

Abstract
Intestinal barrier dysfunction is thought to contribute to the development of multiple organ dysfunction syndrome in sepsis. Although there are similarities in clinical course following sepsis, there are significant differences in the host response depending on the initiating organism and time course of the disease, and pathways of gut injury vary widely in different preclinical models of sepsis. The purpose of this study was to determine whether the timecourse and mechanisms of intestinal barrier dysfunction are similar in disparate mouse models of sepsis with similar mortalities. FVB/N mice were randomized to receive cecal ligation and puncture (CLP) or sham laparotomy, and permeability was measured to fluoresceinisothiocyanate conjugated-dextran (FD-4) six to 48 hours later. Intestinal permeability was elevated following CLP at all timepoints measured, peaking at six to 12 hours. Tight junction proteins claudin 1, 2, 3, 4, 5, 7, 8, 13 and 15, JAM-A, occludin, and ZO-1 were than assayed by Western blot, real-time polymerase chain reaction, and immunohistochemistry 12 hours after CLP to determine potential mechanisms underlying increases in intestinal permeability. Claudin 2 and JAM-A were increased by sepsis whereas claudin-5 and occludin were decreased by sepsis. All other tight junction proteins were unchanged. A further timecourse experiment demonstrated that alterations in claudin-2 and occludin were detectable as early as 1 hour after the onset of sepsis. Similar experiments were then performed in a different group of mice subjected to Pseudomonas aeruginosa pneumonia. Mice with pneumonia had an increase in intestinal permeability similar in timecourse and magnitude to that seen in CLP. Similar changes in tight junction proteins were seen in both models of sepsis although mice subjected to pneumonia also had a marked decrease in ZO-1 not seen in CLP. These results indicate that two disparate, clinically relevant models of sepsis induce a significant increase in intestinal permeability mediated through a common pathway involving alterations in claudin 2, claudin 5, JAM-A and occludin although model-specific differences in ZO-1 were also identified.
INTRODUCTION
Despite advances in medical therapy, sepsis is a major global public health challenge. In the United States alone, over 800,000 patients develop sepsis annually, and between 230,000 and 370,000 die from the disease each year (1). Despite intensive research, therapy for sepsis remains non-specific outside of targeted antimicrobial therapy, with early therapy and supportive care responsible for recent advances in outcomes from sepsis (2).
Sepsis is a complex entity. When trying to understand its underlying pathophysiology, it is important to note that while there are similarities in presentation and clinical course in many septic patients, there are also significant differences in the host response depending on the initiating microorganism and time course of the disease (3). Understanding how much of sepsis is due to a common host response – independent of inciting factors (4) – versus how much is due to the impact of specific exogenous microbial factors or endogenous factors such as genetics or co-morbidities (3;5;6) is critical towards a more robust understanding of the disease.
The gut has long been hypothesized to be “the motor” of critical illness, driving systemic inflammation through a number of disparate feedback and feedforward mechanisms (7). Although the intestinal tract is lined with only a single layer of epithelial cells, it acts as a selective barrier allowing paracellular movement of water, solutes and immune modulating factors while preventing the diffusion of potentially harmful pathogens, toxins, and antigens from the luminal environment into the circulation and mesenteric lymph (8–10). Selective permeability is maintained in large part via the apical tight junction containing numerous claudin isoforms, occludin and ZO-1. Additionally, tight junction-associated protein Junctional Adhesion Molecule-A (JAM-A) plays a critical role in preserving intestinal barrier function (11;12). Tight junctions restrict molecular flux based upon both size and charge, and there are at least two distinct routes of paracellular flux across the tight junction – a high-capacity, size and charge-selective pore pathway and a low-capacity, nonselective leak pathway (9;13). Depending on the function of a tight junction protein, either an increase or a decrease in expression may be associated with hyperpermeability. Notably, there is evidence that pathways of gut injury vary widely between different murine models of sepsis (14).
The integrity of the intestinal barrier can be compromised in both acute and chronic disease states. Multiple studies have previously demonstrated intestinal hyperpermeability in preclinical models of critical illness (6;15–18). Observational studies have also showed evidence of intestinal hyperpermeability in critically ill patients in general (19) and specifically in septic patients (20). Intestinal hyperpermeability has been demonstrated to be associated with alterations in claudins 1, 2, 3, 4, 5 and 8 in colonic epithelial tissue in young (4–6 week old) female C57Bl/6 mice following cecal ligation and puncture (18), a murine model of ruptured appendicitis (21). Importantly, increased permeability is associated with negative outcomes in sepsis as well as other types of noninfectious critical illness (17;22). Increased permeability can result in translocation of bacteria and bacterial products as well as other luminal contents, leading directly or indirectly to distant injury and/or inflammation. Both genetic manipulation and pharmacologic intervention aimed at restoring the intestinal barrier in preclinical studies is associated with decreases in bacterial translocation and systemic cytokines as well as improved survival, although it is not clear whether the observed decrease in mortality is causally related to improvements in intestinal permeability (17;23).
The aim of this study was to determine whether sepsis-induced alterations in intestinal permeability are model-specific or independent and further to characterize the timecourse of intestinal hyperpermeability as well as identify potential mechanisms underlying changes in permeability.
MATERIALS AND METHODS
Animals
Six to twelve week old male and female FVB/N mice were utilized for all experiments. Mice were maintained on a 12 hour light-dark schedule in a specific pathogen-free environment and received standard laboratory mouse chow and water ad libitum. All experiments were performed in accordance with the National Institutes of Health Guidelines for the Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Emory University School of Medicine (Protocol DAR-2002113-110515BN). All surgery (described below) was performed under isoflurane anesthesia and animal suffering was minimized by treating all animals with buprenex post-operatively. Animals were sacrificed at predetermined endpoints using asphyxiation by CO2 or via exsanguination under deep isoflurane anesthesia. Of note, following the induction of sepsis, animals were checked twice daily, and moribund animals were sacrificed using humane endpoints. Moribund animals were identified by a) major organ failure or medical conditions unresponsive to treatment, b) surgical complications unresponsive to immediate intervention or c) clinical or behavioral signs unresponsive to appropriate intervention persisting for 24 hours.
