

Abstract
Membrane‐less organelles in cells are large, dynamic protein/protein or protein/RNA assemblies that have been reported in some cases to have liquid droplet properties. However, the molecular interactions underlying the recruitment of components are not well understood. Herein, we study how the ability to form higher‐order assemblies influences the recruitment of the speckle‐type POZ protein (SPOP) to nuclear speckles. SPOP, a cullin‐3‐RING ubiquitin ligase (CRL3) substrate adaptor, self‐associates into higher‐order oligomers; that is, the number of monomers in an oligomer is broadly distributed and can be large. While wild‐type SPOP localizes to liquid nuclear speckles, self‐association‐deficient SPOP mutants have a diffuse distribution in the nucleus. SPOP oligomerizes through its BTB and BACK domains. We show that BTB‐mediated SPOP dimers form linear oligomers via BACK domain dimerization, and we determine the concentration‐dependent populations of the resulting oligomeric species. Higher‐order oligomerization of SPOP stimulates CRL3SPOP ubiquitination efficiency for its physiological substrate Gli3, suggesting that nuclear speckles are hotspots of ubiquitination. Dynamic, higher‐order protein self‐association may be a general mechanism to concentrate functional components in membrane‐less cellular bodies.
Keywords: isodesmic self‐association, membrane‐less organelle, prostate cancer, speckle‐type POZ protein, ubiquitin ligase
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control; Structural Biology
Introduction
Higher‐order complexes, that is, oligomers in which the number of monomers in a complex is broadly distributed and can be large, have important functions in signal transduction and cell fate decisions (Credle et al, 2005; Korennykh et al, 2009; Yin et al, 2009; Li et al, 2012a; Banjade & Rosen, 2014; Lu et al, 2014; Xu et al, 2014). The inherent size heterogeneity of such higher‐order assemblies generates challenges for their structural and mechanistic characterization, but recent progress has provided insight into their ability to mediate signal amplification, filter noise, and regulate signaling in time and space (Li et al, 2012b; Wu, 2013). Some of these assemblies form via nucleation‐driven polymerization and result in stable complexes, and we are starting to understand the structural basis for their proximity‐enhanced activation (Korennykh et al, 2009; Yin et al, 2009; Li et al, 2012a; Lu et al, 2014). Other assemblies depend on transient interactions and are more labile, generating oligomers with broad size distributions (Rivas et al, 2000; Errington et al, 2012; Canzio et al, 2013). The functions of these more labile higher‐order oligomers are not well understood.
SPOP is a substrate adaptor of a cullin‐3‐RING ubiquitin ligase (CRL3) and serves to recruit substrates to the CRL3 (Zhuang et al, 2009) for subsequent ubiquitination and degradation (Hernández‐Muñoz et al, 2005; Kent et al, 2006; Kwon et al, 2006; Zhang et al, 2006; Li et al, 2008). The Drosophila melanogaster homolog of SPOP, Roadkill/HIB, is essential for early development (Kent et al, 2006). Recent efforts to sequence the cancer genome have identified many mutations in SPOP that are associated with cancers (Lawrence et al, 2014), notably prostate (Kim et al, 2013), breast (Kim et al, 2011), endometrial (Le Gallo et al, 2012), and gastric (Kim et al, 2013) cancers [Appendix Fig S1 and www.cbioportal.org (Cerami et al, 2012; Gao et al, 2013)]. SPOP contains three domains; it recruits substrates through its MATH (meprin and traf homology) domain (Zhuang et al, 2009), forms dimers through its BTB (bric à brac, tramtrack, broad complex) domain (Zhuang et al, 2009; Errington et al, 2012), and forms either dimers or tetramers through its BACK (BTB and C‐terminal Kelch) domain (Errington et al, 2012; van Geersdaele et al, 2013 and Fig 1A). The BTB dimer interface (Fig 1B) has been well characterized in crystal structures of truncated versions of SPOP (Zhuang et al, 2009; Errington et al, 2012). Although the BACK domain (Fig 1C) has been crystallized as a dimer (van Geersdaele et al, 2013), oligomeric states ranging from dimers to tetramers or pentamers have been proposed (Errington et al, 2012; van Geersdaele et al, 2013). Truncated SPOP constructs encoding the BTB and BACK domains self‐associate into higher‐order oligomers (Errington et al, 2012) that possess increased ubiquitination efficiency, supporting the functional importance of oligomerization (Zhuang et al, 2009; Errington et al, 2012). Mutations within the MATH domain perturb interactions with substrates (Geng et al, 2013, 2014; Theurillat et al, 2014; Zeng et al, 2014; Zhang et al, 2015). Mutations within both self‐association domains are also found in cancers [Appendix Fig S1 and www.cbioportal.org (Cerami et al, 2012; Gao et al, 2013)], further supporting a functional role for higher‐order oligomerization, but their pathological mechanism is unclear.
Figure 1. SPOP is composed of three distinct domains.
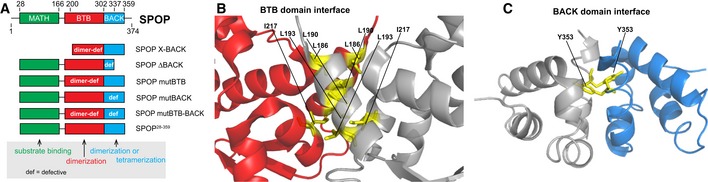
- Cartoon schematic of the SPOP self‐association‐incompetent mutant constructs that form complexes through only one (or neither) of the self‐association domains is shown. The previously described mutation Y353E (van Geersdaele et al, 2013) was introduced into the BACK domain so that self‐association could only occur through BTB‐mediated interactions resulting in SPOP mutBACK; previously described mutations of the conserved BTB interface (Zhuang et al, 2009) resulted in SPOP mutBTB, which self‐associates only through BACK‐mediated interactions. Constructs for expression in mammalian cells encode the full‐length protein; those for expression in bacteria comprise residues 28–359.
- Ribbon diagram of SPOP ΔBACK [PDB ID 3HQI (Zhuang et al, 2009)] showing residues that are mutated to abolish BTB domain self‐association (L186D, L190D, L193D, I217K). The domains are colored as in (A) (MATH, green; BTB, red; and BACK, blue), with one monomer shown in gray for clarity. Introduction of these mutations does not abolish the interaction between SPOP and the CRL because they are not part of the SPOP/cullin‐3 interface (Errington et al, 2012; Zhuang et al, 2009) and because mutation of the BTB domain or deletion of the BACK domain does not prevent SPOP from promoting ubiquitination of substrates (Zhuang et al, 2009; Errington et al, 2012).
- Ribbon diagram of SPOP 270–374 (L273D L282D L285K) dimer crystal structure [PDB ID 4HS2 (van Geersdaele et al, 2013)] showing residue Y353, which is mutated to E to prevent BACK domain self‐association. The domains are colored as in (A) (MATH, green; BTB, red; and BACK, blue), with one monomer shown in gray for clarity.
Membrane‐less cellular bodies are large protein/protein or protein/RNA assemblies. These bodies, in contrast to classic organelles, locally enrich components within defined boundaries despite the lack of an outer membrane. Nucleoli (Brangwynne et al, 2011), P granules (Brangwynne et al, 2009), and stress granules (Molliex et al, 2015; Patel et al, 2015), three membrane‐less organelles, were recently reported to have liquid droplet properties (Brangwynne, 2013), supporting a role for liquid–liquid phase separation in their formation (Li et al, 2012b; Nott et al, 2015; Patel et al, 2015). The recruitment of additional components to these bodies is not well understood but must involve protein/protein interactions (Tourrière et al, 2003). SPOP typically localizes to nuclear speckles (Nagai et al, 1997). However, whether these bodies have liquid character and how higher‐order oligomerization influences the SPOP localization to them is unknown.
Herein, we have used: (i) time‐lapse imaging to ask whether SPOP‐positive bodies have liquid character; (ii) biophysical methods to quantify the heterogeneous size distribution of higher‐order SPOP oligomers; and (iii) mutational analysis and immunofluorescence microscopy to investigate whether the ability to form higher‐order oligomers is linked to SPOP localization in nuclear speckles. We present a model for SPOP function in nuclear speckles and other bodies and speculate how mutations found in cancers may disrupt SPOP function.
Results
SPOP localizes to membrane‐less organelles with liquid droplet properties
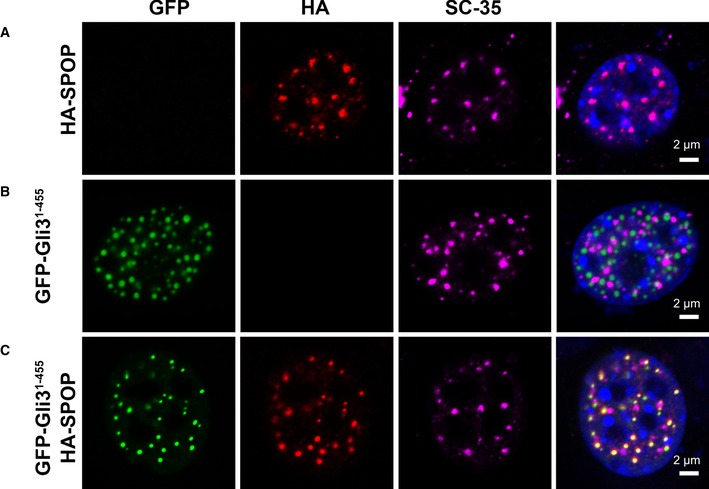
To characterize SPOP localization, we transiently expressed HA‐SPOP in NIH 3T3 cells and observed that SPOP co‐localizes with SC‐35, a marker of nuclear speckles (Fig 2A), in agreement with previous reports (Nagai et al, 1997). SPOP–fluorescent protein fusion constructs produced mislocalized proteins in the cell (Appendix Fig S2), preventing analysis of SPOP dynamics in nuclear speckles. In search of a fluorescent marker of SPOP localization, we asked whether the physiological substrate Gli3 (Wang et al, 2010) is also localized to nuclear speckles. Gli3 is quickly turned over, which led us to use the more stable fragment encompassing residues 1–455, Gli31–455. Interestingly, GFP‐Gli31–455 localized to a different punctate structure that was positive neither for SC‐35 (Fig 2B) nor for typical markers of nucleoli, polycomb bodies, Cajal bodies, or PML bodies (Fig EV1A). When we co‐transfected HA‐SPOP and GFP‐Gli31–455 constructs into NIH 3T3 cells, we observed co‐localization of SPOP and Gli31–455 with each other, but again not with nuclear speckles (Fig 2C). The Gli31–455/SPOP‐positive puncta ranged in size from several hundred nanometers down to the resolution limit of the microscope. We conclude that SPOP can localize to different punctate structures in the nucleus.
Figure 2. SPOP localizes to nuclear speckles or other nuclear bodies.
-
A–CConfocal microscopy images of fixed NIH 3T3 cells transiently expressing (A) full‐length HA‐SPOP, (B) GFP‐Gli31–455, and (C) HA‐SPOP + GFP‐Gli31–455 are shown. DAPI was used to stain the nucleus, SPOP localization was identified using an anti‐HA antibody, nuclear speckle localization was identified using an anti‐SC‐35 antibody, and Gli31–455 localization was identified via GFP fluorescence. SPOP co‐localizes with a marker for nuclear speckles or with the substrate Gli31–455. The areas with overlapping HA and GFP signal contain 75% of the punctate HA signal and 100% of the punctate GFP signal. For expression levels of HA‐SPOP and GFP‐Gli31–455 proteins, see Appendix Fig S3. Transfection efficiencies of pcDNA‐GFP‐Gli31–455 and pcDNA‐HA‐SPOP are 20–25% and 6–10%, respectively. When both constructs are used, ˜70–80% of transfected cells express both constructs (see Appendix Tables S1, S2 and S3).
Source data are available online for this figure.
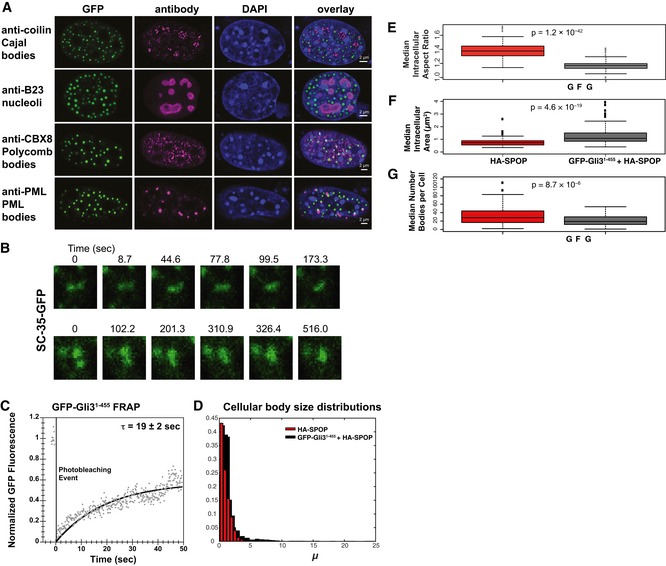
Figure EV1. Size distribution of nuclear puncta.
-
AGli31–455‐positive bodies are not Cajal bodies, nucleoli, polycomb bodies, or PML bodies. NIH 3T3 cells were transfected with GFP‐Gli31–455 and stained with antibodies against coilin (a Cajal marker), B23 (a nucleolus marker), CBX8 (a polycomb marker), or PML (a PML body marker).
-
BNIH 3T3 cells were transfected with a construct expressing SC‐35‐GFP and GFP fluorescence was monitored in live cells. Snapshots at indicated time points show a nuclear speckle fusion event.
-
CNIH 3T3 cells were transfected with a construct expressing GFP‐Gli31–455, and GFP fluorescence was monitored in live cells. Individual nuclear bodies were photobleached, and FRAP was monitored for 1 min. Data were normalized to the maximum and minimum intensity. The mean characteristic recovery time is indicated ± SEM.
-
DHistograms depicting the size distribution of nuclear body areas are shown for cells transfected with HA‐SPOP alone and GFP‐Gli31–455 + HA‐SPOP.
