partially wrap the helix α5 containing amino acid residues E185, E186 and E181, the amidine groups approaching the negatively charged ω-carboxylic groups. On the other hand, given the high number of acidic amino acid residues at this interface, several other modes of interaction may be possible as well, and not just the one suggested in the figure. The homopiperazine-bridged compound 24 did not superimpose with these compounds (not shown); although of high potency, it did not yield high docking scores in this model. Also other bisbenzamidines with inflexible bridge (22, 25) yielded Gold Fitness scores below expectation (see Fig. 2A).
Figure 3.

Binding mode of an overlay of 1 with the most potent analogs (5, 8, 10, 11, 13, 14, 38, and 58—with omission of 24) to the spermine binding site of GluN1 in the Low Po mode. Six amino acid residues of GluN1 known to influence spermine stimulation and the bisbenzamidines are represented as sticks with carbon atoms in coral and green, respectively. Surface representation of 1 is colored light blue.
3.7. Competition or no competition?
Already Reynolds and Aizenman,7 the discoverers of pentamidine as NR antagonist, demonstrated that inhibition of [3H]MK-801 binding by pentamidine was counteracted by the endogenous polyamine spermidine (the shorter-chain homolog of spermine). Since in their hands pentamidine did not only shift der spermidine dose/response curve to the right, but also reduced maximum stimulation, they excluded competitive interaction. Similar data were produced by us for pentamidine and 2 congeners, compounds 10 and 13 (Fig. 1S, supplementary information) in ‘competition’ with spermine. Earlier studies by two of us (MLB, PR)50 had demonstrated that interaction between dicationic longchain inhibitors as diaminododecane (N12N) and pentamidine with spermine-stimulation of [3H]MK-801 binding to native rat membranes was not pronounced enough to be explained by direct competition alone. Also another observation argues against simple direct competition: While polyamine-stimulation of NRs is limited to NRs with GluN2B subunit,18 the prototypic inverse polyamine agonist arcaine inhibits also NRs without GluN2B.51 It also remains to be explained why ‘polyamine inverse agonists’ reduce [3H]MK-801 binding in well washed neuronal membranes (with no effective amounts of the endogenous polyamines spermidine and spermine left). Do they even then drive open to the Low Po mode the GluN1/N2 interface, and against which ‘polyamine-like’ agent? Evidently, spermine and spermidine on the one side and dicationic inhibitors on the other side have additional effects. We interpret our docking data as indication that a considerable part of NR inhibition by bisbenzamidines’ is mediated by targets at the GluN1/2B interface also mediating stimulation by spermine. This includes competition, but leaves ample room for other activities of both compound families.
4. Conclusions
Motivated by the high potency of the homopiperazine-bridged bisbenzamidine 24 as polyamine-sensitive NR inhibitor, we undertook a detailed SAR study with 76 pentamidine analogs prepared in 5 different synthetic laboratories. We identified 9 spermine-sensitive inhibitors with higher potency than 1 (see Fig. S4). Four of them (5, 8, 10, 11) were isosteric to 1, with heteroatom variations in the bridge. Three others had aromatic rings inserted in (58) or attached to the bridge (13, 14), confirming earlier observations pointing to a role for such aromatic rings in target interaction.41–43 Among analogs with shorter bridge, only 38 with 2 ethyl substituents was more potent than 1 (ANOVA, p<0.05). Ironically, the lead homopiperazine 24 stayed the only high-potency bisbenzamidine of this structural type; none of the other homopiperazines came close. In addition, highly significant docking to the GluN1 part of the polyamine-sensitive GluN1/2B interface did not include 24: the docking score for 24 was even lower than that obtained for 1. Thus to provide a theoretical explanation for the particular behavior of 24, we still need more refined docking models based on the molecular landscape of the real (and still unknown) polyamine-sensitive GluN1/2B interface in heterotrimeric NRs constituting the majority in native rat hippocampal and cortical neurons.49
5. Experimental section
All chemicals were purchased from major chemical suppliers as high or highest purity grade and used without further purification. Melting points were determined with an Electrothermal 9001 Digital Melting Point. 1H NMR spectra in solution were recorded with a Bruker Avance DMX 400 or with Varian 300 V NMR S, and chemical shifts δ (ppm) in solutions were referenced to TMS (the solvent is given with the spectral data). Atom numbers are given in Schemes 1 and 2. Elemental analyses or HRMS are provided for new compounds.