Sepsis models
To determine whether permeability was dependent upon type of sepsis, two complementary sepsis models were used: cecal ligation and puncture (CLP, a model of polymicrobial intra-abdominal sepsis) and Pseudomonas aeruginosa pneumonia (a model of monomicrobial extra-abdominal sepsis). CLP was performed according to the method of Baker et al. Of note, we have previously shown that gut integrity is markedly altered in animals undergoing both of these insults with increased apoptosis, decreased proliferation and shortened villus length (15;23;24). Under isoflurane anesthesia, a small midline abdominal incision was made, and the cecum was exteriorized and ligated below the ileocecal valve so as to avoid intestinal obstruction. The cecum was then punctured twice with a 23-guage needle and gently squeezed to extrude a small amount of stool. After replacing the cecum in the abdomen, the abdominal wall was closed in layers. Sham mice were treated identically but their cecums were neither ligated nor punctured.
Animals receiving pneumonia had a 1 cm midline cervical incision and then received an intratracheal injection of 40 μl of P. aeruginosa (Strain 27853, ATCC, Manassas, VA) diluted in normal saline (2–4×107 colony forming units) via a 29 gauge syringe (25). After instillation of bacteria, mice were held vertically for 10 seconds to enhance delivery to the lung. Sham-operated mice were handled identically, but received an intratracheal injection of normal saline only.
In all studies, after the incision was closed, mice received 1 ml of normal saline via subcutaneous injection for fluid resuscitation. Animals also received antibiotics for two days after induction of sepsis. In addition, all operations were performed at a similar time of day (morning) to minimize the potential of diurnal variation on the results.
Intestinal permeability
Mice were gavaged with 0.5 ml of fluoresceinisothiocyanate conjugated-dextran (FD-4, 22 mg/ml, molecular mass 4.4 kDa) five hours prior to sacrifice (6). At time of sacrifice, blood was collected and centrifuged at 13000 rpm at 4°C for 5 minutes. Fifty μl of plasma was then diluted with an equivalent volume of phosphate-buffered saline (pH 7.4), and the concentration of FD-4 was determined using fluorospectrometry (NanoDrop 3300, Thermo Scientific, Wilmington, DE) using an excitation wavelength of 470 nm and an emission wavelength of 515. Serially diluted samples were used as standards, and all samples were run in triplicate.
Everted gut sacs
The proximal and distal small intestine and colon were removed from the mouse abdomen at sacrifice, flushed with ice-cold Krebs-Henseleit Bicarbonate Buffer (KHBB, pH 7.4, 6.9 g/l sodium chloride, 2.1 g/l sodium bicarbonate, 0.35 g/l potassium chloride, 0.145 g/l magnesium sulfate, 0.145 g/l potassium phosphate monobasic, 0.175 g/l calcium chloride dihydrate, and 2 g/l glucose), and then submerged in ice-cold KHBB. One end of the intestine was then ligated with 4-0 silk suture. The intestine was then everted over a thin glass rod, placed on the tip of a gavage feeding needle connected to a 1ml syringe, and secured to the feeding needle with another silk suture. The gut sac was then slightly distended with either 1 ml (small intestine) or 0.5 ml (colon) of KHBB and placed in a container of 100 μg/ml FD-4 in KHBB for 30 minutes. The container was temperature jacketed to 37°C and the buffer gently aerated with 100% oxygen. A 1 ml sample of buffer from the beaker was collected at the beginning and end of the incubation to determine the initial and final mucosal FD-4 concentration, respectively. After 30 minutes, the sac was removed and the fluid inside the sac was collected. The length and diameter of each sac were measured. The fluid was centrifuged at 10,000 rpm for 10 minutes and the supernatant was collected and stored. The fluid was diluted 1:2 with PBS and the concentration of FD-4 was determined by fluorospectometry with an excitation wavelength of 485nm and an emission wavelength of 528nm using serially diluted known concentration samples for standards (26). Permeability was expressed as clearance of FD-4 from mucosa to serosa in nl/min/cm2.
Western blot analysis
Protein expression of tight junction proteins was evaluated by Western blot. Frozen jejunal segments were homogenized in 5× volume of cold homogenization buffer and centrifuged at 10,000 rpm at 4°C for 5 minutes (15;27). After the supernatant was collected, the total protein concentration was determined via the Bradford protein assay. A total of 40μg of protein was then mixed with an equal volume of 2× Laemmli buffer and heated at 95°C for 5 minutes. Samples were then run on polyacrylamide gels (Bio-Rad, Hercules, CA). Samples were then transferred to Immuno-Blot polyvinylidenedifluoride membrane for 2 hours at 80V followed by membranes being blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (Sigma, St. Louis, MO) at room temperature for 60 minutes. This was then incubated overnight with primary antibody in 4°C. The following primary antibodies were used: rabbit anti-claudin-1, anti-claudin-3, anti-claudin-4, anti-claudin-5, anti-claudin-7, anti-claudin-8, anti-claudin-13, anti-claudin-15, anti-occludin, anti-ZO-1, anti-β-actin (Cell Signaling Technology, Danvers MA), anti-claudin-2 and anti-JAM-A (Abcam, Cambridge, MA). Membranes were then washed and incubated for 60 minutes at room temperature with horseradish peroxidase-conjugated goat anti-rabbit (Cell Signaling Technology). Finally, membranes were developed with a chemiluminescent system (Pierce, Rockford, IL) and proteins were detected after exposure to x-ray film. Densitometry was determined by normalizing expression to β-actin. Data were then transformed so sham mice would have a reference value of one for each experiment and the impact of sepsis was measured by the relative fold difference from sham mice.