-
E–GNIH 3T3 cells were transfected with only HA‐SPOP or GFP‐Gli31–455 + HA‐SPOP. Box plots of the median aspect ratio of the (E) GFP‐Gli31–455‐positive nuclear bodies in one cell, (F) of the intracellular median area of nuclear speckles, and (G) of the number of bodies per cell for cells transfected with HA‐SPOP alone and GFP‐Gli31–455 + HA‐SPOP are shown. All three were significantly different (P = 1.2 × 10−42, P = 4.6 × 10−19, and P = 8.7 × 10−6, respectively, according to the Wilcoxon rank‐sum test), consistent with the different nature of distinct nuclear bodies. The medians are indicated as black horizontal lines within the boxes, and boxes enclose values between the first and third quartile. Interquartile range (IQR) is calculated by subtracting the first quartile from the third quartile. All values that lay more than 1.5× IQR lower than the first quartile or 1.5× higher than the third quartile are outliers that are plotted as squares. The smallest and highest values that are not outliers are connected with the dashed line.
Source data are available online for this figure.
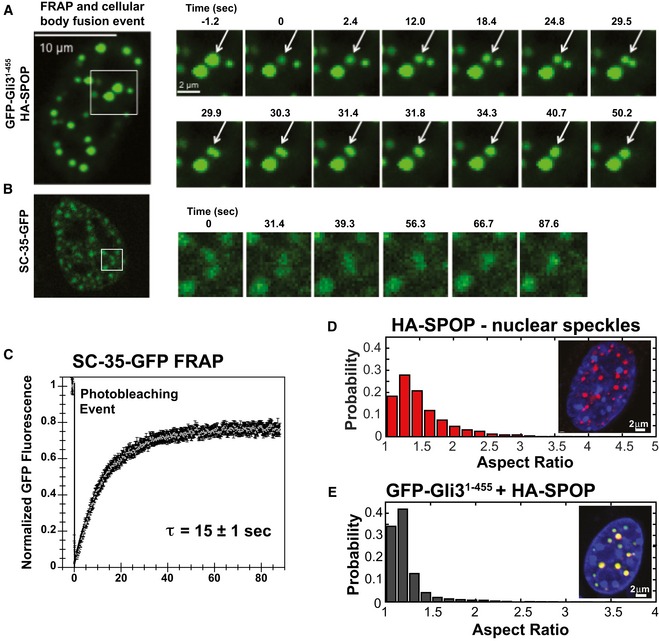
We do not know whether GFP‐Gli31–455 is needed for the formation of Gli31–455‐positive bodies, or whether it is recruited to preexisting bodies, but it can be used to track their material properties. When we observed cells transfected with HA‐SPOP and GFP‐Gli31–455 constructs over time, monitoring GFP fluorescence, the punctate structures sometimes fused with one another into larger structures that were first aspherical and then relaxed into a spherical shape over ~4 s (Fig 3A and Movie EV1). This result indicates that these punctate structures are membrane‐less bodies with liquid‐like properties (Brangwynne et al, 2011). We observed similar behavior for SC‐35‐GFP‐marked nuclear speckles, although the signal was less clear because SC‐35 seems to have a larger diffuse population in addition to the speckle‐localized population (Figs 3B and EV1B).
Figure 3. SPOP nuclear bodies have liquid‐like character.
-
A, BNIH 3T3 cells were (A) co‐transfected with constructs expressing full‐length SPOP and GFP‐Gli31–455 or (B) transfected with a construct expressing SC‐35‐GFP, and GFP fluorescence was monitored in live cells. (A) Snapshots were taken from individual time points as noted in the figure to show photobleaching (at 0 s), recovery, and nuclear body fusion events. The arrow in each panel points to the body that was photobleached and subsequently fuses with another body. These data were not used to calculate the average recovery time as the fusion event precludes accurate measurement of fluorescence recovery. (B) Snapshots at the indicated time points show a nuclear speckle fusion event. Additional images are shown in Fig EV1.
-
CIndividual nuclear speckles were photobleached and FRAP was monitored for 90 s. Data from 45 individual cells and FRAP events were corrected for background, normalized to the minimum and maximum intensity, and the mean is shown with error bars representing the standard error of the mean. The mean characteristic recovery time is indicated ± SEM.
-
D, ENuclear bodies are close to spherical. (D) Aspect ratios for speckles observed in NIH 3T3 cells transfected with constructs expressing HA‐SPOP (151 cells) (D), or GFP‐Gli31–455 and HA‐SPOP (155 cells) (E) were calculated as described previously (Brangwynne et al, 2011). Representative individual images of cells are shown as insets in each panel.
Membrane‐less bodies with liquid‐like properties are characterized by dynamic exchange of components with their surroundings (Li et al, 2012b; Molliex et al, 2015; Nott et al, 2015) and adopt spherical shapes (Brangwynne et al, 2011; Patel et al, 2015). When we photobleached individual nuclear speckles, their fluorescence intensity recovered with a characteristic time of 15 ± 1 s, demonstrating that protein molecules enter and leave the speckles quickly (Fig 3C). Similarly, GFP‐Gli31–455 was dynamically localized to Gli31–455/SPOP‐positive bodies as indicated by FRAP (Fig EV1C).
To examine the shape of the nuclear bodies, we determined their aspect ratio (maximum diameter/minimum diameter), which gives an indication of their sphericity; a perfect circle in the horizontal plane would have a value of 1.0, suggesting a sphere in three dimensions. We observed median aspect ratios (calculated as the median over all cells of the median aspect ratio of the speckles in one cell) of 1.38 (95% CI = 1.36, 1.41) for nuclear speckles (cells transfected with SPOP alone) and 1.17 (95% CI = 1.16, 1.18) for Gli31–455/SPOP‐positive bodies (Fig 3D and E, and Table 1). The majority of all bodies (54%) have an aspect ratio of < 1.3 (Fig 3D and E), indicative of a high degree of circularity in the horizontal plane. The area and the number of bodies observed per cell differed between nuclear speckles and Gli31–455/SPOP‐positive bodies (Fig EV1D–G and Table 1), in agreement with them being distinct nuclear bodies.
Table 1.
Summary of nuclear body statisticsa
| No. of cells | Puncta/cell | Area (μm2) | Aspect ratio | |
|---|---|---|---|---|
| HA‐SPOP | 155 | 28 (25, 31) | 0.750 (0.689, 0.801) | 1.38 (1.36, 1.41) |
| GFP‐Gli31–455 +HA‐SPOP | 151 | 19 (16, 24) | 1.075 (0.994, 1.170) | 1.17 (1.16, 1.18) |
For each cell, the number of bodies, median aspect ratio of the bodies in the cell (intracellular median aspect ratio), and median area of the bodies in the cell (intracellular median area) were determined. The median of each of these variables under each biological condition is given with 95% confidence intervals in parentheses. Cellular body volume could not be calculated as the size of the bodies approaches the limit of optical resolution, and measurements could not be accurately made in the z‐plane.
Together, these results demonstrate that SPOP can localize to nuclear speckles or Gli31–455‐positive bodies, which are both membrane‐less organelles with liquid droplet character. Our results mirror reports that SPOP can localize to different types of nuclear bodies, specifically to polycomb bodies and DNA damage foci (Hernández‐Muñoz et al, 2005), and presumably to PML bodies (Kwon et al, 2006; Jung et al, 2007). Substrate may play a role in recruiting SPOP to these nuclear bodies, but in contrast, SPOP can also recruit substrate to a nuclear body (Kwon et al, 2006). Importantly, all of these compartments are membrane‐less organelles, which may have liquid properties. The liquid behavior of DNA damage foci was recently experimentally supported (Altmeyer et al, 2015; Patel et al, 2015). In conclusion, SPOP localizes to a variety of different nuclear membrane‐less organelles, but has not been found diffusely localized. Therefore, we were interested in the determinants of SPOP localization to nuclear speckles, the liquid compartment to which it typically localizes.
Association of higher‐order oligomers
Membrane‐less organelles are formed through protein/protein and protein/RNA interactions. SPOP is known to form higher‐order oligomers, and therefore, we next explored the nature of SPOP self‐association. The self‐association properties of SPOP, and how these relate to its function, have not been quantitatively described and were not previously investigated for constructs containing all three domains. To understand SPOP self‐association in vitro, we determined the concentration‐dependent behavior of near full‐length SPOP containing all three domains (residues 28–359, referred to as SPOP28–359, Fig 1A) by size‐exclusion chromatography (SEC). The elution volume of the major eluting species of SPOP28–359 decreased strongly with increasing loading concentration (Fig 4A), indicating that SPOP self‐associated into oligomeric species whose size increased concurrently with the protein loading concentration. These observations agree with previous findings for truncated versions of SPOP (Errington et al, 2012). In addition, the elution width increased, demonstrating that SPOP adopts an ensemble of oligomeric species with a broad size distribution. In contrast, the elution volume of a SPOP construct lacking the BACK domain (SPOP ΔBACK) did not show a dependence on loading concentration (Fig EV2A); it eluted at volumes consistent with a dimeric state, in agreement with the BTB domain‐mediated dimer in its crystal structure (Zhuang et al, 2009; Errington et al, 2012). The full‐length protein tended to aggregate and therefore eluded quantitative analysis (Fig EV2B).
Figure 4. Self‐association‐defective SPOP mutants do not localize to nuclear speckles.
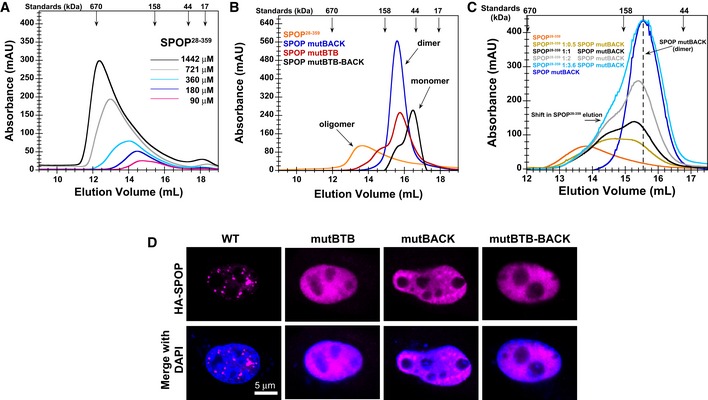
- SEC chromatograms of given loading concentrations of SPOP 28–359 are shown. The concentrations were normalized to the monomer molecular weight, that is, identical concentrations of dimeric SPOP ΔBACK (see Fig EV2) and oligomeric SPOP 28–359 contain identical numbers of protomers.
- SEC chromatograms for SPOP constructs defective in self‐association in one or both oligomerization domains are shown. Proteins were injected at the same loading concentration (533 μM), and the elution volume of globular molecular weight standards is noted above the graph.
- SEC chromatograms of SPOP 28–359 (200 μM), SPOP mutBACK (718 μM), and mixtures of the two (ratios given in the figure, SPOP at 200 μM) are shown. The elution volume of globular molecular weight standards is noted above the graph.
- Constructs for expressing full‐length HA‐SPOP or HA‐SPOP mutants capable of oligomerization through only one (or neither) of the self‐association domains were transfected into NIH 3T3 cells. DAPI was used to stain the nucleus, and SPOP localization was identified using an anti‐HA antibody. Experiments were performed at least twice on four biological samples. Multiple cells were examined, and representative cells are shown. For additional images, see Appendix Fig S4.
Source data are available online for this figure.
Figure EV2. SPOP ΔBACK does not elute in a concentration‐dependent manner and SPOP WT forms aggregates in vitro .
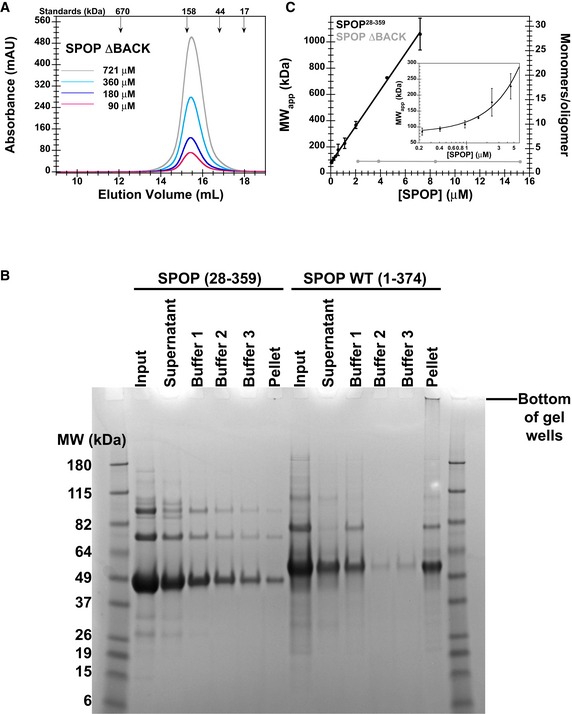
- SEC chromatograms of given loading concentrations of SPOP ΔBACK are shown. The concentrations were normalized to the monomer molecular weight, that is, identical concentrations of dimeric SPOP ΔBACK and oligomeric SPOP 28–359 contain identical numbers of protomers.
- Protein aggregation was assayed by centrifuging protein samples, resuspending soluble pelleted material in buffer three times, and then resuspending the final insoluble pellet in sample loading dye. The ultracentrifugation conditions are expected to pellet some of the larger SPOP 28–359 oligomeric species. These species are readily soluble in fresh buffer and do not represent aggregated material. In contrast, the majority of SPOP FL forms insoluble aggregates that do not dissociate even under extensive dilution, but can be resolubilized in denaturing gel sample buffer. These results show at least very slow off‐rates of SPOP from the aggregates, not only high stability of the aggregates, and are therefore strongly indicative of poor reversibility of aggregation.
- SPOP 28–359 oligomers have no apparent size limit. The apparent molecular weights (calculated from globular standards) of the major eluting species for each injection in Fig 4A and panel (A) are plotted against their elution concentrations and fit to a line. The number of monomers per oligomer was calculated by dividing the apparent molecular weight by the monomer mass and assumes regular packing of the monomer in the oligomer. The average and standard deviation of two independent experiments are shown for SPOP 28–359. Inset shows the same data in a semi‐logarithmic plot to highlight the lower concentrations of SPOP 28–359 assayed.
The apparent molecular weight of the major oligomeric species and the estimated number of monomers per oligomer, both calculated using globular molecular weight standards, depended linearly on the SPOP elution concentration (Fig EV2C). SPOP28–359 oligomerized readily into ~25mers (estimated from standards), despite more than 10‐fold dilution of protein on the size‐exclusion column, with no sign of an upper size limit at the concentrations assayed. This finding suggests that the two oligomerization domains function independently without steric hindrance and that the only limit to SPOP oligomer size is protein availability.