5.1. New syntheses
The bisbenzamidines 9, 11, 15, 40, 43, 44, 71 and 76 were prepared as described below. For the conversion of the precursor bisbenzonitriles into bisbenzamidine hydrochlorides, 3 different procedures were used:
5.1.1. Procedure A
The appropriate dinitrile (5 mmol) was stirred in a sealed flask with a saturated solution of HCl in anhydrous ethanol (25–50 mL) for 20–40 h at rt. The solvent was evaporated in vacuo (or dry diethyl ether was added until a solid was precipitated). The solid residue was filtered, washed with diethyl ether and dried under vacuum over anhydrous CaCl2 for 2–6 h. The crude iminoester obtained in high yield was added to a saturated solution of NH3 in anhydrous ethanol (25–50 mL). The mixture was stirred at rt for 24–48 h in a sealed vessel. The solvent was evaporated in vacuo, and the solid residue was mixed with 10% aq NaOH (10 mL). The precipitate of free amidine was filtered, washed with water, and dried under reduced pressure over anhydrous CaCl2. The white microcrystalline solid was suspended in anhydrous ethanolic HCl (10 mL) and boiled for a few min to obtain the appropriate dihydrochloride salt.
5.1.2. Procedure B
A mixture of hydroxylamine hydrochloride (4.15 g, 60 mmol) and sodium methoxide (3.24 g, 60 mmol) in DMF (15 mL) was heated at 50 °C for 1 h. The precipitate was discarded and the bisbenzonitrile (5 mmol) was added to the filtrate. The so-obtained mixture was heated at 50 °C for 24 h. The reaction medium was then poured into water. The obtained bisbenzamidoxime was collected by filtration and thoroughly washed with water, ethanol, and finally ether. In the next step, a mixture of the bisbenzamidoxime (3 mmol), ammonium formate (1.80 g, 30 mmol), and 10% Pd on carbon (0.3 g) in acetic acid (15 mL) was heated under reflux for 2 h. After cooling the mixture was filtered on a cake of alumina and the filtrate was concentrated under reduced pressure. Addition of a solution of HCl in isopropanol (6 M, 15 mL) afforded a precipitate that was washed with water and ethanol to yield the desired bisbenzamidine (dihydrochloride).
5.1.3. Procedure C
The dinitrile (1 mmol) was allowed to stir overnight in a 1 M solution of LHMDS in THF (20 mL) as described for the conversion of the dinitrile of para-terphenyl to the respective bisamidine.36
5.1.4. Syntheses of 9, 11, and 15
5.1.4.1. 9a 1,5-Bis(1-cyano-3-methoxyphenylamino)pentane
3-Methoxy-4-fluorobenzonitrile (3.02 g, 20mmol), 1,5-diaminopentane dihydrochloride (1.75 g, 10mmol) and anhydrous K2CO3 (11.04 g, 80mmol) were stirred together at 150 °C in NMP (30mL) for 3 h (progress of the reaction was followed by TLC). The hot reaction mixture was poured into ice water (400 mL). The obtained solid was filtered, washed with water and dried. Crystallization from EtOH gave 0.92 g of yellow solid 9a (yield 24%), mp 153–155 °C. 1H NMR (299.87MHz, CDCl3) δ: 1.53–1.55 (m, 2H, Y = CH2), 1.72 (quintet, J = 7.2 Hz, 4H, 9- and 9′-CH2), 3.19 (t, J = 6.9Hz, 4H, 8- and 8′-CH2), 3.85 (s, 6H, 2× RP = OCH3), 4.75 (br s, 2H, 2× RN = NH), 6.50 (d, J = 8.1 Hz, 2H, H-5,-5′), 6.89 (br s, 2H, H-2,-2′), 7.18 (br d, J = 8.1 Hz, 2H, H-6,-6′). For C21H24N4O2 (364.45 g/mol) calcd: C, 69.21; H, 6.64; N, 15.37; found: C, 68.92; H, 6.91; N, 15.03.
5.1.4.2. 9 1,5-Bis(1-amidino-3-methoxyphenylamino)pentane dihydrochloride
This diamidine was obtained by Procedure A from the bisbenzonitrile 9a. The solvent was evaporated in vacuo. Crude product was dissolved in water and then precipitated by addition of dry acetone. Precipitated solid was filtered, washed with acetone and dried to give 9 as a yellow solid (yield 37%), mp 283.5–284.5 °C. 1H NMR (299.87 MHz, DMSO-d6) δ: 1.38–1.40 (m, 2H, Y = CH2), 1.59 (quintet, J = 7.2 Hz, 4H, 9- and 9′-CH2), 3.18 (m, 4H, 8- and 8′-CH2), 3.87 (s, 6H, 2× RP = OCH3), 6.01 (br s, 2H, 2× RN = NH), 6.64 (d, J = 8.4Hz, 2H, H-5,-5′), 7.33 (d, J = 1.5 Hz, 2H, H-2,-2′), 7.46 (dd, J1 = 1.5 Hz, J2 = 8.4Hz, 2H, H-6,-6′), 8.56 (br s, 4H, NH2), 8.90 (br s, 4H, NH2). For C21H30N6O2·2HCl·1½H2O (498.46 g/mol) calcd: C, 50.60; H, 7.02; N, 16.85; found: C, 50.43; H, 7.05; N, 16.48.