Real-time polymerase chain reaction
Total RNA was isolated from jejunal tissue using a kit (RNeasy Mini Kit, Qiagen, Santa Clarita, CA) according to the manufacturer’s protocol. RNA integrity was verified by electrophoresis on a 1.2% agarose gel containing 2.2 M formaldehyde in 1× 3-(N-morpholino)propanesulfonic acid buffer (40 mM 3-(N-morpholino)propanesulfonic acid, pH 7.0; 10 mM sodium acetate; 1 mM EDTA, pH 8.0). cDNA was synthesized from 0.5 μg of total RNA. Claudin-2 and occludin mRNA levels were detected using pre-developed TaqMan primers and probes (Applied Biosystems, Foster City, CA) and run on the ABI StepOnePlus Real-Time PCR system (Applied Biosystems). Samples were run in duplicate and normalized to expression of the endogenous control, glyceraldehyde-3-phosphate (Applied Biosystems). Relative quantification of PCR products were based upon the value differences between the target gene and glyceraldehyde-3-phosphate using the comparative CT method.
Immunohistochemistry
Intestinal sections were deparaffinized, rehydrated and washed with PBS. Samples were incubated with 20% goat serum albumin and then stained with rabbit anti-claudin-2, anti-claudin-5, anti-occludin, anti-ZO-1 (1:1000, Cell Signaling Technology) or anti-JAM-A (1:1000, Abcam) overnight at 4°C. After washing with PBS, samples were incubated with Alexa Fluor® 488 donkey anti-rabbit antibody (1:500, Biolegend, San Diego, CA) for 1 hour at room temperature. After washing in PBS, the samples were counterstained with 4′,6-diamidino-2-phenylindole (Fisher Scientific, Pittsburgh, PA).
Statistical analysis
Data were analyzed using Prism 6.0 (GraphPad San Diego, CA) Data are presented as mean ± SEM. Multi-groups were analyzed via one-way ANOVA, followed by Tukey post-test. Two-way comparisons were tested for normality using the Shapiro-Wilk normality test. Data with Gaussian distribution were compared using the Students T test. Data that did not have Gaussian distribution were compared using the Mann Whitney test. A p value of <0.05 was considered to be statistically significant.
RESULTS
CLP increases intestinal permeability
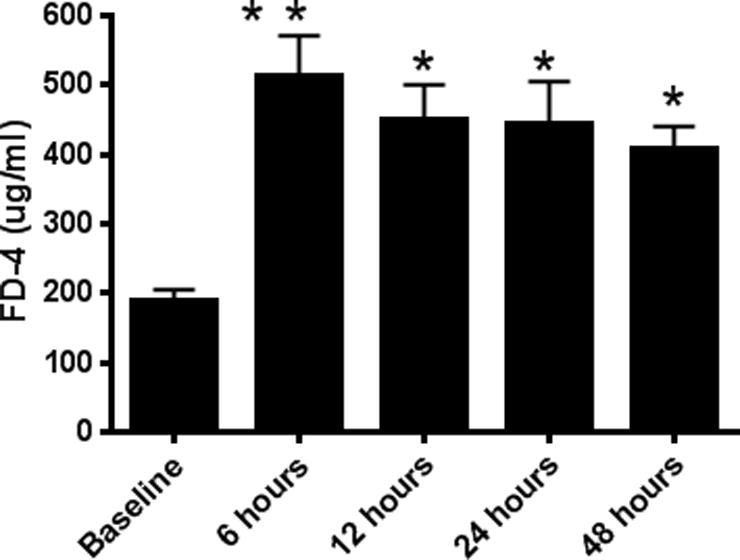
CLP induced a 2–3 fold increase in intestinal permeability as measured by appearance of FD-4 in the blood (Fig. 1). Permeability peaked 6–12 hours following CLP and continued to be elevated as far as 48 hours after the induction of sepsis. To determine which component of the intestine was dominantly responsible for the increased FD-4 seen in the bloodstream of septic mice, ex vivo permeability was measured on everted gut sacs in the proximal and distal small bowel as well as in the colon (Fig. 2A–C). The proximal small bowel – defined as a segment 10 cm distal to the duodenum – showed an increase in permeability whereas the distal small bowel and the colon did not show any alterations in permeability. Based upon these results, further mechanistic experiments examining the intestinal tight junction examined jejunal tissue.
FIG. 1. Intestinal permeability by FD-4 following CLP.
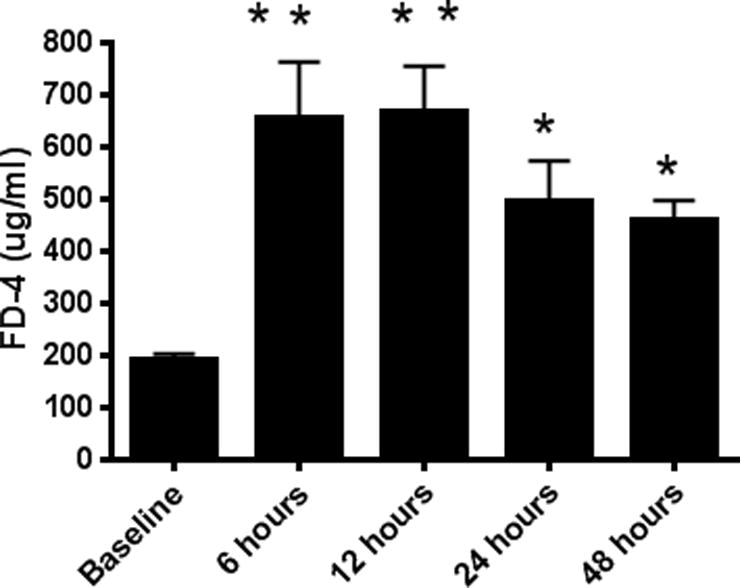
FD-4 levels in the bloodstream were increased compared to baseline at 6 (p<0.001), 12 (p<0.001), 24 (p<0.05), and 48 (p<0.05) hours after the onset of sepsis, indicating increased intestinal permeability (n=9–15/group).