Self‐association‐defective SPOP mutants do not localize to nuclear speckles
Given that cellular clustering can be a consequence of protein self‐association (Kimata et al, 2007; Li et al, 2010), we next asked whether SPOP self‐association influences its ability to localize to nuclear speckles. Mutations that disrupt SPOP oligomerization through the BTB (Zhuang et al, 2009) and BACK domain (van Geersdaele et al, 2013) have been previously described. We purified SPOP harboring mutations in one or both domains and showed by SEC that these proteins form dimers or monomers, respectively, and no longer elute as large, oligomeric species (Fig 4B). Addition of increasing amounts of SPOP mutBACK, which only forms dimers, to SPOP28–359 reduces the size of oligomers formed in a concentration‐dependent manner (Fig 4C), showing that the interactions are reversible, can be outcompeted, and are not the result of non‐native aggregation.
While wild‐type HA‐SPOP localized to nuclear speckles (Fig 4D), SPOP harboring mutations in either of the self‐association domains failed to show punctate localization and instead showed a diffuse distribution throughout the nucleus (Fig 4D and Appendix Fig S4). SPOP mutBACK appeared partially punctate against a background of diffuse protein (Fig 4D and Appendix Fig S4B), suggesting a stronger role for the BTB domain in promoting the correct localization of the protein. Our findings demonstrate that wild‐type SPOP, containing two functional self‐association domains, localizes to nuclear speckles, while the self‐association‐defective mutants do not.
SPOP species formed in cells recapitulate in vitro oligomeric states
Cellular localization of SPOP and each of the mutants suggests that the ability to form higher‐order species promotes the recruitment to nuclear speckles. One additional possibility is that the mutants are not able to bind substrate and that this defect affects recruitment. We performed fluorescence anisotropy binding experiments to assess the ability of SPOP variants to interact with a fragment of the previously described SPOP substrate, Puc, that contains the major SPOP‐binding motif (Zhuang et al, 2009; Pierce et al, 2016). These data (Fig 5A and Table 2) show that all SPOP mutants bind substrate with similar dissociation constants as the WT. The BTB and BACK interface mutations do not disrupt the MATH domain function. Loss of higher‐order oligomerization decreases the valency of SPOP and may affect the interaction with multivalent substrates (Pierce et al, 2016), potentially interfering with recruitment to substrate via the self‐association defect.
Figure 5. SPOP forms higher‐order oligomeric species in cells.
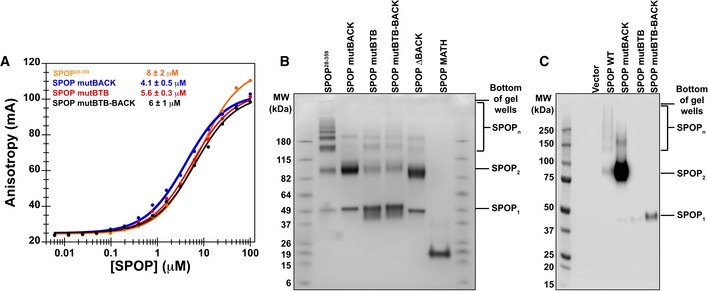
- Substrate binding was assayed by anisotropy using a fluorescently labeled substrate peptide. All SPOP variants bound substrate with an affinity similar to that of SPOP 28–359 (4–8 μM, see Table 2). Experimental data are shown as circles; the solid lines are fits to equation (2) (Roehrl et al, 2004).
- In vitro cross‐linking assays were performed for SPOP 28–359 and each mutant at 30 μM protein with the amide‐specific BS3 cross‐linker. Cross‐linking for SPOP ΔBACK and MATH domain are shown to demonstrate that cross‐linking conditions do not lead to non‐specific cross‐linking of protein species.
- Cross‐linking reactions were performed on whole‐cell lysates from cells expressing wild‐type SPOP, SPOP mutBACK, SPOP mutBTB, or SPOP mutBTB–BACK. SPOP 1, SPOP 2, and SPOP n identify SPOP monomers, dimers, and larger species, respectively. For loading levels, see Appendix Fig S5.
Table 2.
Summary of fluorescence anisotropy analysis for SPOP variants binding to substrate
| K D app (μM)a | |
|---|---|
| SPOP28–359 | 8 ± 2 |
| SPOP mutBACK | 4.1 ± 0.5 |
| SPOP mutBTB | 5.6 ± 0.3 |
| SPOP mutBTB–BACK | 6 ± 1 |
The average and standard deviation of three independent experiments are shown.
Our SEC results show that SPOP forms large oligomeric species with a wide size distribution while mutations to one or both self‐association domains reduce the protein oligomer size to dimers or monomers. We performed in vitro cross‐linking experiments with an amide‐specific cross‐linker and confirmed this observation (Fig 5B). SPOP28–359 forms multiple oligomeric states and is observed as a “ladder” of distinct oligomeric species. The mutants form predominantly dimers (mutBACK) or monomers (mutBTB and mutBTB–BACK). We performed cross‐linking experiments of cell lysates to investigate whether the SPOP variants have the same self‐association properties in cells (Fig 5C and Appendix Fig S5). Indeed, we observed the same behavior for each SPOP construct, with the mutants forming monomers or dimers and SPOP WT forming large species. A small portion of SPOP mutBACK is able to form species larger than a dimer, suggesting some ability to interact with endogenous SPOP and to form larger complexes. This is in agreement with the observation that some punctate localization of this protein is observed (Fig 4D). The wild‐type SPOP complexes range in size from dimers to species that remain in the gel wells and are too large to enter the gel (Fig 5C). Smearing of protein species, rather than a regular laddering appearance, is likely observed because the cellular protein is not cross‐linked in isolation, but is bound to other constituents of nuclear speckles. We conclude that the self‐association‐deficient mutants are able to bind substrate in vitro and form smaller complexes in cells than wild‐type SPOP.
Taken together, the ability of SPOP to form higher‐order SPOP oligomers correlates with its preferential localization to liquid‐like nuclear speckles, whereas self‐association‐deficient SPOP mutants adopt a diffuse distribution in the nucleus. We infer that higher‐order self‐association likely promotes localization to nuclear speckles.
Self‐association of individual domains
SPOP higher‐order oligomers localize to nuclear speckles, but the structural architecture of these oligomers, including the oligomerization state of the BACK domain, is unclear. In addition, while it is known that SPOP forms higher‐order oligomers with a wide size distribution (Errington et al, 2012 and Fig 4A), the populations of oligomers with different sizes are unknown, limiting biophysical and functional characterization. To understand SPOP self‐association quantitatively based on first principles, we determined the oligomeric states and dissociation constants of the BTB and BACK domains individually, using mutant proteins in which one or both self‐association interfaces were mutated (Fig 1A). We used two equilibrium methods: analytical ultracentrifugation (AUC) and composition gradient multi‐angle light scattering (CG‐MALS; for a description of the differences of SEC versus CG‐MALS, see Materials and Methods).
CG‐MALS analysis of SPOP mutBACK, which can only self‐associate through the BTB domain, showed that the protein remained dimeric even at the lowest concentrations assayed (Appendix Fig S6A, gray). Additionally, SPOP28–359 and SPOP ΔBACK both form dimers at the lowest concentrations assayed in SEC (Figs 4A and EV2A, respectively). Together, these results lead to the conclusion that BTB‐mediated dimerization occurred with at least nanomolar affinity. Traditional CG‐MALS, AUC, and SEC experiments are not sensitive at protein concentrations low enough to observe BTB dimer dissociation. We therefore used fluorescence‐detected sedimentation velocity analytical ultracentrifugation (FDS‐AUC) (Zhao et al, 2013b, 2014) to assay BTB domain self‐association. SPOP ΔBACK was labeled with Alexa Fluor 488, and sedimentation velocity AUC experiments were carried out at concentrations of labeled protein from 10 pM to 300 nM (Fig 6A and B). The resulting sedimentation coefficient distributions demonstrate the progressive dissociation of SPOP ΔBACK dimers into monomers at the lower concentrations (Fig 6B). The s‐values were obtained by integration of the sedimentation coefficient distributions, and the resulting binding isotherm (Fig 6C) yielded a K D value of 1.11 nM for BTB self‐association (95% confidence interval, [0.88, 1.40] nM). This result was confirmed in an additional experiment wherein a constant amount of labeled SPOP ΔBACK was titrated with unlabeled material (Appendix Fig S6B–E), yielding a K D value of 1.07 nM [95% confidence interval, (0.69, 1.66) nM]. The results of this second FDS‐AUC control experiment suggest that the fluorescent labeling does not influence BTB self‐association (Zhao et al, 2013c).
Figure 6. The SPOP oligomerization domains dimerize with different affinities.
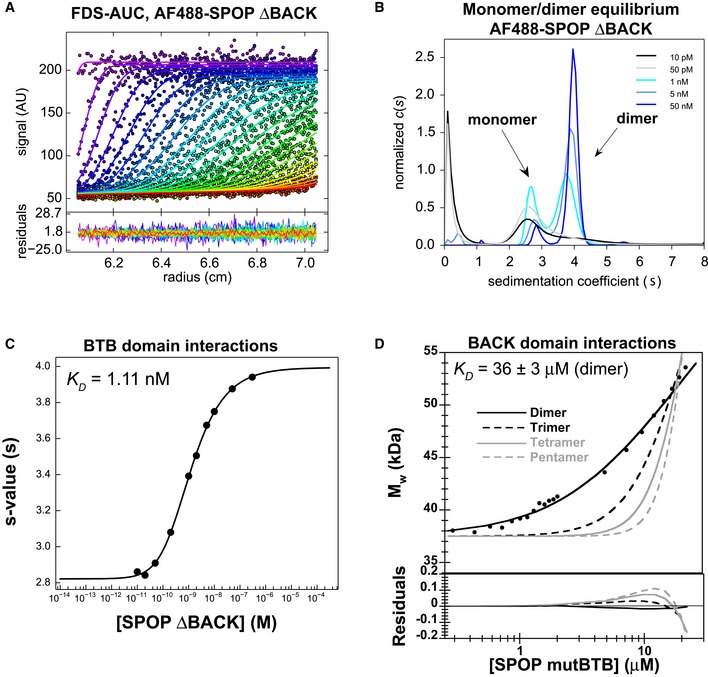
-
A–CThe BTB domain dimerizes with nanomolar affinity. (A) SV‐AUC data for 0.2 nM AF488‐SPOP ΔBACK. Fluorescence scans were collected for 12 h and are plotted against distance from the axis of rotation (circles). The data were subjected to standard c(s) analysis in SEDFIT (Schuck, 2000), and the results of this fit are shown (solid lines, rainbow color scheme). (B) Sedimentation coefficient distributions for a selection of values from the dilution series are shown. (C) The isotherm of weight‐average s‐values versus concentration (black circles) was fit to a dimer self‐association model and reveals a KD of 1.11 nM [95% confidence interval 0.88, 1.40 nM] (solid line).
-
DThe BACK domain dimerizes with micromolar affinity. Experimental weight‐average molar mass (Mw) from CG‐MALS for SPOP mutBTB (black circles) was fitted to a self‐association model (see text for details) for BACK–BACK dimerization (black line) yielding a KD of 36 ± 3 μM (average and standard deviation from three independent experiments). Fits to the monomer/dimer equilibrium model and to alternative monomer–trimer, tetramer, and pentamer equilibrium models are depicted as black solid line and as dashed and gray lines, respectively. Residuals for all models are depicted in the lower panel.
SPOP forms oligomers larger than dimers starting at low micromolar concentrations, and the BTB‐mediated interaction is three orders of magnitude stronger, leading us to the hypothesis that the BACK domain oligomerized with low micromolar affinity. CG‐MALS analysis of SPOP mutBTB, which self‐associates only through BACK‐mediated interactions, showed that this mutant dimerized with a K D value of 36 ± 3 μM (Fig 6D and Table 3). Models for oligomerization into discrete trimer, tetramers, or pentamers (Fig 6D, dotted and gray lines) did not fit the data well, showing that the equilibrium species distribution overwhelmingly consists of monomers and dimers and ruling out previously proposed tetramers or pentamers (Errington et al, 2012) as the major BACK domain oligomerization state. We used sedimentation equilibrium AUC to verify the oligomerization state of SPOP mutBTB (Appendix Fig S6F). The data fit well to a reversible monomer–dimer self‐association model with a calculated dissociation constant of 32.5 μM [95% confidence interval, (17.72, 59.75) μM], which is similar to that determined by CG‐MALS. Together, these results demonstrate that both domains dimerize, which is in agreement with available crystal structures (Zhuang et al, 2009; Errington et al, 2012; van Geersdaele et al, 2013).
Table 3.
Summary of CG‐MALS analysis of SPOP variants
| Model | K D (μM)a | |
|---|---|---|
| SPOP28–359 | Isodesmic | 2.4 ± 0.4 |
| SPOP mutBACK | Monomer–dimer | > 500 |
| SPOP mutBTB | Monomer–dimer | 36 ± 3 |
| SPOP mutBTB–BACK | Monomer–dimer | > 500 |
| SPOP X‐BACK | Monomer–dimer | 118 |
When given, the average and standard deviation of three independent experiments are shown.
First principles self‐association model
Knowledge of the self‐association properties of the individual domains allowed us to construct a self‐association model for full‐length SPOP based upon first principles. SPOP monomers dimerize strongly through the BTB domain at low protein concentrations. We can treat the fully formed BTB dimers as the unit that self‐associates into higher‐order structures because of the more than three orders of magnitude difference in K D values for the BTB and BACK domains. Each dimer has two BACK domains, consistent with an indefinite self‐association model (Fig 7A). We assume that the addition of each unit occurs with the same K D2 as the previous addition, irrespective of the oligomer size, yielding a single dissociation constant. We tested this isodesmic self‐association model (Van Holde & Rossetti, 1967; Na & Timasheff, 1985) using CG‐MALS, which allows rapid, quantitative analysis of macromolecular self‐association at equilibrium and provides insight into size distributions (Attri & Minton, 2005a; Kameyama & Minton, 2006).
Figure 7. Indefinite SPOP self‐association fits to an isodesmic model.
