

Abstract
C9ORF72 expression is reduced in a substantial number of patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), which may contribute to disease pathogenesis. However, its normal molecular function remains unknown. In this issue of The EMBO Journal, Sellier et al (2016) identified a novel protein complex consisting of C9ORF72, WDR41, and SMCR8 that acts as a GDP‐GTP exchange factor (GEF) for RAB8a and RAB39b and is regulated by TBK1, whose partial loss of function also causes ALS and FTD. They further reveal a potential modulatory role for this novel complex in macroautophagy (autophagy), especially in the context of ataxin‐2 toxicity.
Subject Categories: Neuroscience
When GGGGCC (G4C2) repeat expansions in the first intron of C9ORF72 were identified as the most common genetic cause of ALS and FTD (DeJesus‐Hernandez et al, 2011; Renton et al, 2011), nothing was known about the molecular functions of C9ORF72. Since then, breathtaking progress has been made in understanding the effects of this mutation, largely focusing on the toxic products derived from expanded G4C2 repeats (Gitler & Tsuiji, 2016). For instance, the expanded repeats in nuclear RNA foci might misregulate RNA processing by sequestering some RNA‐binding proteins. Moreover, abnormal translation of the sense and antisense repeat RNAs through a repeat‐associated non‐AUG (RAN) translation mechanism gives rise to toxic dipeptide repeat (DPR) proteins. However, expression of C9ORF72 mRNA is also reduced in ALS/FTD patients with C9ORF72 repeat expansions—raising the possibility that compromised C9ORF72 function contributes to disease pathogenesis as well (DeJesus‐Hernandez et al, 2011).
To understand the normal function of C9ORF72, Sellier et al (2016) set out to identify proteins that interact with it directly. Immunoprecipitation from N2A mouse cells followed by tandem mass spectrometry analysis revealed a novel protein complex containing Smcr8 (a DENN domain‐containing protein predicted to function as a GEF), Wdr41 (a WD40 repeat‐containing protein of unknown function), and Rab8a and Rab39b (two small GTPases involved in membrane trafficking), and transfected C9ORF72 (Fig 1). Additional biochemical analysis confirmed the existence of such a protein complex endogenously in mouse brain. Interestingly, RAB8 directly interacts with optineurin and TBK1, mutations in which are implicated in ALS/FTD (Bettencourt & Houlden, 2015). Moreover, in a Drosophila model of FTD linked to chromosome 3, Rab8 is a strong modifier of mutant CHMP2B toxicity (West et al, 2015). Thus, RAB8 may be a signaling hub for several disease proteins in ALS/FTD.
Figure 1. Schematic overview of ALS/FTD disease proteins known to play a role in the autophagy and endosomal–lysosomal pathways.
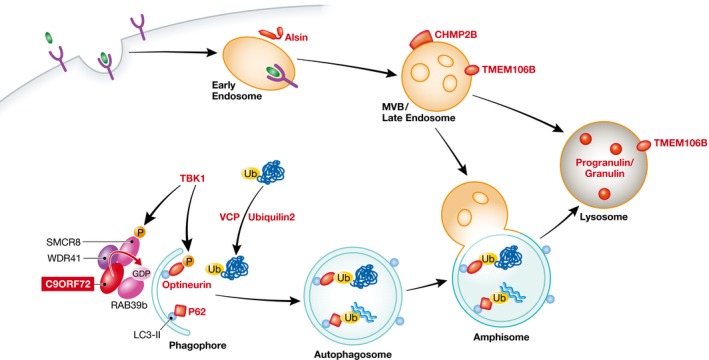
Sellier et al (2016) identified a novel complex consisting of C9ORF72, WDR41, and SMCR8 that serves as a GEF for RAB39b. This complex may play a modulatory role in the initiation of autophagy in certain cell types, but detailed molecular mechanisms remain unclear. Key functions of many other ALS/FTD disease proteins (in red) in some steps of the autophagy and endosomal–lysosomal pathways are also highlighted. MVB, multivesicular body.
What molecular functions might C9ORF72 fulfill in this novel protein complex? Sellier et al (2016) found that the complex composed of C9ORF72, SMCR8, and WDR41 serves as a GEF for RAB8a and RAB39b but not for some other RAB proteins, suggesting a certain degree of specificity. However, C9ORF72 alone does not have any GEF activity even though it has a DENN domain. This complex also interacts with autophagy adaptors p62 and optineurin; however, this interaction seems to be weak and its functional significance needs to be further confirmed.