FIG. 2. Intestinal permeability by everted gut sacs following CLP.
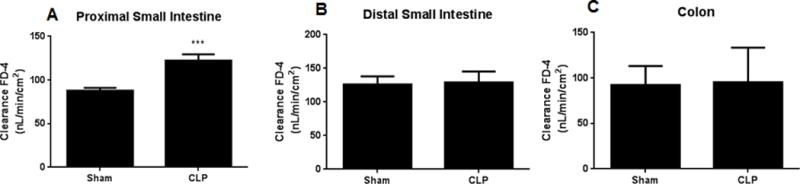
Permeability was higher in the proximal small intestine (10 cm distal to the duodenum) in septic mice vs. sham mice (A, p<0.005, n= 7–8/group). In contrast, permeability was similar between septic mice and sham mice in the distal small intestine and colon (B, C, n=8–9/group).
Effect of CLP on expression of intestinal tight junction proteins
To determine whether alterations in tight junction proteins contributed to intestinal barrier dysfunction in septic mice, protein levels of claudins 1, 2, 3, 4, 5, 7, 8, 13 and 15 (chosen because these are the claudins that have been shown to be present in the intestine (28–30)), JAM-A, occludin, and ZO-1 were analyzed 12 h after CLP. Jejunal levels of claudin-2 and JAM-A were increased in mice subjected to CLP compared to sham mice (Fig. 3A, B). In contrast, jejunal levels of claudin-5 and occludin were decreased in septic mice compared to sham mice (Fig. 3C, D). Similar findings were noted for claudin-2, claudin-5, occludin and JAM-A by immunohistochemistry (Fig. 4). No differences were found between septic mice and sham mice in the other 8 tight junction proteins analyzed by western blotting (Fig. 3E–L).
FIG. 3. Intestinal tight junctions 12 hours after CLP.
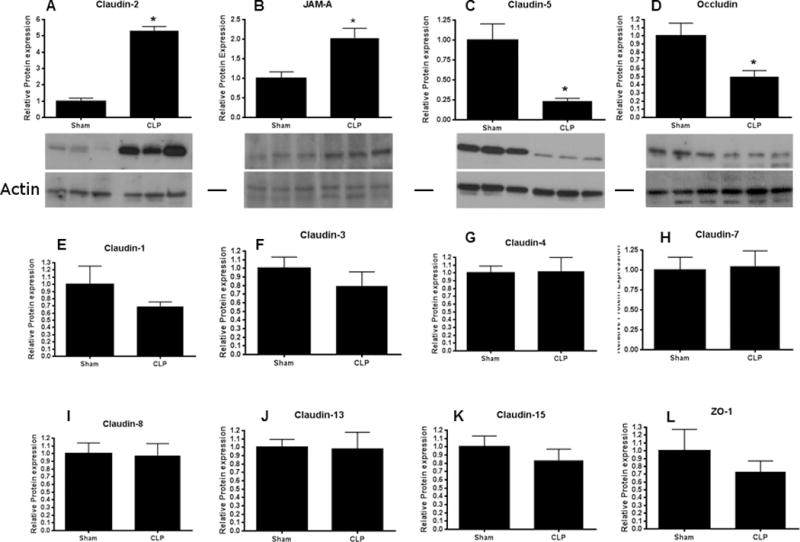
Levels of claudin-2 (A, p<0.05, n=3–5) and JAM-A (B, p<0.05, n=6) were increased after the onset of sepsis while levels of claudin-5 (C, p<0.005, n=6) and occludin (D, p<0.05, n=4–5) were decreased after the onset of sepsis. Representative blots for each are depicted. Actin is shown as a control for equal protein loading in each lane. No statistically significant changes were noted in claudin-1 (E), claudin-3 (F), claudin-4 (G), claudin-7 (H), claudin-8 (I), claudin-13 (J), claudin-15 (K) or ZO-1 (L), n=4–6 for each.
FIG. 4. Histomicrographs of jejunal tight junctions 12 hours following CLP.
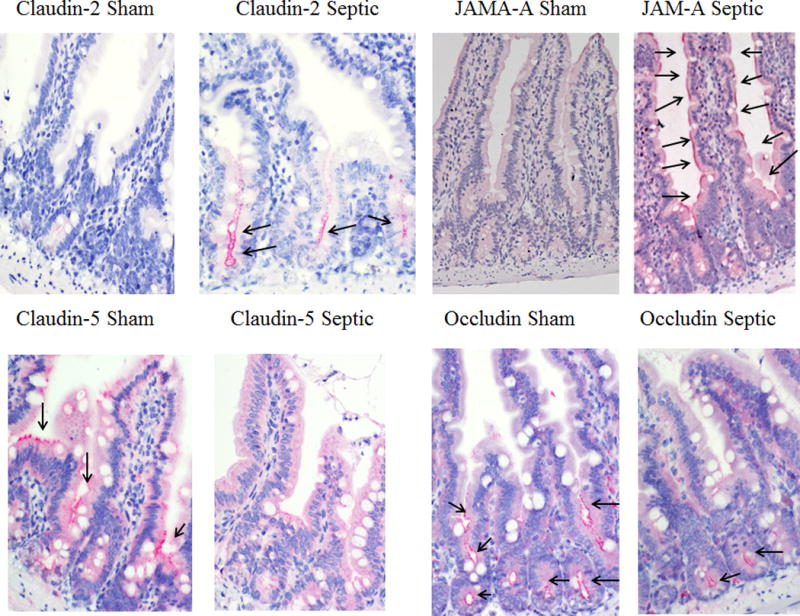
Levels of claudin-2 in the crypt (A) and JAM-A in the villus and crypt (B) were increased while claudin-5 in the upper crypt and lower villus (C) and occludin in the lower villus (D) were decreased in representative histomicrographs. Each tight junction mediator stains red and is highlighted with black arrows in the histomicrographs. Magnification 20×.