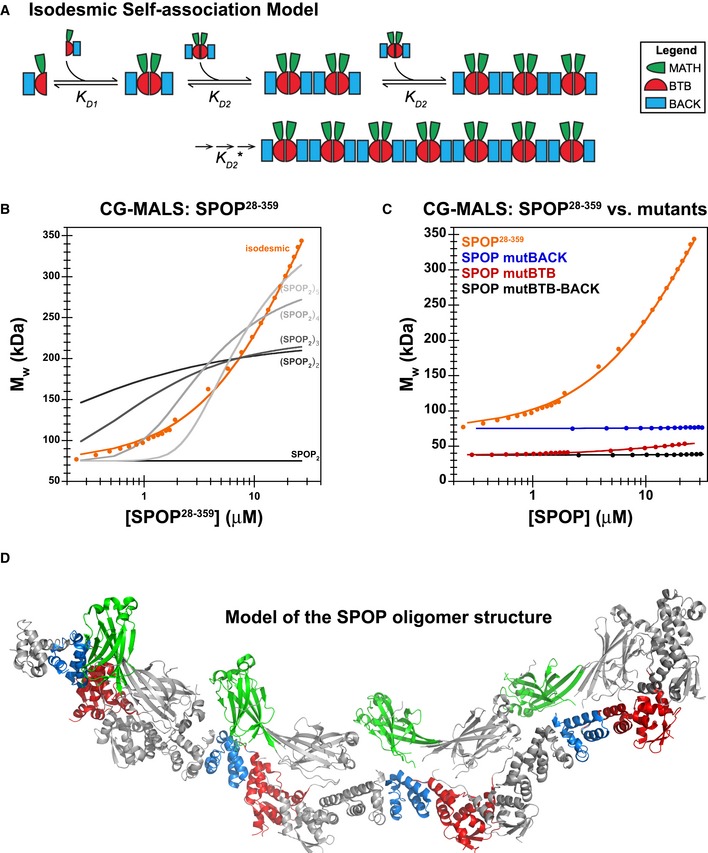
- Cartoon schematic depicting the proposed isodesmic self‐association model. KD 1 and KD 2 represent the domain‐mediated dimerization affinities for the BTB domain and BACK domains, respectively. KD 2 is identical for all association steps independent of oligomer size. At large oligomer sizes, KD 2 may increase due to entropic penalties, resulting in KD 2*. The N‐ and C‐termini contain neither defined domains nor low‐complexity sequences but may add additional self‐association behavior as evidenced by aggregation; these interactions were not dissected due to the poor reversibility of aggregation (Fig EV2B).
- Experimental weight‐average molar mass (Mw) from CG‐MALS for SPOP 28–359 (orange circles) was fitted to an isodesmic self‐association model in which SPOP dimers are the self‐associating unit (orange line). The largest SPOP oligomer taken into account was an undecamer of SPOP dimers [(SPOP 2)11]. The fits from three independent experiments yielded a KD 2 of 2.4 ± 0.4 μM. Lines for fits of the data to self‐association models that assume formation of individual oligomeric species instead of isodesmic self‐association are shown for reference (gray lines).
- Experimental weight‐average molar mass (Mw) from CG‐MALS data (circles) and fits (solid lines) are depicted for each SPOP construct assayed, showing that a progressive increase in SPOP oligomer size is observed only when both self‐association domains are interaction‐competent. This figure is comprised of data shown in panel (B), Fig 6D and Appendix Fig S6A for direct comparison.
- Ribbon diagram illustrating structural model of an octamer SPOP assembly. In a 27 μM SPOP solution, three percent of the oligomeric assemblies would be equal in size or larger. The domains are colored as in panel (A) and Fig 1 (MATH, green; BTB, red; and BACK, blue), with alternating monomers shown in gray for clarity. The model was built using two available crystal structures [PDB ID 3HQI (Zhuang et al, 2009) and 4HS2 (van Geersdaele et al, 2013)], and no further assumptions.
Isodesmic self‐association of SPOP
SPOP28–359 self‐association was assayed at concentrations ranging from 0.11 to 27 μM (Fig 7B) by dilution of a protein stock solution. The apparent molar mass of the solution rapidly re‐equilibrated on a second timescale (Appendix Fig S7), and we therefore assessed the size distribution of SPOP as a function of concentration in equilibrium. At low concentrations, the weight‐average molar mass (Mw) corresponded to dimers, but it increased dramatically with protein concentration, without reaching a plateau, supporting our hypothesis that SPOP self‐associates indefinitely. The CG‐MALS data fit best to a model in which SPOP dimers self‐associate isodesmically with a dissociation constant of 2.4 ± 0.4 μM (Fig 7B, solid orange line and Table 3). This value is an order of magnitude lower than the K D for SPOP mutBTB. We suspect that mutation of domain interfaces leads to overall changes in the SPOP monomer structure that affect the affinity of BACK domain‐mediated self‐association (Appendix Fig S8).
These data also confirmed that the BACK domain dimerizes (see schematic Fig 7A). BACK domain tetramerization would have led to the population of fewer oligomeric states, the sum of which inadequately described the data. In fact, fits of the data to discrete oligomer species models, in which BTB domain‐mediated dimers form discrete tetramers, hexamers, or octamers, did not adequately describe the data (Fig 7B, shades of gray noted in figure).
Mutation of the BACK domain interface, the BTB domain interface, or both led to the abolishment of higher‐order oligomerization (Fig 7C), demonstrating that SPOP self‐association into higher‐order oligomers requires specific interactions through both domains. SPOP mutBTB–BACK, which is mutated in both interfaces, behaves predominantly as a monomer. SPOP mutBACK is observed as a constitutive, BTB‐mediated dimer, while SPOP mutBTB dimerizes with low micromolar affinity.
Although there is not a crystal structure of SPOP that contains all three domains, the two available partial structures from previous studies [the BTB dimer, PDB ID 3HQI (Zhuang et al, 2009) and the BACK dimer, PDB ID 4HS2 (van Geersdaele et al, 2013)] can be used to build a model of the oligomeric species of near full‐length SPOP without further assumptions (Fig 7D). According to this model, SPOP oligomers adopt a fibrillar organization with helical propensity. Because of the snapshot character of the crystal structures, the model is a rigid fibril; however, the domains may be more flexibly tethered in solution and our data show that the SPOP fibrils are labile and adopt a defined distribution of different lengths.
We wondered whether the structural features we describe here are common properties for substrate adaptors of CRLs. Sequence alignment did not reveal other BTB–BACK domain‐containing proteins in humans that could adopt this type of assembly (Fig EV3). The majority of them contain a longer version of the BACK domain that occludes BACK domain dimerization (Fig EV3A and B). Interestingly, sequence similarity shows that a family of BTB–BACK domain‐containing proteins in rodents may be able to form higher‐order oligomers through an isodesmic self‐association mechanism (Fig EV3C and D).
Figure EV3. A family of BTB–BACK domain‐containing proteins in rodents may self‐associate isodesmically.
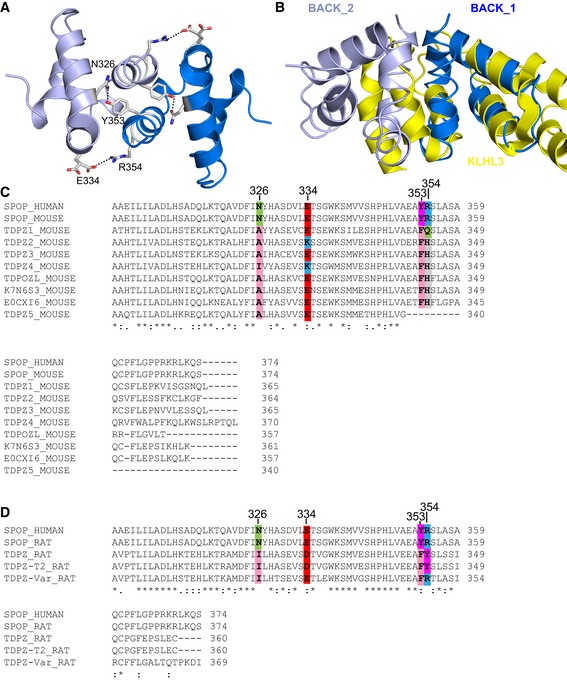
-
AThe BACK domain dimer is stabilized via a hydrogen bond between N326 and Y353 and a salt bridge between E334 and R354. The two BACK domain monomers are shown in different shades of blue.
-
BThe long form of the BACK domain, as exemplified by KLHL3 (shown in yellow, PDB ID 4HXI), occludes the dimerization interface of the short SPOP BACK domain.
-
C, DThe human genome contains only two genes encoding a protein with a BTB and short version of the BACK domain, SPOP and SPOPL, the latter of which does not dimerize because of a sterically hindering insertion. The family of BTB–BACK domain‐containing proteins is expanded in rodents. (C) Alignment of mouse protein sequences containing a BTB and a short BACK domain with human SPOP. The N326/Y353 and E334/R354 interactions are not conserved but could potentially be replaced by hydrophobic/aromatic and negatively charged/histidine interactions, respectively. (D) Alignment of rat protein sequences containing a BTB and a short BACK domain with human SPOP. In the TDPZ variant shown on the bottom, residues E334 and R354 are conserved, potentially enabling a salt bridge. This protein may undergo isodesmic self‐association into higher‐order oligomers.
Quantitative description of SPOP size distribution
To independently demonstrate the population of different SPOP oligomers, we performed native mass spectrometry (MS) analysis of SPOP28–359 (Fig 8A). We directly observed small amounts of SPOP monomers, a high population of SPOP dimers, and additional even‐numbered oligomers up to octamers, in agreement with our model that SPOP dimers are the building blocks for the formation of higher‐order oligomers. Addition of 0.1% formic acid to SPOP28–359 led to the destabilization of the BTB interface and the detection of odd‐ and even‐numbered oligomers (Fig EV4). Together, these data support our conclusion that SPOP oligomerizes isodesmically through the successive self‐association of BTB dimers via the BACK domain.
Figure 8. SPOP forms dynamic higher‐order oligomers that can be analyzed quantitatively.
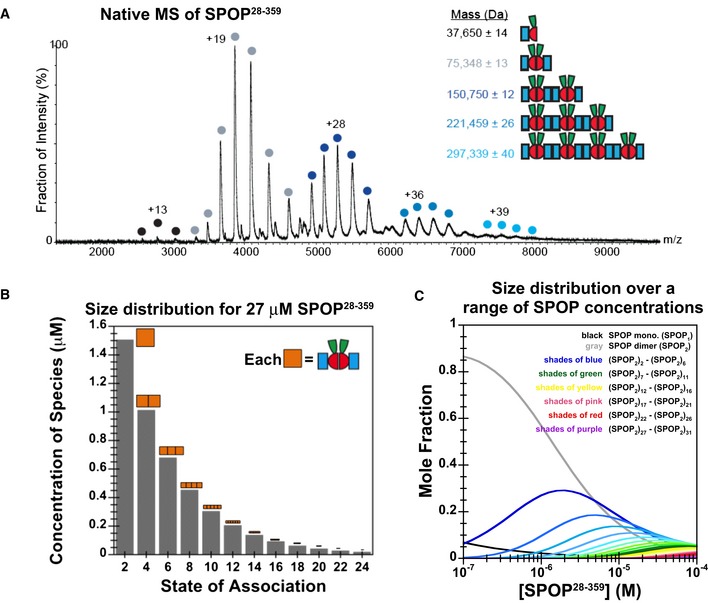
- Native MS spectrum of 30 μM SPOP 28–359 confirms that SPOP self‐associates through addition of dimers. The increase in mass indicates that the dimer is the stable building block within the assemblies.
- Graphical representation of the SPOP concentration of each oligomeric species within a 27 μM SPOP 28–359 sample. The cartoons' sizes are scaled based upon the fraction present within the total sample.
- Concentration dependence of the SPOP oligomer size distribution (mole fraction) from isodesmic modeling of CG‐MALS data.
Figure EV4. Detailed MS analysis of SPOP oligomers.
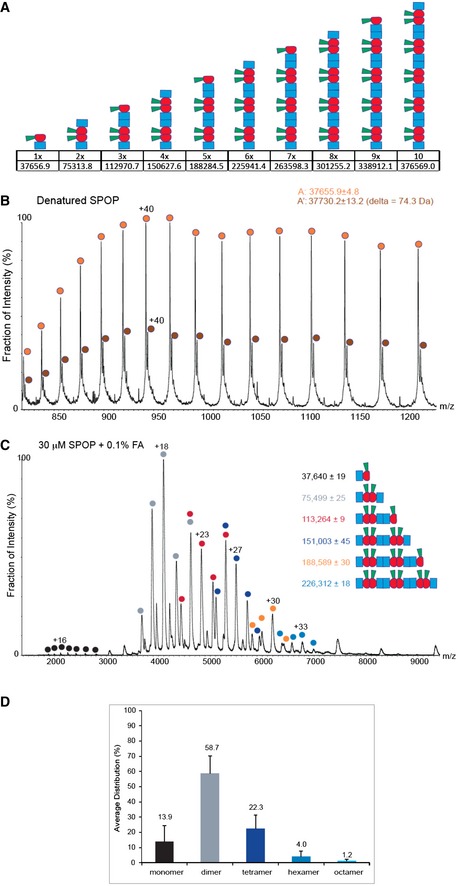
- Schematic representation of putative SPOP assemblies. Under each assembly state, the number of monomers and the theoretical mass are shown.
- Mass spectrum of denatured SPOP. To evaluate the purity of the sample, SPOP (50 pmol) was loaded onto a monolithic column (Rozen et al, 2013) and eluted in a linear gradient of 20–50% ACN over 30 min. The protein eluted as a single peak at 17.6–19.8 min, at approximately 33% ACN. The spectrum shows a major population of 37,656 ± 5 Da (orange circles) and a smaller population of 37,730 ± 13 Da (brown circles). The mass difference may result from β‐mercaptoethanol.
- MS spectrum of SPOP assemblies under partial denaturing conditions. SPOP was analyzed by performing MS after adding of 0.1% formic acid (30 μM) to disturb the non‐covalent interactions. Under these conditions, trimers (red circles) and pentamers (orange circles) were generated. The fact that odd numbers of assemblies appear only under mild denaturing conditions indicates strong interactions between two monomers.
- Size distribution of SPOP oligomers at 30 μM protomer concentration from seven independent measurements.
With a quantitative model of SPOP self‐association in hand, what can we learn about the oligomeric species present in solution? Because addition of dimer building blocks happens isodesmically, the fractional population of oligomeric species present in solution in equilibrium decreases with their size (Oosawa & Kasai, 1962), as illustrated for 27 μM SPOP, the highest concentration assayed in CG‐MALS experiments (Fig 8B). The fractional populations of the assemblies observed in native MS (Figs 8A and EV4D) were slightly lower than those expected based upon solution CG‐MALS experiments, possibly due to reduced ionization efficiency of larger oligomers, but overall in agreement.