The authors went on to examine the cellular functions of the C9ORF72‐containing complex. Knockdown of C9orf72 partially blocked the formation of autophagosomes after activation by torin or rapamycin in cultured primary mouse cortical neurons, consistent with a suggested role for C9ORF72 in autophagy (Farg et al, 2014). This function required RAB39b, since a constitutively active form of RAB39b, but not RAB8a or other RAB proteins, rescued autophagy defects caused by C9orf72 depletion. Accordingly, knockdown of C9orf72 or its binding partners such as Smcr8 or Wdr41 resulted in the accumulation of p62 in rodent neurons. This accumulation was rescued by the expression of the long isoform of C9ORF72, demonstrating that p62 accumulation is indeed a direct consequence of C9orf72 knockdown. Interestingly, the short isoform of C9ORF72 failed to do so, suggesting a distinct function. Moreover, cytoplasmic aggregates of TDP‐43 were observed in some cell types with C9orf72 depletion.
These results in cultured neurons raise several intriguing issues that need to be addressed. First, it seems that C9ORF72 is not essential for autophagy induction but may play a modulatory role in some cell types. Exactly how C9ORF72 and RAB39b do so at the molecular level is unclear. Second, the functional significance of the C9ORF72–RAB8a interaction remains to be further investigated. Third, elevated p62 and a potential autophagy defect were observed in human cortical neurons differentiated from induced pluripotent stem cells of FTD patients with C9ORF72 repeat expansions (Almeida et al, 2013). It will be informative to examine how much of this defect in human neurons results from partial loss of C9ORF72 activity or from DPR protein toxicity. Fourth, p62 accumulation and TDP‐43 aggregates were not reported in C9orf72‐knockout mice (Atanasio et al, 2016; O'Rourke et al, 2016). Thus, compensatory mechanisms may exist when C9orf72 is absent throughout development in vivo.
Another interesting aspect of this study is the exploration of interactions between the C9ORF72‐containing complex and other ALS/FTD disease proteins, such as TBK1, whose haploinsufficiency causes a subset of ALS/FTD cases (Bettencourt & Houlden, 2015). Sellier et al (2016) showed that SMCR8 is a target of TBK1, which also phosphorylates several other targets that function in the autophagy pathway. A form of SMCR8 that mimics a constitutive TBK1 phosphorylation rescued autophagy defects caused by depletion of SMCR8 or TBK1, demonstrating the importance of TBK1‐mediated SMCR8 phosphorylation in mediating the function of the C9ORF72/SMCR8/WDR41 complex in autophagy.
The authors also examined the gene encoding ataxin‐2, in which intermediate expansion of polyglutamine repeats (27–33) increases risk for ALS and FTD (Elden et al, 2010). C9orf72 depletion and simultaneous expression of ataxin‐2 with 30 glutamines, but not each manipulation alone, induced neurodegeneration in cultured rodent neurons and in a zebrafish model. This synergy seems to be specific to ataxin‐2, since C9orf72 depletion did not enhance the toxicity of mutant SOD1 or other disease proteins. The cause of this specificity remains to be further examined. Reducing C9orf72 expression did not significantly affect neuronal viability and had only a partial effect on basal autophagy, raising the possibility that this complex plays a modulatory role. It will be interesting to examine whether loss of C9orf72 also affects the accumulation of DPR proteins, especially in mouse models.
C9orf72‐knockout mice do not show signs of neurodegeneration in the brain (Atanasio et al, 2016; O'Rourke et al, 2016). Remarkably, however, they instead develop progressive splenomegaly and lymphadenopathy, a finding reminiscent of the increased incidence of lymphomas and other tumors caused by partial loss of function of beclin 1, a key factor in autophagy initiation (Yue et al, 2003). More importantly, loss of C9orf72 led to the accumulation of lysosomes and to abnormal immune responses in microglia (O'Rourke et al, 2016), raising the intriguing possibility that partial loss of function of C9ORF72 contributes to neurodegeneration in ALS/FTD patients through a non‐cell‐autonomous mechanism. Future studies are warranted to investigate the cell type‐specific roles of the newly identified C9ORF72/SMCR8/WDR41 complex in the endosomal–lysosomal pathway, which is so closely linked to autophagy and also affected by mutations in several other disease genes such as CHMP2B and GRN.
See also: C Sellier et al (June 2016)
References
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet‐Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol 126: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6: 23204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt C, Houlden H (2015) Exome sequencing uncovers hidden pathways in familial and sporadic ALS. Nat Neurosci 18: 611–613 [DOI] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Miller BL, Dickson DW, Boylan KB, Graff‐Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen‐Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay‐Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM et al (2010) Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD (2014) C9ORF72, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23: 3579–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Tsuiji H (2016) There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res doi: 10.1016/j.brainres.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351: 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari ML, Corbier CJ, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet‐Berguerand N (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 35: 1276–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RJ, Lu Y, Marie B, Gao FB, Sweeney ST (2015) Rab8, POSH, and TAK1 regulate synaptic growth in a Drosophila model of frontotemporal dementia. J Cell Biol 208: 931–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]