Tight junction proteins that were different at 12 hours between sham and septic mice were then assayed earlier to determine how rapidly after the induction of sepsis these changes occurred. Claudin-2 was increased and occludin was decreased as early as 1 hour following CLP (Fig. 5A,B) and the magnitude of difference was similar to that seen at 12 hours In contrast, claudin-5 and JAM-A were similar between sham and septic mice at 1 hour (Fig. 5C, D). All tight junction proteins that were altered at 12 hours were also altered 6 hours following CLP (data not shown). Similar alterations were also seen in mRNA levels with changes persisting for 24 hours after the onset of CLP (data not shown).
FIG. 5. Intestinal tight junctions 1 hour after CLP.
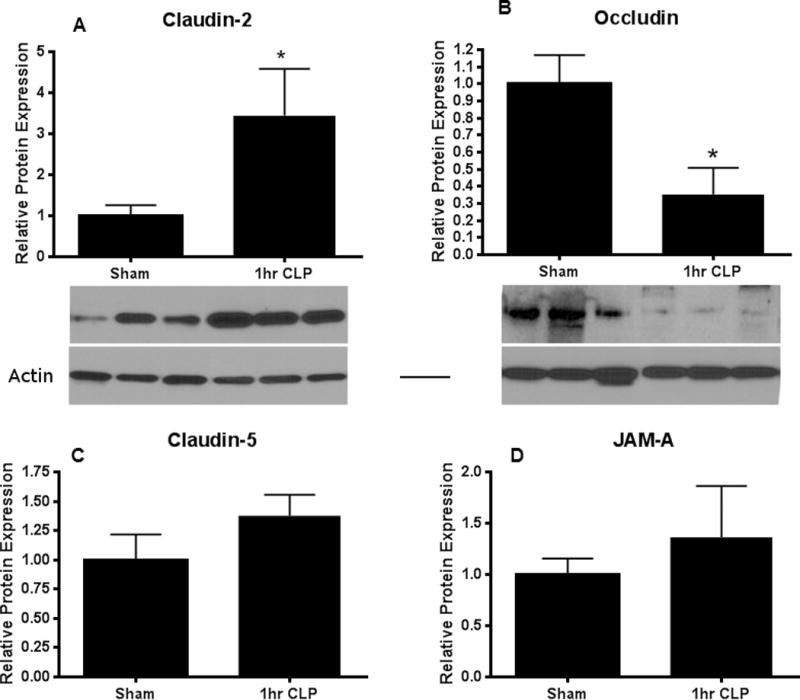
Claudin-2 (A, p<0.05, n=4–6) was increased while occludin (B, p<0.05, n=5–6) was decreased shortly after the onset of sepsis. Representative blots for each are depicted. Actin is shown as a control for equal protein loading in each lane. In contrast, claudin-5 (C) and JAM-A (D) were not altered at this early timepoint (n=5–6 for each).
Effect of pneumonia on intestinal permeability and expression of intestinal tight junction proteins
To determine whether these results were dependent upon the model of sepsis, a different group of animals were subjected to P. aeruginosa pneumonia. Similar to CLP, pneumonia induced a 2–2.5 fold increase in intestinal permeability (Fig. 6). Permeability again peaked at 6 hours and was increased at least 48 hours following the onset of sepsis. All tight junction proteins that were altered by CLP at 12 hours were changed in a similar manner by P. aeruginosa pneumonia (Fig. 7A–D). In addition, ZO-1 was markedly decreased 12 hours following pneumonia (Fig 7E) in contrast to CLP. No differences were found between septic mice and sham mice in the other 7 tight junction proteins analyzed by western blotting (Fig. 7F–L).
FIG. 6. Intestinal permeability following pneumonia.
FD-4 levels in the bloodstream were increased compared to baseline at 6 (p<0.005), 12 (p<0.05), 24 (p<0.05), and 48 (p<0.05) hours after the onset of sepsis (n=6–9/group).
FIG. 7. Intestinal tight junctions 12 hours after pneumonia.
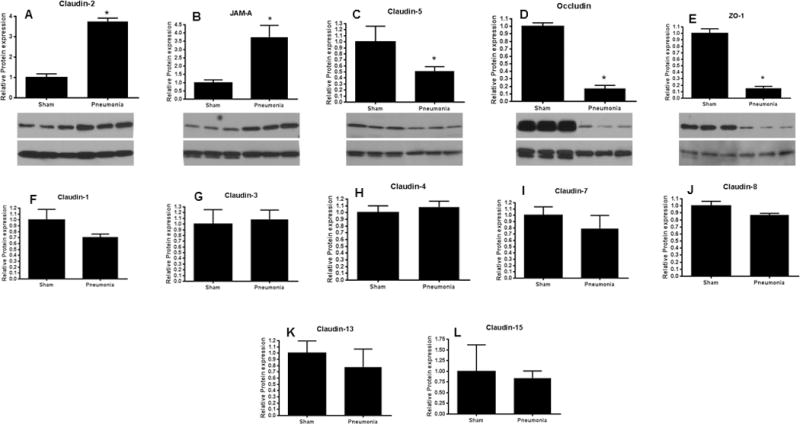
Levels of claudin-2 (A, p<0.005, n=6) and JAM-A (B, p<0.005, n=6) were increased after the onset of sepsis while levels of claudin-5 (C, p<0.05, n=4–6) and occludin (D, p<0.01, n=4–6) were decreased after the onset of sepsis. Unlike CLP, ZO-1 was also decreased following the onset of pneumonia (E, p<0.05, n=5). Representative blots for each are depicted. Actin is shown as a control for equal protein loading in each lane. No statistically significant changes were noted in claudin-1 (F), claudin-3 (G), claudin-4 (H), claudin-7 (I), claudin-8 (J), claudin-13 (K), or claudin-15 (L), n=4–6 for each.