Importantly, the different oligomeric states were now possible to be quantified as a function of protein concentration. At low concentrations, SPOP predominantly forms dimers, but as the protein concentration increases, the fraction of SPOP in dimers decreases and the fraction of SPOP in higher‐order oligomeric species progressively increases (Fig 8C). Only at considerably higher protein concentrations, likely outside of the range that can be biophysically assayed, do we expect that isodesmic association will be dampened due to entropic losses of large oligomers (Chatelier, 1987).
SPOP oligomers are highly dynamic
Our data demonstrate that the oligomeric SPOP species are dynamic in vitro; after a protein concentration change, the SPOP size distribution rapidly re‐equilibrates on the timescale of seconds, as evidenced by the self‐consistent CG‐MALS data, which was acquired by dilution from stock solutions. We wondered whether this dynamic nature is mediated by both the BACK and BTB domains, or whether the high affinity of the latter essentially renders the BTB interface static. We used SEC to monitor the elution profile of individual SPOP mutants (Fig EV5) and that of mixtures of SPOP28–359 with each mutant (Fig EV5B and C). Though populations of mixed oligomeric species are observed, they are significantly smaller in size than for SPOP28–359 alone at equimolar total protein concentration, again demonstrating that self‐association into large higher‐order oligomers requires functional interactions through both domains. Nonetheless, these data suggest that dynamic assembly and disassembly of higher‐order SPOP oligomeric species is primarily mediated by the BACK domain but that the BTB domain can contribute to the observed dynamics at physiological temperatures over longer timescales.
Figure EV5. The dynamic nature of higher‐order SPOP oligomers is mediated by the BACK domain.
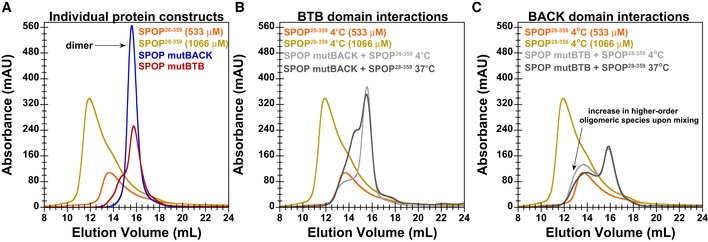
- SEC chromatograms for individual SPOP constructs injected at a loading concentration of 533 μM (and SPOP 28–359 at 1,066 μM) are shown. In agreement with CG‐MALS results, SPOP mutBTB and SPOP mutBACK predominantly form dimers (red and blue lines, respectively).
- SEC chromatogram for mixtures of 533 μM SPOP 28–359 with 533 μM of SPOP mutBACK is shown. When equal amounts of SPOP 28–359 and one of the mutants were mixed at 4°C, we observed that SPOP mutBACK, which cannot participate in typical BACK–BACK interactions, did not increase the population of higher‐order oligomers relative to that of WT only, as evidenced by a similar elution peak from the oligomeric species. This result shows that BTB dimers do not dissociate on the timescale of SEC at low temperature and cannot form hetero‐oligomers with SPOP 28–359. However, incubation at 37°C for 90 min renders BTB domain interactions dynamic enough to allow for BTB domain‐mediated exchange between WT and SPOP mutBACK (dark gray line). The hetero‐oligomers are smaller in size than oligomeric species observed for SPOP 28–359 alone at 533 μM (orange line).
- SEC chromatogram for mixtures of 533 μM SPOP 28–359 with 533 μM of SPOP mutBTB is shown. Conversely, in mixtures of SPOP 28–359 and SPOP mutBTB, which can effectively interact only through the BACK domain, the population of the higher‐order oligomeric species increased relative to that of SPOP 28–359 alone, irrespective of incubation time or temperature (gray lines). However, the mixed oligomers are smaller than SPOP 28–359 oligomers at an equimolar protein loading concentration (golden line).
SPOP oligomerization promotes substrate ubiquitination
SPOP is a substrate adaptor of CRL3, and the BACK domain interfaces are near the residues required for the SPOP/cullin‐3 interaction (Zhuang et al, 2009). Can SPOP form functional complexes with cullin‐3 in its oligomeric form? Crystal structures of subsets of CRL complexes and SPOP can be used to build a model of CRL3SPOP without additional assumptions (Movie EV2). The model suggests that cullin/Rbx1 binding to each SPOP monomer is not impeded by SPOP oligomerization, which is in agreement with experimental data (Theurillat et al, 2014). The helical propensity of the SPOP oligomer places the cullin molecules in a spiral arrangement, with each arm bent toward the helical axis (van Geersdaele et al, 2013).
It has been previously reported that dimerization (Zhuang et al, 2009) and higher‐order oligomerization (Errington et al, 2012) of SPOP stimulate the ubiquitination efficiency of CRL3SPOP. Our in vitro ubiquitination results (Fig EV6) are in agreement with this activity enhancement, although this is the first comparison of the activity of SPOP mutants containing all domains in the context of a fully active, neddylated CRL3. The Hedgehog transcriptional regulator Gli3 is a natural substrate of SPOP (Wang et al, 2010), and we used a fragment encompassing residues 1–455 as our model substrate. When we used self‐association‐competent SPOP28–359, neddylated CRL3SPOP ubiquitinated Gli31–455 efficiently in less than 10 min (Fig EV6). In contrast, SPOP mutants that are able to self‐associate through only one (or none) of the dimerization domains showed severely reduced ubiquitination efficiency, even after incubation for 30 min. SPOP mutBACK, which forms stable dimers at the 2 μM CRL3SPOP concentration used in the assay, was slightly more active than either SPOP mutBTB or SPOP mutBTB–BACK, which are both mostly monomeric under these conditions.
Figure EV6. Higher‐order SPOP self‐association promotes ubiquitination in vitro .
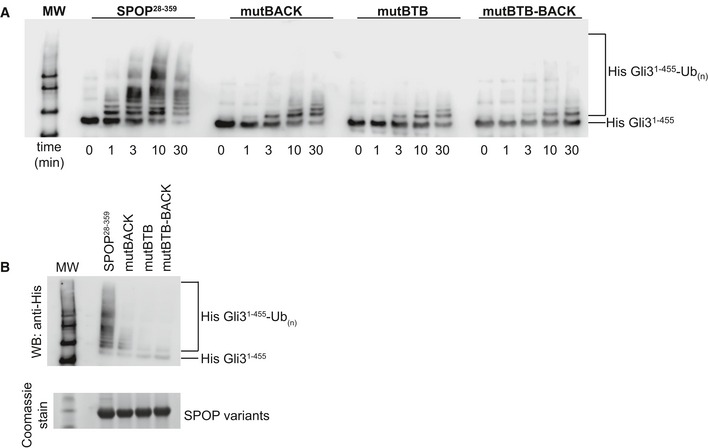
- In vitro ubiquitination assays with CRL3SPOP were performed as described previously (Zhuang et al, 2009) using His‐Gli31–455 as a substrate, SPOP 28–359, and each of the self‐association‐incompetent mutants. The reaction was monitored for 30 min, and time points taken are indicated below the Western blot. Ubiquitination efficiency was monitored by immunoblotting with an anti‐His antibody.
- Reactions were performed as in (A) and monitored for 30 min. Equal amounts of SPOP variants were used as shown in the SDS–PAGE gel.
Self‐association‐defective SPOP mutant affects Drosophila wing patterning
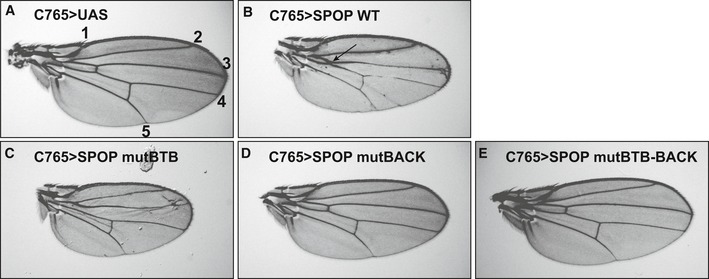
SPOP is known to regulate Hedgehog (Hh) pathway signaling through ubiquitination of GLI transcription factors (Kent et al, 2006; Hatayama & Aruga, 2012). We therefore chose to test the importance of SPOP oligomerization in vivo by monitoring Hh‐dependent wing patterning using the Drosophila melanogaster model system. Hh signaling patterns the anterior wing blade and dictates the distance between longitudinal veins (LV) 3 and 4 (de Celis, 2003). We expressed wild‐type or mutant SPOP in the developing wing under the control of the C765‐Gal4 epithelial driver and monitored for changes in LV3–LV4 intervein space in adult wings. Vector control wings show normal vein patterning (Fig 9A). Expression of wild‐type SPOP (Fig 9B) caused a proximal LV3–LV4 fusion (arrow) with reduction of intervein space. This is suggestive of attenuated Hedgehog signaling, presumably due to increased degradation of endogenous Ci, the Drosophila GLI ortholog. In contrast, expression of SPOP mutBTB (Fig 9C) triggered LV3–LV4 widening that is typical of a moderate Hh gain of function. This is consistent with this mutant interacting with endogenous HIB, the Drosophila SPOP ortholog, and acting in a dominant‐negative manner to interfere with Ci degradation. The BTB and BACK domain interfaces of mouse SPOP and HIB are highly conserved. Interestingly, expression of SPOP mutBACK and mutBTB–BACK (Fig 9D and E, respectively) failed to induce wing‐patterning defects, indicating that these mutants do not likely impact the function of endogenous HIB. These results demonstrate that the higher‐order oligomer forming wild‐type SPOP down‐regulates Hh signaling, whereas SPOP mutBTB interferes with the function of endogenous HIB in vivo.
Figure 9. The self‐association‐deficient SPOP mutant mutBTB has a substrate degradation defect in vivo .
-
AUAS vector served as a control and yielded a normal wing. Longitudinal veins are denoted by numbers 1–5.
-
BExpression of SPOP WT resulted in a modest Hh loss‐of‐function phenotype with fusion of LV3 and LV4 (arrow).
-
CmutBTB acts in a dominant‐negative manner, as evidenced by a modest Hh gain‐of‐function phenotype with expansion of the LV3–LV4 intervein space.
-
D, EExpression of (D) mutBACK and (E) mutBTB–BACK does not induce a wing‐patterning defect.
Discussion
Here, we show that nuclear speckles have liquid‐like character. Additionally, we provide a link between higher‐order SPOP oligomers in cells and their localization in these nuclear speckles. We show that SPOP self‐associates indefinitely via the cooperation of its tandem self‐association domains, the BTB and the BACK domains, and we quantify the resulting wide size distribution of higher‐order oligomers. The self‐association‐deficient BTB mutant demonstrates a dominant‐negative functional defect in flies.
Model for physiological function of higher‐order oligomerization of SPOP
Based on our results, we present a model in which tandem self‐association domains in SPOP promote the recruitment of SPOP to nuclear speckles, which may serve as hotspots for CRL3SPOP‐mediated ubiquitination (Fig 10). The tandem self‐association domains promote the formation of higher‐order SPOP oligomers. Smaller oligomeric SPOP species are diffusely distributed, and higher‐order oligomers preferentially localize to nuclear speckles. Dynamic SPOP BACK domain‐mediated association and dissociation promotes exchange between the speckles and the diffuse pool. We propose that the high local concentrations of substrate and of oligomeric components of the enzymatic ubiquitination cascade promote efficient, proximity‐enhanced ubiquitination, not only in nuclear speckles, but likely also in other SPOP‐positive liquid compartments in the nucleus.
Figure 10. Higher‐order SPOP oligomers localize to nuclear speckles.
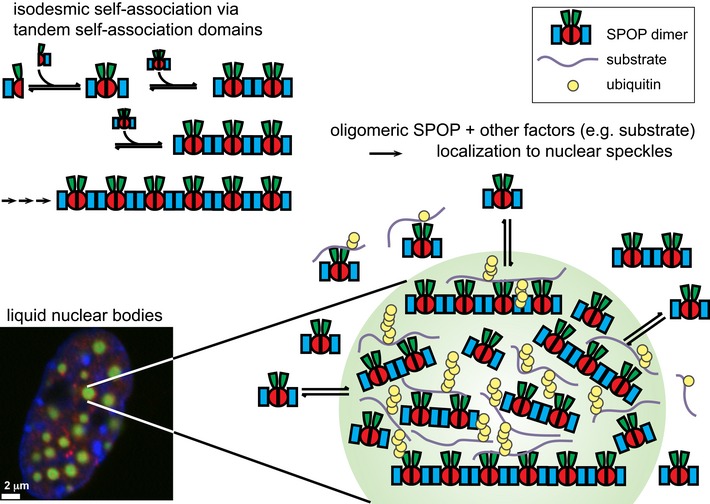
We propose a model in which the ability of SPOP to form higher‐order oligomers promotes its localization to nuclear speckles. Smaller oligomeric SPOP species are diffusely distributed, but higher‐order oligomers preferentially localize to speckles. Dynamic BACK domain association mediates exchange between higher‐order/punctate structures and the diffuse pool. SPOP interacts with a number of binding partners, including other components of the CRL and substrates, and can likely recruit them to punctate structures (Kwon et al, 2006; Chen et al, 2009). We speculate that nuclear speckles serve as hotspots of SPOP‐mediated ubiquitination by concentrating SPOP oligomers, substrates, and other components locally.
Self‐association behavior of large, labile oligomers can be determined
SPOP forms higher‐order oligomers (i.e., their oligomeric state is not restricted to a specific size) through an isodesmic self‐association mechanism. SPOP was previously shown to form higher‐order oligomers (Errington et al, 2012), but neither the structural organization of the oligomeric assemblies nor their size distribution was understood. We find that SPOP BTB dimers serve as building blocks for the formation of higher‐order structures through BACK domain‐mediated self‐association (cartoon schematic, Fig 7A). The apparent equilibrium constants are identical for each addition of the unit building block, independent of the oligomeric size already present (Fig 7A and B) (Van Holde & Rossetti, 1967). The presence of both domains is required for the formation of large oligomeric species (Fig 7C) that apparently have no size limit (Figs 4A and 7B).
