

Abstract
An intronic expansion of GGGGCC repeats within the C9ORF72 gene is the most common genetic cause of amyotrophic lateral sclerosis and frontotemporal dementia (ALS‐FTD). Ataxin‐2 with intermediate length of polyglutamine expansions (Ataxin‐2 Q30x) is a genetic modifier of the disease. Here, we found that C9ORF72 forms a complex with the WDR41 and SMCR8 proteins to act as a GDP/GTP exchange factor for RAB8a and RAB39b and to thereby control autophagic flux. Depletion of C9orf72 in neurons partly impairs autophagy and leads to accumulation of aggregates of TDP‐43 and P62 proteins, which are histopathological hallmarks of ALS‐FTD. SMCR8 is phosphorylated by TBK1 and depletion of TBK1 can be rescued by phosphomimetic mutants of SMCR8 or by constitutively active RAB39b, suggesting that TBK1, SMCR8, C9ORF72, and RAB39b belong to a common pathway regulating autophagy. While depletion of C9ORF72 only has a partial deleterious effect on neuron survival, it synergizes with Ataxin‐2 Q30x toxicity to induce motor neuron dysfunction and neuronal cell death. These results indicate that partial loss of function of C9ORF72 is not deleterious by itself but synergizes with Ataxin‐2 toxicity, suggesting a double‐hit pathological mechanism in ALS‐FTD.
Keywords: C9ORF72, autophagy, neurodegeneration, ALS‐FTD
Subject Categories: Neuroscience
Introduction
Amyotrophic lateral sclerosis (ALS), a motor neuron degenerative disease, and frontotemporal dementia (FTD), a presenile dementia affecting frontal and temporal brain regions, share clinical, genetic, and pathological overlap and are now considered in some cases as manifestations of a similar disease continuum (Lomen‐Hoerth et al, 2002; Ringholz et al, 2005; Neumann et al, 2006). A notion that is emphasized by the identification of expanded GGGGCC repeats within the first intron of the C9ORF72 gene as the most common inherited cause for both ALS and FTD (DeJesus‐Hernandez et al, 2011; Renton et al, 2011; Gijselinck et al, 2012; Majounie et al, 2012).
Three non‐exclusive mechanisms by which expanded GGGGCC repeats cause neuron degeneration have been proposed. First, sense and antisense transcripts containing expanded GGGGCC repeats accumulate in nuclear RNA aggregates, which recruit specific RNA‐binding proteins, thereby potentially inhibiting their functions (Almeida et al, 2013; Donnelly et al, 2013; Lagier‐Tourenne et al, 2013; Mizielinska et al, 2013). Various proteins have been reported to bind to GGGGCC RNA repeats, but their roles in pathogenesis are, yet, to be determined (Lee et al, 2013; Mori et al, 2013a; Cooper‐Knock et al, 2014; Haeusler et al, 2014). The second potential mechanism for neurotoxicity of CCGGGG expansions is a form of non‐canonical protein translation termed repeat‐associated non‐ATG (RAN) translation (Zu et al, 2013). Indeed, expanded GGGGCC repeats are RAN‐translated in all six sense and antisense frames, resulting in expression of five different dipeptide repeats containing proteins (DPRs or DRPs, also named C9RANT), which form inclusions throughout the brain of patients with C9‐ALS/FTD (Ash et al, 2013; Gendron et al, 2013; Mori et al, 2013b; Zu et al, 2013), as well as in mice expressing expanded CCGGGG repeats (Chew et al, 2015; O'Rourke et al, 2015; Peters et al, 2015). These DPRs were recently shown to be toxic in neuronal cell cultures and in Drosophila models through alteration of the nucleocytoplasmic transport (Kwon et al, 2014; May et al, 2014; Mizielinska et al, 2014; Wen et al, 2014; Zhang et al, 2014, 2015; Freibaum et al, 2015; Jovičić et al, 2015; Tao et al, 2015). Third, decreased expression of C9ORF72 mRNA expression levels in C9‐ALS/FTD patients (DeJesus‐Hernandez et al, 2011; Gijselinck et al, 2012; Almeida et al, 2013; Waite et al, 2014; van Blitterswijk et al, 2015), as well as motor deficit caused by knockdown of C9orf72 in zebrafish (Ciura et al, 2013), suggests that haploinsufficiency of C9ORF72 may participate in neuronal degeneration. However, the absence of neuronal phenotypes in mouse depleted of C9orf72 expression in brain or in neurons (Lagier‐Tourenne et al, 2013; Koppers et al, 2015), as well as the absence of ALS/FTD patients with null alleles or missense mutations in C9ORF72, strongly argues against a sole loss of function of C9ORF72 as the cause of ALS‐FTD.