DISCUSSION
This study demonstrates that there appears to be a common mode of sepsis-induced hyperpermeability with increases in claudin-2 and JAM-A and decreases in claudin-5 and occludin, with changes in the tight junction appearing as early as 1 hour following sepsis. However, there also appears to be a model-specific component to intestinal hyperpermeability with a marked reduction in ZO-1 in animals subjected to P. aeruginosa pneumonia, without statistically significant alterations in this tight junction mediator following CLP. In addition, intestinal permeability is increased as early as 6 hours following murine sepsis, with hyperpermeability persisting at least 48 hours.
Our data that tight junction alterations were similar – but not identical – between CLP and pneumonia, suggests that most alterations in tight junctions are common in critical illness although there also appears to be a component that is model specific. This is consistent with data on apoptotic mediators following sepsis, in which both commonalities and disease-specific differences exist (14). It is not entirely clear why these differences exist between models. We have previously demonstrated that the inflammatory response to a septic insult varies depending on organism studied, kinetics of mortality and microbial burden (3). Each of these are potentially important here since a) the monomicrobial infection with P. aeruginosa is obviously different from the polymicrobial infection seen in CLP, b) mortality is more rapid following pneumonia vs. CLP and c) even though blood cultures are positive in both infections, the number of organisms recovered is typically different. Further, tight junctions can be altered by either direct host-microbial interactions or indirectly via cytokines and other secondary mediators. We speculate that the differences seen relate more to indirect causes (more hypoxia, hypoperfusion in the pneumonia model) than to direct host-microbial interaction, and believe this is important to clarify in future experiments since understanding these differences may be crucial if intestinal integrity is to be considered as a potential therapeutic target in the future. It should also be noted that superimposing co-morbidities such as chronic alcohol usage further exacerbate specific abnormalities in sepsis-induced alterations in tight junction levels, and future investigations may help elucidate the impact of co-morbidities in sepsis-induced hyperpermeability (6).
In addition to examining the question of whether alterations in gut barrier function are model-specific or represent a common host response, an equally important part of this study is that we believe this is the most comprehensive study of intestinal permeability and intestinal tight junctions in sepsis to date. There are, however, multiple previous publications examining the impact of sepsis or endotoxemia on intestinal tight junctions. Li et al examined colonic tight junctions following CLP (18). They found that claudins 1, 3, 4, 5, and 8 had alterations in cellular localization, claudin 2 was markedly upregulated and occludin and claudins were displaced from raft fractions to non-raft fractions six to 24 hours following CLP. This was similar to findings from the same group examining intestinal permeability and tight junctions in ulcerative colitis (31). While some of our results are similar (notably the increase in claudin 2 and decrease in occludin), some are not. The most likely rationale for this is that we examined different areas of the intestine, in that we examined jejunum as opposed to colonic tissue based upon our findings with the everted gut sacs that permeability is different predominantly in the proximal small intestine. There are clear geographical differences in protein expression and distribution within the intestine, so it is not surprising that tight junctions might be differentially expressed and respond differently to sepsis. Further, different strains were used between the experiments (C57Bl/6 vs. FVB/N), and our study examined both genders and used slightly older mice.
Occludin, ZO-1 and claudin 4 have also been shown to be decreased following CLP in rats in a study examining the impact of berberine pre-treatment on sepsis (these were the only tight junctions assayed) (32). Further, male Balb/C mice subjected to endotoxemia have increased permeability with decreases in occludin and ZO-1 in the ileum 12 hours after injection of LPS (33). Notably, stimulation of the vagus nerve ameliorated these changes and inhibited the upregulated activity of both myosin light chain kinase and nuclear factor kappa B.
It is important to note that sepsis increased some tight junction proteins in this study (claudin-2 and JAM-A) while decreasing other tight junction proteins (claudin-5 and occludin in both models, ZO-1 in pneumonia). The impact of an alteration of a tight junction protein on permeability is determined by its role in regulating the paracellular space although whether a tight junction protein augments or inhibits permeability can vary even within a single family. For instance, within the claudin family, claudins can broadly be divided into sealing or pore-forming (9;13). As such, an increase in a pore-forming protein such as claudin-2 would directly lead to hyperpermeability and worsened barrier function while a decrease in a sealing protein such as claudin-5 would cause the same result in a mechanistically complementary manner. Our findings are therefore consistent with known opposing functions of these two members of the large (at least 24 member) claudin family as are our the findings that decreases in occludin and ZO-1 – both important regulators in the “tightness” of the tight junction – are associated with increased permeability. In contrast, our finding that the transmembrane tight junction protein JAM-A is increased following sepsis is not as easily explained since JAM-A diminishes paracellular permeability, as evidenced by the observation that intestinal permeability is higher in JAM-A knockout mice (12). However, barrier function taken as a whole is the result of a complex interplay of numerous tight junction proteins, and it is most appropriate to consider the changes in all tight junction proteins seen in sepsis as an aggregate readout of numerous influences as opposed to examining each protein in isolation. This is analogous to the marked upregulation in gut epithelial apoptosis caused by sepsis which is associated with simultaneous increases in both pro- and anti-apoptotic mediators (14) and to sepsis in general where there is a simultaneous upregulation of both pro- and anti-inflammatory cytokines.