5.1.4.3. 11a 1,5-Bis[(1-cyanophenyl)N-methylamino]-3-oxapentane
The bisbenzonitrile 11a was obtained from 2 equiv 4-fluorobenzonitrile and 1 equiv 2,2′-methylaminodiethyl ether with K2CO3 in DMF (Scheme 1) as a white solid, mp 88.2–89.4 °C. 1H NMR (299.87 MHz, CDCl3) δ: 2.98 (s, 6H, 2× RN = CH3), 3.53–3.62 (m, 8H, 8-, 8′, 9 and 9′-CH2), 6.62–6.65 (m, 4H, H-5,-3,-5′,-3′), 7.42–7.45 (m, 4H, H-6,-2,-6′,-2′). HRMS (ESI-ToF) M+Na C20H22N4O+Na, calcd: m/z 357.1691; found: m/z 357.1697.
5.1.4.4. 11 1,5-Bis[(1-amidinophenyl)N-methylamino]-3-oxapentane
This diamidine was obtained by Procedure A from the dinitrile 11a. The white solid was filtered off and dried at 80 °C for 1 h to obtain 0.356 g of 11 (yield 90%) mp 276–279 °C (dec). 1H NMR (299.87 MHz, DMSO-d6) δ: 2.98 (s, 6H, 2× RN = CH3), 3.57–3.60 (m, 8H, H-8,-9,-8′,-9′), 6.79–6.82 (m, 4H, H-5,-3,-5′,-3′), 7.70–7.73 (m, 4H, H-6,-2,-6′,-2′), 8.48 (br s, 4H, NH2), 8.87 (br s, 4H, NH2). For C20H28N6O·2HCl (441.40 g/mol) calcd: C, 54.42; H, 6.85, N 19.04; found: C, 54.28; H, 6.96; N, 18.89.
5.1.4.5. 15a N,N-Bis[2-(1-cyano-3-methoxyphenoxy)ethyl]-4-methylbenzenesulfonamide
3-Methoxy-4-hydroxybenzonitrile (1.49 g, 10 mmol) and N,N-bis(2-chloroethyl)-4-methylbenzenesulfonamide (1.48 g, 5 mmol) in NMP (20 mL) were stirred at 145 °C together with anhydrous K2CO3 (2.76 g, 20 mmol) (progress of the reaction was followed by TLC). After 6 h the reaction mixture was poured into ice water (300 mL). The obtained solid was filtered, washed with water and dried. The crude product was dissolved in hot EtOH and filtered hot. The precipitate was purified by column chromatography (Merck Silica gel 60, 230–400 mesh ASTM, eluent CH2Cl2:MeOH—98:2) to afford 1.31 g of 15a (yield 50%), mp 177–179 °C. 1H NMR (400.13 MHz, CDCl3) δ: 2.34 (s, 3H, 11-CH3), 3.61 (t, J = 6.0 Hz, 4H, 9- and 9′-CH2), 3.69 (s, 6H, 10- and 10′-OCH3), 4.24 (t, J = 6.0 Hz, 4H, 8- and 8′-CH2), 6.82 (d, J = 8.4 Hz, 2H, H-5,-5′), 6.98 (d, J = 2 Hz, 2H, H-2,-2′), 7.16–7.21 (m, 4H, H-6,-6′,-3″,-5″), 7.64–7.66 (m, 2H, H-2″,-6″). For C27H27N3O6S (521.60 g/mol) calcd: C, 62.17; H, 5.22; N, 8.06; S, 6.15; found: C, 62.03; H, 5.47; N, 7.86; S, 6.32.
5.1.4.6. 15 N,N-Bis[2-(1-amidino-3-methoxyphenoxy)ethyl]-4-methylbenzenesulfonamide dihydrochloride
This diamidine was obtained by Procedure A from the bisbenzonitrile 15a. The solvent was evaporated in vacuo to give 15 as a yellow solid (yield 86%), mp 233–234 °C. 1H NMR (299.87 MHz, DMSO-d6) δ: 2.36 (s, 3H, 11-CH3), 3.65 (t, J = 5.7 Hz, 4H, 9- and 9′-CH2), 3.80 (s, 6H, 10- and 10′-OCH3), 4.27 (t, J = 5.7 Hz, 4H, 8- and 8′-CH2), 7.11– 7.14 (m, 2H, H-5,-5′), 7.36–7.39 (m, 2H, H-3″,-5″), 7.53–7.55 (m, 4H, H-6,-2,-6′,-2′), 7.75–7.78 (m, 2H, H-2″,-6″), 9.12 (br s, 4H, NH2), 9.39 (br s, 4H, NH2). For C27H33N5O6S·2HCl·H2O (646.60 g/mol) calcd: C, 50.16; H, 5.77; N, 10.83; S, 4.96; found: C, 50.36; H, 5.94; N, 10.67, S, 4.80.
5.1.5. Syntheses of 40, 43, and 44
40, 43 and 44 were prepared via the bisbenzonitriles 40a, 43a and 44a. For their synthesis, 4-fluorobenzonitrile (4.84 g, 40 mmol), the appropriate diamine (20 mmol), and K2CO3 (4.52 g, 40 mmol) in DMF (25 mL) were heated under reflux for 8 h. The mixture was then poured into water. The desired bisbenzonitrile was collected by filtration and thoroughly washed with water, ethanol, and finally ether. The bisbenzonitriles were converted to the bisbenzamidines by Procedure B.