Despite these advances, little is known about the normal molecular and cellular functions of C9ORF72. Bioinformatics analysis predicts that C9ORF72 contains DENN (Differentially Expressed in Normal and Neoplastic cells) domains characteristic of Rab GDP/GTP exchange factors (GEFs) (Levine et al, 2013; Zhang et al, 2012). Rab GTPases are monomeric guanine nucleotide‐binding proteins that switch between two conformational states, an inactive form bound to GDP and an active form bound to GTP. GEF proteins catalyze the conversion of Rab proteins from GDP‐bound to GTP‐bound form, thereby activating Rab functions. Rab GTPases regulate many steps of membrane traffic, including vesicle formation, vesicle movement, and membrane fusion. Consistent with a function of C9ORF72 as a GEF protein, C9ORF72 was reported to interact with Rab1, Rab5, Rab7, and Rab11 and its depletions leads to endocytosis and autophagy dysfunctions (Farg et al, 2014).
Macroautophagy, named autophagy thereafter, is a catabolic process that engulfs cytoplasmic constituents within a double‐membrane vesicle named autophagosome, which is directed to the lysosome for degradation and recycling. Hence, autophagy plays an essential role in homeostasis by providing energy and recycling cellular components, but also by facilitating lysosomal degradation of intracellular pathogens, defective organelles, and aggregates of misfolded proteins. In that aspect, autophagy is crucial for normal brain function (Hara et al, 2006; Komatsu et al, 2006), and autophagy dysfunction has been reported in various neurodegenerative diseases, including Alzheimer's, Parkinson's, and Huntington's diseases, as well as ALS and FTD (review in Wong & Cuervo, 2010; Nixon, 2013).
A better comprehension of C9ORF72 functions is essential to understand its possible implication in ALS‐FTD. Here, we found that C9ORF72 forms a complex with WDR41 and SMCR8, a DENN protein previously identified within the interactome of autophagy (Behrends et al, 2010). We found that the complex formed by C9ORF72, SMCR8, and WDR41 interacts and acts as a GDP/GTP exchange factor for RAB8a and RAB39b, which are Rab GTPases involved in vesicle trafficking and autophagy (Pilli et al, 2012; Seto et al, 2013; Sato et al, 2014).
Next, we found that decreased expression of C9ORF72 partly inhibits autophagy and leads to accumulation of cytoplasmic aggregates of P62/SQSTM1 and of TDP‐43, thus recapitulating two histopathological hallmarks of ALS‐FTD patients (Neumann et al, 2006; Al‐Sarraj et al, 2011). Also, SMCR8, but not C9ORF72 or WDR41, is phosphorylated by ULK1 or TBK1, which are kinases regulating autophagy (Chan et al, 2007; Hara et al, 2008; Thurston et al, 2009; Wild et al, 2011; Pilli et al, 2012; Matsumoto et al, 2015; Heo et al, 2015; Lazarou et al, 2015). Importantly, either a mutant of SMCR8 mimicking a constitutive TBK1‐dependent phosphorylation or a constitutively active mutant of RAB39b that does not require GEF activity can correct alteration of autophagy caused by depletion of TBK1 or C9ORF72. These results suggest that TBK1, C9ORF72 complex, and RAB39b belong to a common pathway regulating autophagy in neurons.