This study has a number of limitations. All studies were performed in a single inbred mouse strain. Considering the importance of murine genetics on the host response (34), it is unclear if the results are generalizable to other strains. Although four timepoints were examined, it is not clear how early sepsis induces hyperpermeability and how long alterations in barrier function exist. While we showed the permeability is increased as early as six hours following sepsis, some tight junction proteins were elevated as early as one hour following sepsis. We did not assay permeability earlier, since FD4 was gavaged 5 hours prior to sacrifice, and examining permeability one hour after sepsis would have required gavaging FD-4 four hours prior to the onset of CLP or pneumonia, introducing a confounding factor. Further, although alterations in the tight junction can mediate barrier function, the experiments performed cannot prove that the alterations seen in tight junction following sepsis are mechanistically responsible for hyperpermeability, nor can they determine the relative contributions of the claudin-2, claudin-5, JAM-A, occludin and ZO-1. The experiments also do not examine the role of nutrition in intestinal hyperpermeability, since starvation, in and of itself, induces gut barrier dysfunction which can be ameliorated via intestinal alkaline phosphatase (35). While all animals in this study had free access to food and water throughout, it is possible that oral intake was decreased after the onset of sepsis. Finally, the experiments do not elucidate the molecular mechanisms through which sepsis induces alterations in the tight junction.
Despite these limitations, this study provides a comprehensive view of how sepsis impacts intestinal permeability across a significant time continuum and how tight junctions are impacted by different models of sepsis. Claudin 2, JAM-A, claudin-5 and occludin were impacted in a similar fashion by both CLP and pneumonia (although changes do not begin in a synchronous manner) whereas ZO-1 was altered by pneumonia alone. Further work is required to determine if ameliorating sepsis-induced alterations in the gut barrier will lead to improved survival following sepsis.
Acknowledgments
Source of Funding: This work was supported by funding from the National Institutes of Health (GM072808, GM095442, GM104323, GM109779, GM113228)
Footnotes
Conflicts of Interest: Craig Coopersmith was the President of the Society of Critical Care Medicine when this work was submitted.
References
- 1.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 2.Levy MM, Rhodes A, Phillips GS, Townsend SR, Schorr CA, Beale R, Osborn T, Lemeshow S, Chiche JD, Artigas A, Dellinger RP. Surviving sepsis campaign: association between performance metrics and outcomes in a 7.5-year study. Crit Care Med. 2015;43:3–12. doi: 10.1097/CCM.0000000000000723. [DOI] [PubMed] [Google Scholar]
- 3.McConnell KW, McDunn JE, Clark AT, Dunne WM, Dixon DJ, Turnbull IR, Dipasco PJ, Osberghaus WF, Sherman B, Martin JR, Walter MJ, Cobb JP, Buchman TG, Hotchkiss RS, Coopersmith CM. Streptococcus pneumoniae and Pseudomonas aeruginosa pneumonia induce distinct host responses. Crit Care Med. 2010;38:223–241. doi: 10.1097/CCM.0b013e3181b4a76b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fry DE. The generic septic response. Crit Care Med. 2008;36:1369–1370. doi: 10.1097/CCM.0b013e31816a11e9. [DOI] [PubMed] [Google Scholar]
- 5.Grunwell JR, Weiss SL, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, Checchia PA, Shanley TP, Bigham MT, Fitzgerald J, Howard K, Frank E, Harmon K, Wong HR. Differential expression of the Nrf2-linked genes in pediatric septic shock. Crit Care. 2015;19:327. doi: 10.1186/s13054-015-1052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoseph BP, Breed E, Overgaard CE, Ward CJ, Liang Z, Wagener ME, Lexcen DR, Lusczek ER, Beilman GJ, Burd EM, Farris AB, Guidot DM, Koval M, Ford ML, Coopersmith CM. Chronic alcohol ingestion increases mortality and organ injury in a murine model of septic peritonitis. PLoS ONE. 2013;8(5):e62792. doi: 10.1371/journal.pone.0062792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med. 2014;20:214–223. doi: 10.1016/j.molmed.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 9.Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. 2013;11:1075–1083. doi: 10.1016/j.cgh.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nalle SC, Aimee KH, Edelblum KL, Joseph NE, Singh G, Khramtsova GF, Mortenson ED, Savage PA, Turner JR. Recipient NK cell inactivation and intestinal barrier loss are required for MHC-matched graft-versus-host disease. Sci Transl Med. 2014;6:243ra87. doi: 10.1126/scitranslmed.3008941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khounlotham M, Kim W, Peatman E, Nava P, Medina-Contreras O, Addis C, Koch S, Fournier B, Nusrat A, Denning TL, Parkos CA. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. 2012;37:563–573. doi: 10.1016/j.immuni.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrone EE, Jung E, Breed E, Dominguez JA, Liang Z, Clark AT, Dunne WM, Burd EM, Coopersmith CM. Mechanisms of methicillin-resistant Staphylococcus aureus pneumonia-induced intestinal epithelial apoptosis. Shock. 2012;38:68–75. doi: 10.1097/SHK.0b013e318259abdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominguez JA, Samocha AJ, Liang Z, Burd EM, Farris AB, Coopersmith CM. Inhibition of IKKbeta in Enterocytes Exacerbates Sepsis-Induced Intestinal Injury and Worsens Mortality. Crit Care Med. 2013;41:e275–e285. doi: 10.1097/CCM.0b013e31828a44ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rupani B, Caputo FJ, Watkins AC, Vega D, Magnotti LJ, Lu Q, Xu dZ, Deitch EA. Relationship between disruption of the unstirred mucus layer and intestinal restitution in loss of gut barrier function after trauma hemorrhagic shock. Surgery. 2007;141:481–489. doi: 10.1016/j.surg.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Wang P, Su Q, Wang S, Wang F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE. 2012;7:e34946. doi: 10.1371/journal.pone.0034946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Zhang Q, Wang C, Liu X, Li N, Li J. Disruption of tight junctions during polymicrobial sepsis in vivo. J Pathol. 2009;218:210–221. doi: 10.1002/path.2525. [DOI] [PubMed] [Google Scholar]