5.1.5.1. 40a 1,3-Bis[(1-cyanophenyl)N-methylamino]propane
This bisbenzonitrile (75%) was obtained from the appropriate diamine N,N′-dimethylpropylenediamine as a white solid, mp 139–140 °C. 1H NMR (300 MHz, CDCl3) δ: 1.92 (m, 2H, CH2), 3.01 (s, 6H, N-CH3), 3.43 (t, J = 7 Hz, 4H, N-CH2), 6.62 (d, J = 9 Hz, 4H, H-5,-3, H-5′,-3′), 7.46 (d, J = 9 Hz, 4H, H-6,-2, H-6′,-2′). IR: 2945, 2909, 2893, 2206, 1602, 1520 cm−1. HRMS (ESI-ToF) MH+ C19H21N4, calcd: m/z 305.1766; found: m/z 305.1767.
5.1.5.2. 40 1,3-Bis[(1-amidinophenyl)N-methylamino]propane dihydrochloride
From the nitrile precursor 40a, Procedure B yielded the desired bisbenzamidine 40 (55%) as a white solid, mp >300 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ: 1.73 (m, 2H, CH2), 3.00 (s, 6H, N-CH3), 3.51 (t, J = 7 Hz, 4H, N-CH2), 6.82 (d, J = 9 Hz, 4H, H-5,-3,-5′,-3′), 7.81 (d, J = 9 Hz, 4H, H-6,-2,-6′,-2′), 8.85 (br s, 4H, NH2), 9.09 (br s, 4H, NH2). IR: 3130, 3040, 2970, 1682, 1609, 1487, 1393 cm−1. HRMS (ESI-ToF) MH+ C19H27N6, calcd: m/z 339.2297; found: m/z 339.2300.
5.1.5.3. 43a 1,7-Bis[(1-cyanophenyl)amino]heptane
This bisbenzonitrile (85%) was obtained from the appropriate diamine 1,7-diaminoheptane as a white solid, mp 136–137 °C. 1H NMR (300 MHz, CDCl3) δ: 1.40–1.60 (br m, 10H, CH2), 3.15 (t, J = 7 Hz, 4H, N-CH2), 4.13 (br s, 2H, NH), 6.53 (d, J = 9 Hz, 4H, H-5,-3, H-5′,-3′), 7.41 (d, J = 9 Hz, 4H, H-6,-2, H-6′,-2′). IR: 3384, 2937, 2875, 2853, 2198, 1603, 1524 cm−1. HRMS (ESI-ToF) MH+ C21H25N4, calcd: m/z 333.2079; found: m/z 333.2070.
5.1.5.4. 43 1,7-Bis[(1-amidinophenyl)amino]heptane dihydrochloride
From the nitrile precursor 43a, Procedure B yielded the desired bisbenzamidine 43 (40%) as a white solid, mp >270– 272 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ: 1.30–1.60 (br m, 10H, CH2), 3.08 (t, J = 7 Hz, 4H, N-CH2), 6.65 (d, J = 9 Hz, 4H, H-5,-3,-5′,-3′), 7.67 (d, J = 9 Hz, 4H, H-6,-2,-6′,-2′), 8.68 (br s, 4H, NH2), 8.90 (br s, 4H, NH2). IR: 3298, 3091, 2931, 2842, 1660, 1606, 1352 cm−1. HRMS (ESI-ToF) MH+ C21H31N6, calcd: m/z 367.2610; found: m/z 367.2607.
5.1.5.5. 44a 1,8-Bis[(1-cyanophenyl)amino]octane
This bisbenzonitrile (80%) was obtained from the appropriate diamine 1,8-diaminooctane as a white solid, mp 152–153 °C.1H NMR (300 MHz, CDCl3) δ: 1.37–1.62 (br m, 12H, CH2), 3.14 (t, J = 7 Hz, 4H, N-CH2), 4.14 (br s, 2H, NH), 6.53 (d, J = 9 Hz, 4H, H-5,-3, H-5′,-3′), 7.41 (d, J = 9 Hz, 4H, H-6,-2, H-6′,-2′). IR: 3383, 2927, 2852, 2203, 1605, 1526 cm−1. HRMS (ESI-ToF) MH+ C22H27N4, calcd: m/z 347.2236; found: m/z 347.2224.
5.1.5.6. 44 1,8-Bis[(1-amidinophenyl)amino]octane dihydrochloride
From the nitrile precursor 44a, Procedure B yielded the desired bisbenzamidine 44 (25%) as a white solid, mp >263– 267 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ: 1.30–1.60 (br m, 12H, CH2), 3.08 (t, J = 7 Hz, 4H, N-CH2), 6.64 (d, J = 9 Hz, 4H, H-5, -3,-5′,-3′), 7.67 (d, J = 9 Hz, 4H, H-6,-2,-6′,-2′), 8.68 (br s, 4H, NH2), 8.89 (br s, 4H, NH2). IR: 3291, 3112, 2927, 2851, 1637, 1608, 1494, 1464, 1351 cm−1. HRMS (ESI-ToF) MH+ C22H33N6, calcd: m/z 381.2767; found: m/z 381.2759.