Finally, we found that decreased expression of C9ORF72 potentiates the aggregation and toxicity of Ataxin‐2 with intermediate length of polyglutamine expansions (Ataxin‐2 Q30x) but not of Ataxin‐2 with normal polyQ length (Ataxin‐2 Q22x). This is relevant as intermediate size of polyglutamine expansions in Ataxin‐2 is a genetic modifier of ALS‐FTD (Elden et al, 2010; Daoud et al, 2011; Ross et al, 2011; Van Damme et al, 2011; Lattante et al, 2014).
In conclusion, these results provide a molecular and cellular function for C9ORF72 as a GEF protein regulating autophagy, but also support a double‐hit mechanism in ALS‐FTD, where the sole haploinsufficiency of C9ORF72 might not be sufficient to cause neuronal cell death but may synergize the neurodegeneration caused by accumulation of toxic proteins, including Ataxin‐2 with intermediate polyQ length.
Results
C9ORF72 forms a complex with the WDR41 and SMCR8 proteins
To better characterize the function of C9ORF72, we performed a proteomic analysis to identify potential interactants of C9ORF72. Of technical interest, transfection of Flag‐tagged human C9ORF72 cDNA resulted in low expression of C9ORF72 protein. Bioinformatics analysis indicated that mRNA sequences of human or mouse C9ORF72 present an excess of rare codons and negative RNA cis element that would impair expression of C9ORF72. Thus, we cloned an optimized sequence of human C9ORF72 cDNA expressing the exact same amino acid sequence, but in which codon usage was optimized and negative RNA cis elements were removed. Neuronal N2A mouse cells were transfected with HA‐Flag‐tagged optimized C9ORF72 and tandem‐tag purification followed by nano‐LC‐MS/MS analysis identified various proteins, including Rab8a, Rab39b, Smcr8, Wdr41, P62, Hsc70, Hsp90, and Bag3 (Fig 1A and Table EV1). Bioinformatics analysis of this putative C9ORF72 interactome using the DAVID database (NIAID, NIH) predicted a potential association with ALS disease (P‐value of 4.3 × 10−2), and analysis of KEGG, GO, and Reactome biological pathways using Gene Set Enrichment Analysis (GSEA, Broad institute) revealed significant enrichment for adaptive immune system (FDR q‐value, 2.6 × 10−6), activation of NF‐κB (FDR q‐value, 9.8 × 10−5), formation of phagosome and autophagosome (FDR q‐values of 8.8 × 10−5 and 1.4 × 10−4, respectively), vesicle‐mediated transport (FDR q‐value, 6.7 × 10−3), and proteasome (FDR q‐value, 6.3 × 10−3).
Figure 1. C9ORF72 in complex with SMCR8 and WDR41 is a GEF for RAB8 and RAB39.