We demonstrate that the BACK domain dimerizes with micromolar affinity (Figs 6D and 7B, and Appendix Fig S6F). Our LS and AUC data of SPOP mutBTB as well as the CG‐MALS data of SPOP28–359 exclude the possibility that the major oligomeric state of the BACK domain are tetramers or pentamers (Errington et al, 2012). Knowledge of the properties for each individual domain (Fig 6) helped us to construct the isodesmic self‐association model (Fig 7A), but we were also able to fit the experimental CG‐MALS data without prior knowledge of the dissociation constants and oligomeric states of the individual domains, demonstrating the analytical power of CG‐MALS.
Careful biophysical characterization has previously allowed the elucidation of isodemic self‐association behavior for several other proteins, including insulin (Jeffrey et al, 1976), FtsZ (Rivas et al, 2000), Phage ϕ29 protein p6 (Abril et al, 1997), chicken deoxy hemoglobin D (Rana & Riggs, 2011), human apolipoprotein C‐II (Yang et al, 2012), tubulin (Frigon & Timasheff, 1975), and for the assembly of heterochromatin protein 1 with nucleosomes in controlling heterochromatin spread (Canzio et al, 2013). Isodesmic self‐association may be a general mechanism for creating large, labile polymers that can disaggregate readily when required. The inherent size heterogeneity of labile higher‐order oligomers poses challenges because the contribution of the different oligomeric states to function cannot be assigned. Herein, we present rigorous biophysical analysis of higher‐order SPOP oligomers. We quantify the fractional population of different oligomeric SPOP species at a given concentration and are able to demonstrate how these populations change as a function of SPOP concentration. These results will enable future detailed functional and structural characterization.
Higher‐order oligomerization promotes localization to liquid nuclear speckles
Liquid cellular bodies lack an outer membrane but enrich specific components and allow dynamic exchange with the surrounding nucleoplasm or cytoplasm. Protein/protein and protein/RNA interactions promote assembly of and recruitment to these cellular bodies (Tourrière et al, 2003). Their liquid nature is thought to be generated via the process of liquid demixing phase separation (Brangwynne et al, 2009; Li et al, 2012b; Molliex et al, 2015; Nott et al, 2015; Patel et al, 2015), which leads to large, micrometer‐sized assemblies above threshold macromolecular concentrations. We show that nuclear speckles have liquid droplet properties and that they, together with nucleoli (Brangwynne et al, 2011), P granules (Brangwynne et al, 2009), and stress granules (Molliex et al, 2015), belong to the category of liquid membrane‐less organelles. These recent discoveries suggest that many micrometer‐sized cellular bodies may have liquid properties.
SPOP was previously known to localize to nuclear speckles (Nagai et al, 1997) and other nuclear bodies (Hernández‐Muñoz et al, 2005), but the molecular basis for its recruitment was unclear. Our findings strongly suggest that the presence of two functional self‐association domains in SPOP promotes its localization to nuclear speckles (Fig 4). Mutations disrupting the self‐association ability of one or both domains result in diffuse localization, supporting the notion that the localization of SPOP depends on specific domain interactions, rather than nonspecific aggregation. In addition, we demonstrate through cross‐linking experiments that SPOP forms higher‐order oligomers not only in vitro but also in cells (Fig 5B and C). These higher‐order oligomers are recruited to nuclear speckles although the mechanism is unknown. While isodesmic self‐association of SPOP promotes SPOP localization to nuclear speckles, it is not a mechanism that promotes liquid demixing phase separation because it leads to a gradual shift to larger oligomeric species with increasing protein concentration (Fig 8C), not to the switch‐like formation of a protein‐rich phase. However, the labile properties of SPOP oligomers, that is, the fact that dimer building blocks can dissociate and re‐associate readily, may be conducive to dynamic exchange within liquid assemblies. Enrichment of SPOP in nuclear speckles may create hotspots for CRL3SPOP‐mediated ubiquitination.
Potential roles for SPOP self‐association in oncogenesis
The new level of insight we provide into physiological SPOP function promises to be useful for understanding SPOP‐driven oncogenesis. SPOP cancer mutations are not usually accompanied by loss of heterozygosity (Barbieri et al, 2012), raising the possibility that mutant and WT SPOP form mixed oligomers. The dominant‐negative phenotype observed for the SPOP BTB domain mutant in fly wings supports this hypothesis. A number of mutations in the BTB and BACK domains of SPOP have been found in cancer patients [Appendix Fig S1 and www.cbioportal.org (Cerami et al, 2012; Gao et al, 2013)], but their functional impact has not been determined. We speculate that they may affect SPOP self‐association and have effects on both substrate turnover and cellular localization. In agreement with this proposal, cellular mislocalization of SPOP into the cytoplasm has been shown to drive tumorigenesis via targeting of an unnatural set of substrates (Li et al, 2014).
Our work provides a link between the size distribution of SPOP oligomers, protein function, and cellular localization. Tandem self‐association domains may represent a general architecture for concentrating proteins in liquid bodies and for their recruitment to large signaling assemblies with high activity.
Materials and Methods
Protein expression and purification
A synthetic, codon‐optimized gene for Gli31–455 was purchased from GeneArt (Life Technologies) and introduced into an expression vector using the Gateway technology (Life Technologies). Gli31–455 was expressed as a His6‐tagged fusion protein in BL21‐Gold (DE3) cells (Agilent) in LB medium. Cells were lysed in 50 mM Tris pH 8, 500 mM NaCl, and 2 mM β‐mercaptoethanol (β‐ME) with a microfluidizer. Because His6‐Gli31–455 was in inclusion bodies, the soluble lysate was discarded, the insoluble fraction was washed with lysis buffer + 0.1% Triton X‐100, and the protein was extracted with 6 M guanidine hydrochloride (GndHCl), 50 mM Tris pH 8, 30 mM imidazole, and 2 mM β‐ME. The cleared lysate was loaded onto a HisTrap column (GE Healthcare), washed with extraction buffer followed by 50 mM Tris pH 8, 500 mM NaCl, and 2 mM β‐ME, and eluted in the wash buffer with a gradient from 30 mM to 500 mM imidazole. Fractions were pooled, concentrated, and subjected to SEC on a Superdex 200 16/60 column (GE Healthcare) equilibrated with 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM DTT, and 0.005% Tween‐20.
His‐SUMO‐SPOP28–359 was expressed in BL21 RIPL cells in ZYM‐5052 autoinduction media (Studier, 2005). Cells were lysed in 50 mM Tris pH 8.0, 500 mM NaCl, 30 mM imidazole, 2 mM β‐ME, and Complete protease inhibitor cocktail (Roche) with a microfluidizer. The cleared lysate was loaded onto a gravity Ni‐NTA column, washed with lysis buffer, and eluted in 50 mM Tris pH 8.0, 500 mM NaCl, 300 mM imidazole, and 2 mM β‐ME. The eluted protein was cleaved with TEV protease while dialyzing against 50 mM Tris pH 8.0, 500 mM NaCl, and 2 mM β‐ME with incubation at room temperature for 4 h followed by incubation at 4°C for an additional 60 h. The dialysate was passed back over Ni‐NTA resin, equilibrated with dialysis buffer, to remove His‐SUMO and uncleaved protein. The flow‐through was concentrated and subjected to SEC on a Superdex 200 16/60 column (GE Healthcare) equilibrated with 25 mM Tris pH 7.5, 150 mM NaCl, and 5 mM DTT. Mutants were generated by performing site‐directed mutagenesis to produce the following mutations: SPOP mutBACK, Y353E; SPOP mutBTB, L186D, L190D, L193D, I217K; and SPOP mutBTB–BACK, L186D, L190D, L193D, I217K, Y353E (Zhuang et al, 2009; van Geersdaele et al, 2013) and were expressed and purified as described above.
Size‐exclusion chromatography
Protein samples (50 μl) were injected onto a Superose 6 (GE Healthcare) gel filtration column equilibrated with 25 mM Tris pH 7.6, 150 mM NaCl, and 5 mM DTT. The elution volume was monitored and converted into an apparent molecular weight (MWapp) based upon calibration with globular protein standards (Bio‐Rad Gel Filtration Standard). The injection loop was overfilled with 1.2 times its volume to ensure consistent injection volumes.
SPOP ΔBACK AF488 labeling
Purified protein (2 mg) was buffer‐exchanged into 0.1 M HEPES pH 7.5 with 50 mM NaCl and concentrated to 350 μl. Dimethylformamide (20 μl) was added to 1 mg Alexa Fluor 488 carboxylic acid succinimidyl ester (Molecular Probes). Protein and fluorophore solutions were incubated at 37°C for 5 min. Fluorophore was added to the protein solution and gently pipetted to mix. The protein–fluorophore mixture was incubated in the dark at room temperate for 2 h and then overnight at 4°C. P6 DG resin (Bio‐Rad) was hydrated in 0.1 M HEPES pH 7.5 with 50 mM NaCl, and the fine particles were poured off three times. The resin was poured into a column and allowed to pack by gravity flow. After incubation, the protein was purified on the desalting column to separate labeled protein from excess fluorophore. The labeled protein was then further purified by ion‐exchange chromatography with a HiTrap Q column (GE Healthcare). Fractions containing labeled protein (AF488‐SPOP ΔBACK) were pooled, concentrated, and stored at −80°C. The calculated labeling efficiency was 76% per monomer, but mass spectrometry analysis (St. Jude Children's Research Hospital Proteomics Facility) of the labeled protein revealed that two sites per monomer are available for labeling, suggesting that overall labeling efficiency is lower.
Fluorescence‐detected analytical ultracentrifugation (FDS‐AUC)
Sedimentation velocity analytical ultracentrifugation (SV‐AUC) experiments were conducted in an Optima XL‐A analytical ultracentrifuge (Beckman Coulter) with the fluorescence detection system (AVIV Biomedical), following standard protocols (Zhao et al, 2013a,c). All samples were prepared in a freshly made working buffer of 25 mM Tris pH 7.6, 150 mM NaCl, and 5 mM DTT at 20°C. To enable work in the picomolar concentration range (Zhao et al, 2014), cell assemblies with double‐sector 12‐mm charcoal‐filled Epon centerpieces and quartz or sapphire windows were filled with working buffer and screened in an initial blank FDS‐SV run to ensure no obvious contamination with fluorescent signal. Buffer was then taken out, and SPOP samples were inserted into the cell assemblies. A dilution series with AF488‐SPOP ΔBACK (10 pM – 300 nM) was examined, and, as a control (Zhao et al, 2013c), a titration series with the mixture of the labeled (0.2 nM) and unlabeled SPOP ΔBACK (0.3 nM – 10 μM) was used in the FDS‐SV experiments. In order to avoid surface adsorption of SPOP ΔBACK, 0.1 mg/ml bovine serum albumin (BSA) was added in all protein samples as a carrier protein. In order to quantify the signal contribution from BSA, an additional reference cell filled with BSA alone was added (Zhao et al, 2014). To ensure the same hydrodynamic behavior of the labeled and unlabeled SPOP ΔBACK, we carried out SV experiments in parallel with the conventional Rayleigh interference and absorbance optics with samples in the micromolar range, following a standard protocol (Zhao et al, 2013a). Furthermore, a control experiment was carried out to establish that the presence of BSA did not impact the s‐value of AF488‐SPOP ΔBACK. For the FDS experiments, radial calibration and the setup of focusing depth and PMT voltage were performed at 650 g (3,000 rpm). The focal depth was set to 4 mm, and, in order to obtain sufficient fluorescent signal for the samples with different concentrations of the fluorescently labeled protein, a range of PMT settings were used (e.g. 60% and gain 8 for 10 pM, 40% and gain 1 for 300 nM). Then, equilibration was carried out for 2 h with the rotor at rest, followed by acceleration from 0 to 182,000 g (50,000 rpm). Fluorescence scans were acquired continuously for a period of 12 h.
Analysis of sedimentation velocity data
The FDS‐SV data were sorted according to the sample position, PMT voltage, and gains using the Fluorescence Tools set in SEDFIT version 14.4 (Zhao et al, 2013b), and subjected to standard c(s) analysis (Schuck, 2000) in SEDFIT. Integration between 1 and 6 S was carried out to generate the isotherm analysis of signal‐weighted sedimentation coefficients, s w. The signal contribution of BSA was subtracted from the low concentration samples where its signal is significant, as described previously (Zhao et al, 2013c, 2014). The resulting s w isotherm was loaded into SEDPHAT and fit with the homo‐dimerization model for nonlinear least square analysis,
| (1) |
in which s 1 and s 2 are the s‐values for monomer and dimer, respectively, c 1 and c tot denote the molar concentration for monomer and protomer, respectively, and K 12 indicates the equilibrium association constant (K 12 = 1/K D) (Zhao et al, 2013c). K 12, s 1, and s 2 were optimized in the fit, and the error intervals of the best‐fit values were determined using error surface projection at a 95% confidence level. All plots were generated with GUSSI (kindly provided by Dr. Chad Brautigam, UT Southwestern).
Sediment equilibrium analytical ultracentrifugation (SE‐AUC)
Experiments were carried out in a ProteomeLab XL‐I analytical ultracentrifuge with a four‐hole rotor (Beckman An‐60Ti) and cells containing quartz windows and charcoal‐filled Epon double‐sector center pieces (Beckman Coulter, Fullerton, CA). AUC was performed in 25 mM Tris pH 7.6, 150 mM NaCl, and 1 mM TCEP buffer for SPOP mutBTB. The partial specific volume at 20°C and the molecular weight of the protein were calculated based on its amino acid composition using the software SEDFIT (https://sedfitsedphat.nibib.nih.gov/software/default.aspx). Buffer from the size‐exclusion column was used as the ultracentrifugation buffer and as an optical reference.
Sedimentation equilibrium for SPOP mutBTB at a rotor temperature of 20°C was attained at increasing rotor speeds of 5,900 g (9,000 rpm) (for 42 h), 12,300 g (13,000 rpm) (for 32 h), and 35,200 g (22,000 rpm) (for 20 h) (Zhao et al, 2013a). Loading protein concentrations were between 1.60 and 16.57 μM (130 μl), and absorbance distributions were recorded at 280 nm in 0.001 cm radial intervals with 20 replicates for each point. Global least squares modeling was performed at multiple rotor speeds with the software SEDPHAT (https://sedfitsedphat.nibib.nih.gov/software/default.aspx) using the reversible monomer–dimer self‐association model (Zhao et al, 2013a).