5.1.6. Syntheses of 71 and 76
The bisbenzamidines with 3 (71) and 4 aromatic nuclei in a row (76) were prepared by first synthesizing, by Pd-catalyzed condensation, their dinitrile precursors 71a and 76a, respectively, and by converting them to the desired bisbenzamidines by Procedure C.
5.1.6.1. 71a 5,5″-Dicyano-2,2′:5′,2″-terthiophene
To a round-bottom flask were added 3.76 g (20 mmol) 5-bromo-2-cyanothiophene in 70 mL 1,4-dioxane, 0.46 g (2 mol %) Pd(PPh3)4 and 4.12 g (10 mmol) 2,5-bis(trimethylstannyl) thiophene. The mixture was allowed to reflux overnight. The solvent was removed under reduced pressure. Hexane was added to the residue and the precipitate was filtered. The solid was dissolved in 150 mL CH2Cl2 and washed with 5% NaF. The organic layer was dried over Na2SO4. The solvent was evaporated, the product was dried under reduced pressure at 50 °C (12 h) yielding 71a as orange needles (2.10 g, 70%); mp 206–7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.94 (d, 2H, J = 4 Hz), 7.58 (s, 2H), 7.53 (d, 2H, J = 4 Hz). MS (FAB): m/z 298 (M+). Anal. calcd for C14H6N4S3: C, 51.51; H, 1.85; N, 17.16; found: C, 51.44; H, 2.12; N, 17.27.
5.1.6.2. 71 5,5″-Diamidino-2,2′:5′,2″-terthiophene dihydrochloride
This diamidine was obtained from the dinitrile 71a by Procedure C as a brown solid (0.31 g, 76%); mp >320 °C (dec). 1H NMR (400 MHz, DMSO-d6/50 °C) δ: 9.66 (br s, 4H), 9.16 (br s, 4H), 8.11 (d, 2H, J = 4.2 Hz), 7.60 (d, 2H, J = 4.2 Hz), 7.57 (s, 2H). MS (FAB): m/z 333 (M++1). Anal. calcd for C14H12N4S3·2HCl·¼H2O: C, 41.02; H, 3.56; N, 13.66; found: C, 41.01; H, 3.62; N, 13.55.
5.1.6.3. 76b 4-(5-Bromopyridin-2-yl)benzonitrile
2,5-Dibromopyridine (1.18 g, 5 mmol) was coupled to 4-cyanophenylboronic acid (0.88 g, 6 mmol) with Pd(PPh3)4 (0.231 g, 0.2 mmol) as described for linking together the dinitrile of para-terphenyl.36 Finally, 0.9 g 76b (70%) was obtained: mp 174–175 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.90–8.03 (m, 3H), 8.14–8.24 (m, 3H), 8.80 (s, 1H). MS (m/z, rel. int.); 259 (M+, 100), 179 (70), 152 (55).
5.1.6.4. 76a 4,4′-[(3,3′Bipyridine)-6,6′-diyl]dibenzonitrile
Homocoupling of the above bromo nitrile 76b (0.78 g, 3 mmol) with Pd(PPh3)4 (0.231 g, 0.2 mmol) and hexa-n-butylditin (1.04 g, 1.8 mmol) was achieved as described for other ring systems.52 After washing the precipitate with ether, 0.82 g 76a was obtained (77%): mp >300 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ: 7.96 (d, J = 8.1 Hz, 4H), 8.20–8.37 (m, 8H), 9.18 (s, 2H). MS (m/z, rel int.); 358 (M+, 100), 330 (5), 256 (10), 179 (70).
5.1.6.5. 76 4,4′-[(3,3′Bipyridine)-6,6′-diyl]dibenzamidine tetrahydrochloride
This diamidine was obtained from the dinitrile 76a by Procedure C with 81% yield: mp >300 °C (dec). 1H NMR (400 MHz, D2O/DMSO-d6) δ: 8.00 (s, 4H), 8.23–8.43 (m, 8H), 9.15 (s, 2H). MS (m/z, rel int.); 393 (M++1, 95), 376 (8), 359 (5), 197 (100). Anal. calcd for C24H20N6·4HCl· 2½H2O: C, 49.41; H, 4.99; N, 14.40; found: C, 49.18; H, 4.67; N, 14.22.