- 19.Puleo F, Arvanitakis M, Van GA, Preiser JC. Gut failure in the ICU. Semin Respir Crit Care Med. 2011;32:626–638. doi: 10.1055/s-0031-1287871. [DOI] [PubMed] [Google Scholar]
- 20.Klaus DA, Motal MC, Burger-Klepp U, Marschalek C, Schmidt EM, Lebherz-Eichinger D, Krenn CG, Roth GA. Increased plasma zonulin in patients with sepsis. Biochem Med (Zagreb) 2013;23:107–111. doi: 10.11613/BM.2013.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- 22.Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, Hammer AM, Morris NL, Li X, Eberhardt JM, Gamelli RL, Kennedy RH, Choudhry MA. Burn Injury Alters the Intestinal Microbiome and Increases Gut Permeability and Bacterial Translocation. PLoS ONE. 2015;10:e0129996. doi: 10.1371/journal.pone.0129996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez JA, Vithayathil PJ, Khailova L, Lawrance CP, Samocha AJ, Jung E, Leathersich AM, Dunne WM, Coopersmith CM. Epidermal growth factor improves survival and prevents intestinal injury in a murine model of pseudomonas aeruginosa pneumonia. Shock. 2011;36:381–389. doi: 10.1097/SHK.0b013e31822793c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark JA, Clark AT, Hotchkiss RS, Buchman TG, Coopersmith CM. Epidermal growth factor treatment decreases mortality and is associated with improved gut integrity in sepsis. Shock. 2008;30:36–42. doi: 10.1097/shk.0b013e31815D0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox AC, Breed ER, Liang Z, Clark AT, Zee-Cheng BR, Chang KC, Dominguez JA, Jung E, Dunne WM, Burd EM, Farris AB, Linehan DC, Coopersmith CM. Prevention of Lymphocyte Apoptosis in Septic Mice with Cancer Increases Mortality. J Immunol. 2011;187:1950–1956. doi: 10.4049/jimmunol.1003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alam MA, Al-Jenoobi FI, Al-Mohizea AM. Everted gut sac model as a tool in pharmaceutical research: limitations and applications. J Pharm Pharmacol. 2012;64:326–336. doi: 10.1111/j.2042-7158.2011.01391.x. [DOI] [PubMed] [Google Scholar]
- 27.Fox AC, Robertson CM, Belt B, Clark AT, Chang KC, Leathersich AM, Dominguez JA, Perrone EE, Dunne WM, Hotchkiss RS, Buchman TG, Linehan DC, Coopersmith CM. Cancer causes increased mortality and is associated with altered apoptosis in murine sepsis. Crit Care Med. 2010;38:886–893. doi: 10.1097/CCM.0b013e3181c8fdb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita H, Chiba H, Yokozaki H, Sakai N, Sugimoto K, Wada T, Kojima T, Yamashita T, Sawada N. Differential expression and subcellular localization of claudin-7, -8, -12, -13, and -15 along the mouse intestine. J Histochem Cytochem. 2006;54:933–944. doi: 10.1369/jhc.6A6944.2006. [DOI] [PubMed] [Google Scholar]
- 29.Tamagawa H, Takahashi I, Furuse M, Yoshitake-Kitano Y, Tsukita S, Ito T, Matsuda H, Kiyono H. Characteristics of claudin expression in follicle-associated epithelium of Peyer’s patches: preferential localization of claudin-4 at the apex of the dome region. Lab Invest. 2003;83:1045–1053. doi: 10.1097/01.lab.0000078741.55670.6e. [DOI] [PubMed] [Google Scholar]
- 30.Clark JA, Gan H, Samocha AJ, Fox AC, Buchman TG, Coopersmith CM. Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G471–G479. doi: 10.1152/ajpgi.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, Zhang Q, Zhang M, Wang C, Zhu Z, Li N, Li J. Effect of n-3 polyunsaturated fatty acids on membrane microdomain localization of tight junction proteins in experimental colitis. FEBS J. 2008;275:411–420. doi: 10.1111/j.1742-4658.2007.06210.x. [DOI] [PubMed] [Google Scholar]
- 32.Li GX, Wang XM, Jiang T, Gong JF, Niu LY, Li N. Berberine prevents damage to the intestinal mucosal barrier during early phase of sepsis in rat through mechanisms independent of the NOD-like receptors signaling pathway. Eur J Pharmacol. 2014;730:1–7. doi: 10.1016/j.ejphar.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhou H, Liang H, Li ZF, Xiang H, Liu W, Li JG. Vagus nerve stimulation attenuates intestinal epithelial tight junctions disruption in endotoxemic mice through alpha7 nicotinic acetylcholine receptors. Shock. 2013;40:144–151. doi: 10.1097/SHK.0b013e318299e9c0. [DOI] [PubMed] [Google Scholar]
- 34.Cauvi DM, Williams MR, Bermudez JA, Armijo G, De MA. Elevated expression of IL-23/IL-17 pathway-related mediators correlates with exacerbation of pulmonary inflammation during polymicrobial sepsis. Shock. 2014;42:246–255. doi: 10.1097/SHK.0000000000000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamarneh SR, Mohamed MM, Economopoulos KP, Morrison SA, Phupitakphol T, Tantillo TJ, Gul SS, Gharedaghi MH, Tao Q, Kaliannan K, Narisawa S, Millan JL, van der Wilden GM, Fagenholz PJ, Malo MS, Hodin RA. A novel approach to maintain gut mucosal integrity using an oral enzyme supplement. Ann Surg. 2014;260:706–714. doi: 10.1097/SLA.0000000000000916. [DOI] [PMC free article] [PubMed] [Google Scholar]