CG‐MALS data collection and analysis
Light scattering and absorbance at 280 nm measurements were collected using a Calypso system (Wyatt Technology Corporation) consisting of a software‐controlled multiple syringe pump to create the concentration gradient and a DAWN HELEOS multi‐angle light scattering photometer and a UV–vis detector (Agilent Technologies) to collect data from the incoming sample stream. Static light scattering data were collected at 14 scattering angles as a function of protein concentration. For each injection into the light scattering detector, the solution was allowed to come to equilibrium for 60 s. Protein concentration was monitored simultaneously by UV. Equilibrium MALS versus concentration data were analyzed as described by Attri et al, and data obtained at all angles in a single experiment were combined for subsequent model generation (Attri et al, 2010). Simple monomer–dimer, monomer–trimer, etc. interactions and isodesmic self‐association models available in the CALYPSO software (Wyatt Technology Corporation) were used to model the data. In a solution, in which the different scattering species X i correspond to different association states of a single protein, the theory of Rayleigh scattering from multicomponent solutions yields the concentration‐dependent Rayleigh ratio R
in which M x is the molar mass of protein X and [X i] is the concentration of the species X i. R is normalized to an optical constant K* defined as
where n 0 denotes the refractive index of the solvent, λ0 the vacuum wavelength of incident light (690 nm), N A, Avogadro's number, and dn/dc the specific refractive increment of SPOP.
The concentrations of each species are related to the equilibrium constants and total protein concentration. This results in the following equations for typical monomer–dimer, monomer–trimer, monomer–i‐mer equilibria:
In the equations above, i = 1 represents the free monomer, the total molar concentration [X]total is known at each gradient injection, and R(0)/K* is measured. Nonlinear least square optimization is used to obtain a single K A value that fits the data across the entire concentration range of interest.
To describe isodesmic self‐association, we used equations previously described by Attri et al (2010):
The only assumption entered into the model was the fact that SPOP dimers were treated as self‐associating building blocks. The model was broken off at n = 12 (SPOP2)12, a dodecamer of SPOP dimers, because populations of larger oligomeric species did not contribute to the fit significantly. Fitting parameters were the K A and the molecular weight of the building block, which was in agreement with a SPOP dimer. For the calculation of size distributions, we explicitly modeled the monomer–dimer equilibrium that coexists with the isodesmic equilibrium and influences the concentrations of available dimer building blocks for isodesmic self‐association.
For SPOP28–359, models that describe the formation of a mixture of discrete oligomeric states revealed that each of these states is important for the fit of the data. The association constants for their formation are highly correlated, effectively turning the model into an isodesmic model.
Size‐exclusion chromatography versus CG‐MALS
To gain insight into the large protein species that contribute to the formation of this type of cellular structure, we set out to quantify SPOP self‐association. Higher‐order oligomers, which lack a discrete oligomeric size and instead adopt a range of different sizes in equilibrium, are notoriously challenging to characterize biophysically and structurally because of their inherent heterogeneity. In SEC, directed flow competes with diffusion and the associated continuous dilution of the sample leads to constant re‐equilibration of systems that exhibit fast exchange on the timescale of the method (Stevens & Schiffer, 1981). Therefore, SEC precludes equilibrium analysis of self‐association. The difference in apparent molecular weight calculated from SEC data (Figs 4A and EV2C) and weight‐average molar mass calculated from CG‐MALS data (Fig 7B) is due to the differences in deriving this value. SEC relies upon globular standards to translate the elution volume, a measure of the hydrodynamic radius, into apparent molecular weight and, therefore, is affected by the dependence of the hydrodynamic radius on the protein conformation. SEC also cannot account for the mixture of oligomeric species present at a given concentration. Light scattering provides an accurate and precise measurement of the weight‐average molar mass (Attri & Minton, 2005b).
Native MS
Prior to MS analysis, SPOP was diluted to 30 μM with 150 mM ammonium acetate (pH 7.5) and 5 mM DTT and buffer‐exchanged into the same buffer, using Bio‐Spin columns (Bio‐Rad). Typically, aliquots of 2 μl of protein sample were electrosprayed from gold‐coated borosilicate capillaries prepared in‐house, as previously described (Kirshenbaum et al, 2010). Nanoflow electrospray ionization MS experiments were then conducted under non‐denaturing conditions on a QToF Q‐Star Elite instrument (MDS Sciex, Canada), modified for improved transmission of large non‐covalent complexes. Assays were performed in positive ion mode. Conditions were optimized to enable the ionization and removal of adducts, without disrupting the non‐covalent interactions of the proteins tested. The following experimental parameters were used: ionspray voltage, 1,250 V; declustering potential, 180 V; focusing potential, 250 V; second declustering potential, 5 V; and collision energy, 60 V. Spectra are shown with minimal smoothing and without background subtraction.
Fluorescence anisotropy assays
A peptide comprising residues 91–107 from the protein Puckered [amino acid sequence Ac‐ENLACDEVTSTTSSSST‐NH2 (Zhuang et al, 2009)] and N‐terminally labeled with fluorescein was purchased from GenScript. Assays were performed in 20 mM Tris pH 7.6, 150 mM NaCl, 5 mM DTT, and 0.01% Triton X‐100. Serial dilutions of SPOP, from 0.006 to 100 μM, were prepared on a 384‐well plate (Greiner BioOne). Peptide was added for a final concentration of 40 nM into each well. Anisotropy was measured using a CLARIOstar plate reader (BMG LABTECH). K D values were obtained by fitting experimental data to the following equation, adapted from Roehrl et al,
| (2) |
where Puc is the total concentration of peptide, SPOP is the total concentration of SPOP, I max is the maximum anisotropy, and I min is the minimum anisotropy (Roehrl et al, 2004). Three independent experiments were performed for each SPOP, and the average and standard deviation are reported.
In vitro ubiquitination assays
Gli31–455 ubiquitination was assayed in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2, 5 mM ATP, 1 mM DTT, and 2 mg/ml BSA at room temperature at time points from 0 to 30 min. The reaction mixture contained ubiquitinating enzymes at final concentrations of 0.25 μM UBA1 (E1), 8 μM UbcH5B (E2), 2 μM NEDD8‐CUL3‐Rbx1 (E3), 2 μM SPOP28–359 or mutant (substrate adaptor), 75 μM ubiquitin (Boston Biochem, cat. # U‐100H), and 5 μM His6‐tagged Gli31–455. E1, E2, and E3 were purified as described previously (Zhuang et al, 2009). The substrate and products were visualized by Western blotting with anti‐His antibody (Thermo Scientific, cat. #MA1‐21315).
SPOP aggregation assays
SPOP28–359 or WT samples were prepared at 35 μM. The samples were centrifuged at 195,000 g (60k rpm) for 60 min at 20°C. The supernatant was decanted, and the remaining pellet was washed three times with buffer (25 mM Tris pH 7.6, 150 mM NaCl, 5 mM DTT). After the washes, the remaining pelleted material was resuspended in sample loading dye. This experiment was repeated twice and a representative gel is shown.
In vitro cross‐linking reactions
Protein samples were prepared at 30 μM in the following buffer: 20 mM HEPES pH 7.5, 150 mM NaCl, and 5 mM DTT. The amine cross‐linker BS3 (bis(sulfosuccinimidyl)suberate, Fisher Scientific) was added for a final concentration of 0.3 mM. Reactions were incubated at room temperature for 30 min. The reactions were quenched by the addition of 100 mM Tris pH 7.6 and were incubated at room temperature for at least 15 min prior to taking samples for analysis on a gel. Reactions were performed for SPOP ΔBACK and SPOP MATH (protein containing the MATH domain only) to show that cross‐linking conditions are not excessive. As expected, SPOP ΔBACK forms dimers and no cross‐linking is observed for SPOP MATH. This experiment was repeated three times and a representative gel is shown.
Cellular cross‐linking assays
V5‐tagged constructs were generated by performing site‐directed mutagenesis using the HA‐SPOP construct described above. Constructs were expressed in NIH 3T3 cells as described previously (Carroll et al, 2012). NIH 3T3 cells were obtained from ATCC (cat# 1658) and grown in mycoplasma‐free conditions. Cell lysis and Western blot analysis were performed 48 h after transfection, as described previously (Carroll et al, 2012) with minor modifications. Cells were lysed in the following: 25 mM HEPES pH 7.5, 150 mM NaCl, 1% NP‐40, 1% sodium deoxycholate, 0.2% SDS, 1× protease inhibitor (Roche), and 0.5 mM DTT. Equal amounts of protein lysate (based upon total protein concentration) were used in the cross‐linking reactions. BS3 was added for a final concentration of 2.5 mM, and reactions were incubated at room temperature for 30 min. The reactions were quenched by the addition of 300 mM Tris pH 7.6 and were incubated at room temperature for at least 15 min prior to taking samples for analysis on a gel. Anti‐V5 antibody (1:5,000; Invitrogen, cat# R96025) was used to visualize SPOP. This experiment was repeated three times for a total of four replicates. Representative Western blots are shown.
Immunofluorescence
The plasmid for expression of full‐length HA‐mouse SPOP (which is identical in protein sequence to human SPOP) was obtained from Hans Haecker (St. Jude Children's Research Hospital). Mutants were generated by site‐directed mutagenesis, introducing the same mutations as used for recombinant proteins above. The Gli3 coding sequence was amplified from cDNA, attB sites added via PCR, and the gene product cloned into a pcDNA 6.2 EmGFP vector via Gateway cloning (Life Technologies). The pCNMV6‐AC‐SC‐35‐GFP plasmid was purchased from OriGene (RG209842). Constructs were expressed in NIH 3T3 cells as described previously (Carroll et al, 2012). NIH 3T3 cells were obtained from ATCC (cat# 1658) and grown in mycoplasma‐free conditions. Fixation, immunostaining, and image analysis were performed 48 h after transfection, as described previously (Carroll et al, 2012), and images were collected at 60× magnification. Images at 10× magnification were used to calculate transfection efficiencies (Appendix Tables S1, S2 and S3). Rat anti‐HA antibody (1:250; Clone 3F10, Roche, cat# 11867423001), GFP fluorescence, mCherry fluorescence, or ReAsH (Fisher Scientific) was used to visualize SPOP, GFP fluorescence was used to visualize Gli3, mouse anti‐SC‐35 antibody (1:250; Abcam, cat# ab11826), rabbit anti‐coilin antibody (1:200; Santa Cruz Biotechnology, cat# SC32860), mouse anti‐B23 antibody (1:200, Santa Cruz Biotechnology, cat# SC56622), mouse anti‐CBX8 antibody (1:200, Santa Cruz Biotechnology, cat# SC374332), and mouse anti‐PML antibody (1:200, EMD Millipore, cat# 05‐718) were used to visualize nuclear speckles, Cajal bodies, nucleoli, polycomb bodies, and PML bodies, respectively. DAPI (Life Technologies, cat# D1306) was used to stain the nucleus. Anti‐mouse Alexa Fluor 555 (1:1,000, Life Technologies), anti‐rat Alexa Fluor 647 or Alexa Fluor 488 (1:1,000, Life Technologies), and anti‐rabbit Alexa Fluor 647 were used as secondary antibodies. For Fig 2, anti‐rat Alexa Fluor 488 was used as secondary antibody when SPOP was transfected alone, and anti‐rat Alexa Fluor647 was used when HA‐SPOP was transfected along with GFP‐Gli31–455. The images in Fig 2 are pseudocolored. For Fig EV1, anti‐rabbit Alexa Fluor 555 was used as secondary antibody to detect coilin, and anti‐mouse Alexa Fluor 555 was used to detect B23, CBX8, and PML.
Aspect ratio calculations were performed as described previously (Brangwynne et al, 2011), and iMaris (Oxford Instruments) was used to calculate nuclear body diameters and areas. Bodies in 151 cells transiently expressing HA‐SPOP (to mark nuclear speckles) and 155 cells transiently expressing HA‐SPOP and GFP‐Gli31–455 were analyzed. For each cell, the number of bodies, median aspect ratio of the bodies in the cell (intracellular median aspect ratio), and median area of the bodies in the cell (intracellular median area) were determined. Further statistical analyses were performed on these three variables of cell‐level data. Confidence intervals for the median of each of these variables under each biological condition (SPOP and SPOP+GLI31–455) were determined by inversion of the sign test. The Wilcoxon rank‐sum test was used to compare the median of each of these three variables across the two biological conditions. No multiple testing adjustments were performed. These analyses were performed using R statistical computing software (www.r-project.org). The number of cells imaged and median values (with 95% confidence intervals) are presented in Table 1.
Cells used in live cell imaging were grown under the same conditions as above, transfected with either SC‐35‐GFP, or both HA‐SPOP and GFP‐Gli31–455, and imaged at 63× magnification. FRAP events were corrected for background, normalized to the minimum and maximum intensity. FRAP data were fit to a single exponential rise, with the y‐intercept set at zero to account for the photobleaching event, to calculate the recovery rates. For SC‐35‐GFP‐transfected cells, the average from 45 FRAP events and cells is shown with respect to the standard error of the mean. The mean characteristic recovery time is indicated ± SEM. For GFP‐Gli31–455/SPOP‐transfected cells, one individual FRAP event is shown and the rate of recovery presented is an average of 5 independent FRAP events in independent cells. The mean and standard deviation of the characteristic recovery time are indicated. The FRAP experiments were performed on a Multimodal 3i Marianas systems configured with Yokagowa CSU‐X spinning disk scan heads, and the analyses were performed with Slidebook 6.0 software and Microsoft Excel.
Fusion of cellular bodies was observed in four independent GFP‐Gli31–455/SPOP‐transfected cells (71 cells were imaged for 51 s each), and photobleaching is not required for this event to occur. Fusion of nuclear speckles was observed in five independent SC‐35‐GFP‐transfected cells (out of 70 imaged).
Expression levels of HA‐SPOP and GFP‐Gli31–455
NIH 3T3 cells were transfected with empty vector pcDNA, pcDNA‐GFP‐Gli31–455, pcDNA‐HA‐SPOP, pcDNA‐Gli31–455 + pcDNA‐HA‐SPOP. Two days post‐transfection, cells were lysed in RIPA + 0.1% SDS lysis buffer for 30 min and supernatants were collected after centrifugation for 30 min at 12,000 g. Equal amounts (~35 μg) of protein were loaded onto a 4–15% gradient gel, and Western blotting was performed using the following antibodies to detect HA, GFP, and tubulin, respectively: rat anti‐HA and anti‐rat HRP, rabbit anti‐GFP and anti‐rabbit IR800, and mouse anti‐tubulin and anti‐mouse IR680.