5.2. [3H]MK-801 binding
For binding experiments with [3H]MK-801, membranes were prepared from rat frontoparietal cortex and hippocampus in 50 mM Tris acetate buffer (pH 7.0) as described.41 [3H]MK-801 (5 nM) was bound in glass vials at 23 °C for 3 h in 10 mM Tris acetate buffer (pH 7.0) in presence of 10 µM glutamate and glycine to membranes corresponding to about 1 mg wet tissue per vial (triplicates). Under these conditions, the affinity of the radioligand to its binding site was 13.3 nM. Test compounds were tested at 3–4 widely spaced concentrations achieving inhibitions from 10% to 90% of specific binding. Non-specific binding (NB) was obtained in presence of 100 µM (S)-ketamine. Bound radioligand was separated by filtration over glass fiber filters pre-soaked in 0.3% polyethyleneimine (washing buffer had rt). Filters were shaken with toluene-based scintillation cocktail for 30 min and counted in a betacounter. Sensitivity of inhibition of [3H]MK-801 binding to 30 µM spermine was defined as ratio of IC50s, one obtained in presence, the other in absence of 30 µM spermine.
5.3. [3H]Ifenprodil binding
The same membrane preparation was also used for [3H]ifenprodil binding experiments. [3H]Ifenprodil (5 nM) was bound in glass vials (triplicates) as described.20 In brief, incubations were conducted for 2 h on ice in 50 mM Tris·HCl (pH 7.4) in presence of 2 µM GBR 12909 (to block σ-sites). Under these conditions, the affinity of the radioligand to its binding site was 42 nM. Test compounds were tested at 3–4 widely spaced concentrations achieving inhibitions from 10% to 90% of specific binding. NB was obtained with 10 µM Ro 25-6981. Bound radioligand was separated by filtration over glass fiber filters pre-soaked in 0.3% PEI (washing buffer was ice cold; background rises fast if PEI solution is exhausted by repeated use). Filters were shaken with toluene-based scintillation cocktail for 30 min and counted in a betacounter.
5.4. Inhibition data analysis
Inhibition of [3H]MK-801 and of [3H]ifenprodil binding by test compounds was evaluated by fitting the results to the function y = A * IC50h/(IC50h + xh) + NB (A, specific binding in absence of inhibitor; x, inhibitor concentration; h, Hill coefficient) by iterative non-linear curve fitting53 using OriginPro 9.0.
5.5. Modeling
We observed molecular level interactions between the NR and 45 bisbenzamidine analogs (those with the highest spermine-sensitivities, Fig. S1) by docking them to putative spermine and ifenprodil binding sites of the NR. NR heterodimers GluN1/2B can adopt High Po and Low Po modes;18 Po stands for open probability). In the High Po mode, the acidic domains of the lower lobes of GluN1 and GluN2B are in close apposition, whereas in the Low Po mode, these domains are far apart as seen in the crystal of NR in complex with ifenprodil. Initial coordinates for the Low Po mode from the species Rattus norvegicus were obtained from the protein data bank (3Q41.pdb and 3QEL.pdb for GluN1-1a and GluN2B, respectively).21 We also took into account the recently published crystal structures of whole length GluN1/2B heterodimeric channels;54,55 we superimposed the 2014 to the 2011 structures and saw only very slight deviations. We stayed with the older structure because of its better resolution. Docking was calculated separately either to the acidic domain of GluN1 (contributed by amino acid residues D170, E181, E185, E186, F340, and E342) or to the acidic domain of GluN2B (amino acid residues E191, W197, E198, E201, D206, D210 and D21318,21,22), with the conformation in the Low Po mode. In addition, these compounds were also docked to the ifenprodil binding site, again in the Low Po mode as in the crystal structure. Initial structures of all compounds selected for docking were generated using the builder module of Maestro (Schrodinger, LLC, New York) followed by energy minimization using the OPLS 2005 force field. Compounds were docked using the GOLD docking software56 which is based on a genetic algorithm. This method allows partial flexibility of the protein and full range of ligand conformational flexibility. The default docking parameters were used for all the calculations, and the dockings produced with the ChemPLP scoring functions were rescored and re-ranked using GoldScore.
Supplementary Material
Acknowledgments
The following students contributed to the binding data: Tanja Wernle, Franziska Bender, Natalie Caspari, Dagmar Pretsch, Stephan Holzer, Andreas Pribitzer, Marie-Lena Müller, Katharina Blutsch, Turac Karaca, Julia Hnat. Computer Modeling was supported by the NIH-National Institute on Minority Health and Health Disparities (NIMHD) Grant No. 2G12MD007595, and by the Louisiana Cancer Research Consortium.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.06.012.
References and notes
- 1.Baker N, de Konig HP, Mäser P, Horn D. Trends Parasitol. 2013;29:110. doi: 10.1016/j.pt.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Queener SF. J. Med. Chem. 1995;38:4739. doi: 10.1021/jm00024a001. [DOI] [PubMed] [Google Scholar]
- 3.Valle M, Price RW, Nilsson A, Heyes M, Verotta D. Brain. 2004;127:1047. doi: 10.1093/brain/awh130. [DOI] [PubMed] [Google Scholar]
- 4.Kandanearatchi A, Brew BJ. FEBS J. 2012;279:1366. doi: 10.1111/j.1742-4658.2012.08500.x. [DOI] [PubMed] [Google Scholar]
- 5.Munde M, Ismail MA, Arafa R, Peixoto P, Collar CJ, Liu Y, Hu L, David-Cordonnier M-H, Lansiaux A, Bailly C, Boykin DW, Wilson WD. J. Am. Chem. Soc. 2007;129:13732. doi: 10.1021/ja074560a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Żołek T, Maciejewska D. Eur. J. Med. Chem. 2010;45:1991. doi: 10.1016/j.ejmech.2010.01.047. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds IJ, Aizenman E. J. Neurosci. 1992;12:970. doi: 10.1523/JNEUROSCI.12-03-00970.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood DH, Hall JE, Rose BG, Tidwell RR. Eur. J. Pharmacol. 1998;353:97. doi: 10.1016/s0014-2999(98)00386-0. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Qiu L, Li M, Dürrnagel S, Orser BA, Xiong Z-G, MacDonald JF. Neuropharmacology. 2010;58:1045. doi: 10.1016/j.neuropharm.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Boer TP, Nalos L, Stary A, Kok B, Houtman MJC, Antoons G, van Veen TAB, Beekman JDM, de Groot BL, Opthof T, Rook MB, Vos MA, van der Heyden MAG. Brit. J. Pharmacol. 2010;159:1532. doi: 10.1111/j.1476-5381.2010.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang TL, Mayence A, Vanden Eynde JJ. Bioorg. Med. Chem. 2014;22:1983. doi: 10.1016/j.bmc.2014.02.049. [DOI] [PubMed] [Google Scholar]
- 12.Paoletti P. Eur. J. Neurosci. 2011;33:1351. doi: 10.1111/j.1460-9568.2011.07628.x. [DOI] [PubMed] [Google Scholar]
- 13.Guillemin GJ. FEBS J. 2012;279:1356. doi: 10.1111/j.1742-4658.2012.08485.x. [DOI] [PubMed] [Google Scholar]
- 14.Sanderson L, Dogruel M, Rodgers J, De Koning HP, Thomas SA. J. Pharmacol. Exp. Ther. 2009;329:967. doi: 10.1124/jpet.108.149872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao B, Huang TL, Zhang Q, Jackson L, Queener SF, Donkor IO. Eur. J. Med. Chem. 1999;34:531. [Google Scholar]
- 16.Tao B, Huang TL, Sharma TA, Reynolds IJ, Donkor IO. Bioorg. Med. Chem. Lett. 1999;9:1299. doi: 10.1016/s0960-894x(99)00184-5. [DOI] [PubMed] [Google Scholar]
- 17.Berger ML, Noe CR. Current Topics in Medicinal Chemistry. Trivandrum: Research Trends; 2003. pp. 51–64. [Google Scholar]
- 18.Mony L, Zhu S, Carvalho S, Paoletti P. EMBO J. 2011;30:3134. doi: 10.1038/emboj.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong EHF, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7104. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto K, Mantione CR, Spada MR, Neumeyer JL, London ED. Eur. J. Pharmacol. 1994;266:67. doi: 10.1016/0922-4106(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 21.Karakas E, Simorowski N, Furukawa H. Nature. 2011;475:249. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomitori H, Suganami A, Saiki R, Mizuno S, Yoshizawa Y, Masuko T, Tamura Y, Nishimura K, Toida T, Williams K, Kashiwagi K, Igarashi K. J. Pharmacol. Exp. Ther. 2012;343:82. doi: 10.1124/jpet.112.192286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maciejewska D, Kaźmierczak P, Żabiński J, Wolska I, Popis S. Monatsh. Chem. 2006;137:1225. [Google Scholar]
- 24.Żabiński J, Maciejewska D, Wolska I. J. Mol. Struct. 2010;984:68. [Google Scholar]
- 25.Żabiński J, Maciejewska D, Kaźmierczak P. J. Mol. Struct. 2009;923:132. [Google Scholar]
- 26.Maciejewska D, Żabiński J, Kaźmierczak P, Rezler M, Krassowska-Świebocka B, Collins MS, Cushion MT. Eur. J. Med. Chem. 2012;48:164. doi: 10.1016/j.ejmech.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maciejewska D, Żabiński J, Kaźmierczak P, Wójciuk K, Kruszewski M, Kruszewska H. Bioorg. Med. Chem. Lett. 2014;24:2918. doi: 10.1016/j.bmcl.2014.04.075. [DOI] [PubMed] [Google Scholar]
- 28.Tidwell RR, Jones SK, Geratz JD, Ohemeng KA, Cory M, Hall JE. J. Med. Chem. 1990;33:1252. doi: 10.1021/jm00166a026. [DOI] [PubMed] [Google Scholar]