Fly crosses
UAS‐SPOP transgenic flies were generated by injecting transgenes and targeting them to the 2R‐51D landing site using the PhiC31 system (Bischof et al, 2007). Bestgene Inc performed the injections. The C765‐Gal4 stock was obtained from the Bloomington stock center. It derives from an Oregon R strain with white mutation. Transgenes carry the mini‐white gene to give red eye color. Fly stocks were maintained at 18°C on Jazz agarose (Fisher Scientific), and crosses were performed at 25°C using standard techniques. Genotypes tested were w;C765‐Gal4/P{w,UAS‐empty} (control), w;C765‐Gal4/P{w,UAS‐SPOP}, w;C765‐Gal4/P{UAS‐SPOP mutBTB}, w;C765‐Gal4/P{UAS‐SPOP mutBACK}, w;C765‐Gal4/P{UAS‐SPOP mutBTB–BACK}. For wing analyses, crosses were performed at least twice and ~50 progeny, that is, 2–3‐day‐old female flies, were analyzed. Representative wings from adult flies were mounted on glass slides using DPX imaging medium. Images were acquired on a Zeiss Stemi 2000‐C11 microscope with a Zeiss AxioCam ICc3 camera.
Author contributions
TM and MRM developed the project, analyzed the data, and wrote the paper with assistance from all of the authors. MRM collected SEC data. JL generated the mutants, and MRM and JL purified all proteins. WJE and GGP provided reagents and collected preliminary data. MRM, JL, and AN collected and analyzed CG‐MALS and AUC data, and SK analyzed CG‐MALS data. MRM, JLP, SM, RMK, JPT, and SKO collected and analyzed cellular data. SM and SKO collected and analyzed fly data. HZ and PS collected and analyzed FDS‐AUC data. GB‐N and MS collected and analyzed MS data. TM collected in vitro ubiquitination data. SP computed statistics on nuclear bodies.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 9
Acknowledgements
We thank BA Schulman, WK Pierce, EW Martin, and CR Grace for valuable discussions and N Clark for preliminary data collection. We also thank H Haecker (St. Jude Children's Research Hospital). This work was funded by a V Foundation Scholar Grant (T.M.), R01GM112846 (T.M.), R01GM101087 (S.K.O.), the National Cancer Institute Cancer Center Support Grant P30CA21765 (at St. Jude Children's Research Hospital), the American Lebanese Syrian Associated Charities (to J.P.T., S.K.O., and T.M.), and the Intramural Program of the National Institute of Biomedical Imaging and Bioengineering (P.S.). Microscopy images were acquired at the Cell & Tissue Imaging Center at St. Jude Children's Research Hospital.
The EMBO Journal (2016) 35: 1254–1275
References
- Abril AM, Salas M, Andreu JM, Hermoso JM, Rivas G (1997) Phage phi29 protein p6 is in a monomer‐dimer equilibrium that shifts to higher association states at the millimolar concentrations found in vivo . Biochemistry 36: 11901–11908 [DOI] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grøfte M, Rask M‐BD, Streicher W, Jungmichel S, Nielsen ML, Lukas J (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP‐ribose). Nat Commun 6: 8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attri AK, Minton AP (2005a) New methods for measuring macromolecular interactions in solution via static light scattering: basic methodology and application to nonassociating and self‐associating proteins. Anal Biochem 337: 103–110 [DOI] [PubMed] [Google Scholar]
- Attri AK, Minton AP (2005b) Composition gradient static light scattering: a new technique for rapid detection and quantitative characterization of reversible macromolecular hetero‐associations in solution. Anal Biochem 346: 132–138 [DOI] [PubMed] [Google Scholar]
- Attri AK, Fernández C, Minton AP (2010) pH‐dependent self‐association of zinc‐free insulin characterized by concentration‐gradient static light scattering. Biophys Chem 148: 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S, Rosen MK (2014) Phase transitions of multivalent proteins can promote clustering of membrane receptors. Elife 3: e04123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J‐P, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae S‐S, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T et al (2012) Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44: 685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ‐line‐specific phiC31 integrases. Proc Natl Acad Sci USA 104: 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324: 1729–1732 [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Mitchison TJ, Hyman AA (2011) Active liquid‐like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci USA 108: 4334–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP (2013) Phase transitions and size scaling of membrane‐less organelles. J Cell Biol 203: 875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Liao M, Naber N, Pate E, Larson A, Wu S, Marina DB, Garcia JF, Madhani HD, Cooke R, Schuck P, Cheng Y, Narlikar GJ (2013) A conformational switch in HP1 releases auto‐inhibition to drive heterochromatin assembly. Nature 496: 377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CE, Marada S, Stewart DP, Ouyang JX, Ogden SK (2012) The extracellular loops of Smoothened play a regulatory role in control of Hedgehog pathway activation. Development 139: 612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Celis JF (2003) Pattern formation in the Drosophila wing: the development of the veins. BioEssays 25: 443–451 [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelier RC (1987) Indefinite isoenthalpic self‐association of solute molecules. Biophys Chem 28: 121–128 [DOI] [PubMed] [Google Scholar]
- Chen M‐H, Wilson CW, Li Y‐J, Law KKL, Lu C‐S, Gacayan R, Zhang X, Hui C‐C, Chuang P‐T (2009) Cilium‐independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 23: 1910–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle JJ, Finer‐Moore JS, Papa FR, Stroud RM, Walter P (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA 102: 18773–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington WJ, Khan MQ, Bueler SA, Rubinstein JL, Chakrabartty A, Privé GG (2012) Adaptor protein self‐assembly drives the control of a cullin‐RING ubiquitin ligase. Structure 20: 1141–1153 [DOI] [PubMed] [Google Scholar]
- Frigon RP, Timasheff SN (1975) Magnesium‐induced self‐association of calf brain tubulin. II. Thermodynamics. Biochemistry 14: 4567–4573 [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geersdaele LK, Stead MA, Harrison CM, Carr SB, Close HJ, Rosbrook GO, Connell SD, Wright SC (2013) Structural basis of high‐order oligomerization of the cullin‐3 adaptor SPOP. Acta Crystallogr D Biol Crystallogr 69: 1677–1684 [DOI] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, Blattner M, Lonard DM, Demichelis F, Coarfa C, Rubin MA, Zhou P, O'Malley BW, Mitsiades N (2013) Prostate cancer‐associated mutations in speckle‐type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci USA 110: 6997–7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, Bond R, He B, Coarfa C, Mitsiades N (2014) Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 74: 5631–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatayama M, Aruga J (2012) Gli protein nuclear localization signal. Vitam Horm 88: 73–89 [DOI] [PubMed] [Google Scholar]
- Hernández‐Muñoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y, van Lohuizen M (2005) Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci USA 102: 7635–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey PD, Milthorpe BK, Nichol LW (1976) Polymerization pattern of insulin at pH 7.0. Biochemistry 15: 4660–4665 [DOI] [PubMed] [Google Scholar]
- Jung Y‐S, Kim H‐Y, Lee YJ, Kim E (2007) Subcellular localization of Daxx determines its opposing functions in ischemic cell death. FEBS Lett 581: 843–852 [DOI] [PubMed] [Google Scholar]
- Kameyama K, Minton AP (2006) Rapid quantitative characterization of protein interactions by composition gradient static light scattering. Biophys J 90: 2164–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent D, Bush EW, Hooper JE (2006) Roadkill attenuates Hedgehog responses through degradation of Cubitus interruptus . Development 133: 2001–2010 [DOI] [PubMed] [Google Scholar]
- Kim B, Nam HJ, Pyo KE, Jang MJ, Kim IS, Kim D, Boo K, Lee SH, Yoon J‐B, Baek SH, Kim JH (2011) Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3‐SPOP E3 ubiquitin ligase complex. Biochem Biophys Res Commun 415: 720–726 [DOI] [PubMed] [Google Scholar]
- Kim MS, Je EM, Oh JE, Yoo NJ, Lee SH (2013) Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. APMIS 121: 626–633 [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata‐Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K (2007) Two regulatory steps of ER‐stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol 179: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshenbaum N, Michaelevski I, Sharon M (2010) Analyzing large protein complexes by structural mass spectrometry. J Vis Exp 40: e1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer‐Moore J, Zhang C, Shokat KM, Stroud RM, Walter P (2009) The unfolded protein response signals through high‐order assembly of Ire1. Nature 457: 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, Baek SH, Chiba T, Tanaka K, Bang OS, Joe CO, Chung CH (2006) BTB domain‐containing speckle‐type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3‐based ubiquitin ligase. J Biol Chem 281: 12664–12672 [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC, NIH Intramural Sequencing Center (NISC) Comparative Sequencing Program , Hieter P, Mullikin JC, Merino MJ, Bell DW (2012) Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin‐remodeling and ubiquitin ligase complex genes. Nat Genet 44: 1310–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Liang Y‐Y, Feng X‐H, Tsai SY, Tsai M‐J, O'Malley BW (2008) Essential phosphatases and a phospho‐degron are critical for regulation of SRC‐3/AIB1 coactivator function and turnover. Mol Cell 31: 835–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Korennykh AV, Behrman SL, Walter P (2010) Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA 107: 16113–16118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y‐S, Damko E, Moquin D, Walz T, McDermott A, Chan FK‐M, Wu H (2012a) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng H‐C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang Q‐X, Nixon BT, Rosen MK (2012b) Phase transitions in the assembly of multivalent signalling proteins. Nature 483: 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L, Zhuang M, Hu S, Li X, Zhou L, Li X, Calabrese MF, Watson ER, Prasad SM, Rinker‐Schaeffer C, Eggener SE et al (2014) SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell 25: 455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 156: 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na GC, Timasheff SN (1985) Velocity sedimentation study of ligand‐induced protein self‐association. Meth Enzymol 117: 459–495 [DOI] [PubMed] [Google Scholar]
- Nagai Y, Kojima T, Muro Y, Hachiya T, Nishizawa Y, Wakabayashi T, Hagiwara M (1997) Identification of a novel nuclear speckle‐type protein. SPOP FEBS Lett 418: 23–26 [DOI] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett‐Jones DP, Pawson T, Forman‐Kay JD, Baldwin AJ (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57: 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosawa F, Kasai M (1962) A theory of linear and helical aggregations of macromolecules. J Mol Biol 4: 10–21 [DOI] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Pierce WK, Grace CR, Lee J, Nourse A, Marzahn MR, Watson ER, High AA, Peng J, Schulman BA, Mittag T (2016) Multiple weak linear motifs enhance recruitment and processivity in SPOP‐mediated substrate ubiquitination. J Mol Biol 428: 1256–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana MS, Riggs AF (2011) Indefinite noncooperative self‐association of chicken deoxy hemoglobin D. Proteins 79: 1499–1512 [DOI] [PubMed] [Google Scholar]
- Rivas G, López A, Mingorance J, Ferrándiz MJ, Zorrilla S, Minton AP, Vicente M, Andreu JM (2000) Magnesium‐induced linear self‐association of the FtsZ bacterial cell division protein monomer. The primary steps for FtsZ assembly. J Biol Chem 275: 11740–11749 [DOI] [PubMed] [Google Scholar]
- Roehrl MHA, Wang JY, Wagner G (2004) A general framework for development and data analysis of competitive high‐throughput screens for small‐molecule inhibitors of protein‐protein interactions by fluorescence polarization. Biochemistry 43: 16056–16066 [DOI] [PubMed] [Google Scholar]
- Rozen S, Tieri A, Ridner G, Stark A‐K, Schmaler T, Ben‐Nissan G, Dubiel W, Sharon M (2013) Exposing the subunit diversity within protein complexes: a mass spectrometry approach. Methods 59: 270–277 [DOI] [PubMed] [Google Scholar]
- Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens FJ, Schiffer M (1981) Computer simulation of protein self‐association during small‐zone gel filtration. Estimation of equilibrium constants. Biochem J 195: 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW (2005) Protein production by auto‐induction in high density shaking cultures. Protein Expr Purif 41: 207–234 [DOI] [PubMed] [Google Scholar]
- Theurillat J‐PP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, Moch H, Privé GG, Carr SA, Garraway LA (2014) Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP‐mutant prostate cancer. Science 346: 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J (2003) The RasGAP‐associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160: 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Van Holde KE, Rossetti GP (1967) A sedimentation equilibrium study of the association of purine in aqueous solutions. Biochemistry 6: 2189–2194 [DOI] [PubMed] [Google Scholar]
- Wang C, Pan Y, Wang B (2010) Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full‐length activators but not their repressors. Development 137: 2001–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H (2013) Higher‐order assemblies in a new paradigm of signal transduction. Cell 153: 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, la Cruz de MJ, Borkowski B, Zhang X, Chen ZJ, Jiang Q‐X (2014) Structural basis for the prion‐like MAVS filaments in antiviral innate immunity. Elife 3: e01489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Griffin MDW, Binger KJ, Schuck P, Howlett GJ (2012) An equilibrium model for linear and closed‐loop amyloid fibril formation. J Mol Biol 421: 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Lin S‐C, Lamothe B, Lu M, Lo Y‐C, Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, Lenardo MJ, Darnay BG, Wu H (2009) E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol 16: 658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Wang Y, Lu Q, Chen J, Zhang J, Liu T, Lv N, Luo S (2014) SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J Exp Clin Cancer Res 33: 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang L, Wang B, Ou C‐Y, Chien C‐T, Jiang J (2006) A hedgehog‐induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 10: 719–729 [DOI] [PubMed] [Google Scholar]
- Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, Dong Y, Huang H, Yu L, Wang C (2015) Endometrial cancer‐associated mutants of SPOP are defective in regulating estrogen receptor‐α protein turnover. Cell Death Dis 6: e1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, Schuck P (2013a) Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci 71: Chapter 20: Unit 20.12.1–20.12.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Casillas E, Shroff H, Patterson GH, Schuck P (2013b) Tools for the quantitative analysis of sedimentation boundaries detected by fluorescence optical analytical ultracentrifugation. PLoS ONE 8: e77245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lomash S, Glasser C, Mayer ML, Schuck P (2013c) Analysis of high affinity self‐association by fluorescence optical sedimentation velocity analytical ultracentrifugation of labeled proteins: opportunities and limitations. PLoS ONE 8: e83439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Mayer ML, Schuck P (2014) Analysis of protein interactions with picomolar binding affinity by fluorescence‐detected sedimentation velocity. Anal Chem 86: 3181–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP, Schulman BA (2009) Structures of SPOP‐substrate complexes: insights into molecular architectures of BTB‐Cul3 ubiquitin ligases. Mol Cell 36: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 9