- 29.Huang TL, Tao B, Quarshie Y, Queener SF, Donkor IO. Bioorg. Med. Chem. Lett. 2001;11:2679. doi: 10.1016/s0960-894x(01)00541-8. [DOI] [PubMed] [Google Scholar]
- 30.Cushion MT, Walzer PD, Collins MS, Rebholz S, Vanden Eynde JJ, Mayence A, Huang TL. Antimicrob. Agents Chemother. 2004;48:4209. doi: 10.1128/AAC.48.11.4209-4216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vamecq J, Maurois P, Pages N, Bac P, Stables JP, Gressens P, Stanicki D, Vanden Eynde JJ. Eur. J. Med. Chem. 2010;45:3101. doi: 10.1016/j.ejmech.2010.03.044. [DOI] [PubMed] [Google Scholar]
- 32.Vanden Eynde JJ, Mayence A, Huang TL, Collins MS, Rebholz S, Walzer PD, Cushion MT. Bioorg. Med. Chem. Lett. 2004;14:4545. doi: 10.1016/j.bmcl.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 33.Huang TL, Vanden Eynde JJ, Mayance A, Collins MS, Cushion MT, Rattendi D, Londono I, Mazumder L, Bacchi CJ, Yarlett N. Bioorg. Med. Chem. Lett. 2009;19:5884. doi: 10.1016/j.bmcl.2009.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarak I, Marjanovic M, Piantanida I, Kralj M, Karminski-Zamola G. Eur. J. Med. Chem. 2011;46:2807. doi: 10.1016/j.ejmech.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Boykin DW, Kumar A, Xiao G, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR. J. Med. Chem. 1998;41:124. doi: 10.1021/jm970570i. [DOI] [PubMed] [Google Scholar]
- 36.Ismail MA, Arafa RK, Brun R, Wenzler T, Miao Y, Wilson DW, Generaux C, Bridges A, Hall JE, Boykin DW. J. Med. Chem. 2006;49:5324. doi: 10.1021/jm060470p. [DOI] [PubMed] [Google Scholar]
- 37.Mallena S, Lee MP, Bailly C, Kumar A, Boykin DW, Wilson WD. J. Am. Chem. Soc. 2004;126:13659. doi: 10.1021/ja048175m. [DOI] [PubMed] [Google Scholar]
- 38.Giordani F, Munde M, Wilson WD, Ismail MA, Kumar A, Boykin DW, Barrett MP. Antimicrob. Agents Chemother. 2014;58:1793. doi: 10.1128/AAC.02024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzales JL, Stephens CE, Wenzler T, Brun R, Tanious FA, Wilson WD, Barszcz T, Werbovetz KA, Boykin DW. Eur. J. Med. Chem. 2007;42:552. doi: 10.1016/j.ejmech.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds IJ, Zeleski DM, Rothermund KD, Hartnett KA, Tidwell R, Aizenman E. Eur. J. Pharmacol. 1993;224:175. doi: 10.1016/0922-4106(93)90023-3. [DOI] [PubMed] [Google Scholar]
- 41.Berger ML, Schödl C, Noe CR. Eur. J. Med. Chem. 1998;33:3. [Google Scholar]
- 42.Berger ML, Bitar AY, Waitner MJ, Rebernik P, O’Sullivan MC. Bioorg. Med. Chem. Lett. 2006;16:2837. doi: 10.1016/j.bmcl.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Sharma TA, Carr AJ, Davis RS, Reynolds IJ, Hamilton AD. Bioorg. Med. Chem. Lett. 1998;8:3459. doi: 10.1016/s0960-894x(98)00631-3. [DOI] [PubMed] [Google Scholar]
- 44.Donkor IO, Berger ML. Bioorg. Med. Chem. Lett. 1997;7:1455. [Google Scholar]
- 45.Kew JNC, Kemp JA. J. Physiol. 1998;512:17. doi: 10.1111/j.1469-7793.1998.017bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Traynelis SF, Hartley M, Heinemann SF. Science. 1995;268:873. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- 47.Berger ML, Pohler T, Schadt O, Stanger M, Rebernik P, Scholze P, Noe CR. Chem Med Chem. 2013;8:82. doi: 10.1002/cmdc.201200470. [DOI] [PubMed] [Google Scholar]
- 48.Tovar KR, McGinley MJ, Westbrook GL. J. Neurosci. 2013;33:9150. doi: 10.1523/JNEUROSCI.0829-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen KB, Ogden KK, Yuan H, Traynelis SF. Neuron. 2014;81:1084. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berger ML, Rebernik P. J. Pharmacol. Exp. Ther. 1999;289:1584. [PubMed] [Google Scholar]
- 51.Sharma TA, Reynolds IJ. J. Pharmacol. Exp. Ther. 1999;289:1041. [PubMed] [Google Scholar]
- 52.Ismail MA, Boykin DW. Synth. Commun. 2011;41:319. doi: 10.1080/00397910903537364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson ML, Faunt LM. Methods Enzymol. 1992;210:1. doi: 10.1016/0076-6879(92)10003-v. [DOI] [PubMed] [Google Scholar]
- 54.Karakas E, Furukawa H. Science. 2014;344:992. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee C-H, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. Nature. 2014;511:191. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.