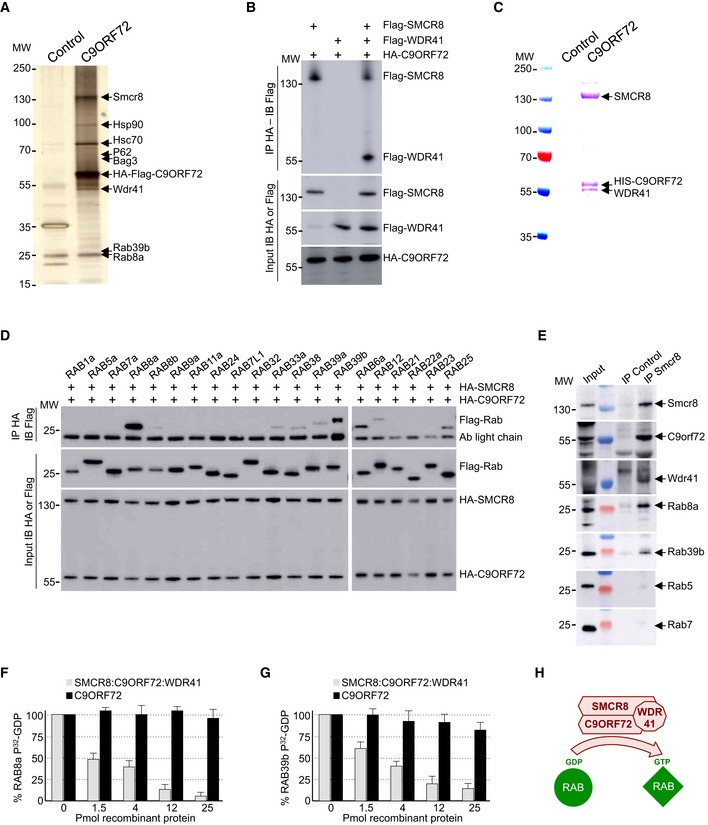
- Silver staining of proteins extracted from N2A mouse neuronal cells expressing Flag‐HA‐tagged C9ORF72 and captured through consecutive anti‐Flag and anti‐HA affinity purification steps.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged C9ORF72 and/or Flag‐tagged SMCR8 and/or Flag‐tagged WDR41.
- Coomassie blue staining of Nickel‐NTA affinity purification of HIS‐C9ORF72, SMCR8, and WDR41 co‐expressed in baculovirus‐infected insect cells.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged C9ORF72 and HA‐tagged SMCR8 with various Flag‐tagged Rab GTPases.
- Immunoblot against endogenous C9orf72, Wdr41, Rab8a, Rab39b, Rab5, and Rab7 of control (IgG alone) or endogenous Smcr8 immunoprecipitated from adult mouse brain.
- α‐32P‐radiolabelled GDP release from GST‐tagged purified RAB8a as a function of increased concentration of either recombinant purified C9ORF72 alone or in complex with SMCR8 and WDR41.
- Identical GDP release assay as in (F) but using recombinant purified GST‐tagged RAB39b instead of RAB8a.
- Schematic representation of the C9ORF72 complex acting as a Rab‐guanine nucleotide exchange factor.
Western blotting on tandem‐tag‐purified proteins confirmed association of endogenous Rab8a, Rab39b, Smcr8, Wdr41, P62, Hsc70, and Bag3 with transfected HA‐Flag‐tagged C9ORF72 (Fig EV1A). RAB8 and RAB39 are Rab GTPases involved in vesicle trafficking and autophagy (Pilli et al, 2012; Seto et al, 2013; Sato et al, 2014). WDR41 is a 52‐kDa protein of unknown function that contains a protein–protein interaction domain consisting of six WD40 repeats. Smith–Magenis syndrome chromosome region candidate 8 (SMCR8) is a 105‐kDa protein that, similar to C9ORF72, contains a divergent Differentially Expressed in Normal and Neoplasia (DENN) domain (Zhang et al, 2012; Levine et al, 2013). Interestingly, SMCR8 has been found to interact with the ULK1 kinase complex, which initiates autophagy (Behrends et al, 2010). P62, encoded by the SQSTM1 gene, binds to poly‐ubiquitinated proteins and targets these proteins for autophagy degradation. Finally, HSC70, encoded by the HSPA8 gene, is a constitutively expressed heat‐shock protein that among various functions interacts with BAG3 (BCL2‐associated athanogene 3) and the ubiquitin ligase STUB1 (also known as CHIP) to initiate aggrephagy, also named chaperone‐assisted selective autophagy (CASA), which is the selective autophagy of chaperone‐bound misfolded or aggregated proteins (Gamerdinger et al, 2009; Arndt et al, 2010). In contrast, we found no interaction of C9ORF72 with other putative candidates identified by proteomic analysis, such as Senataxin, TDP‐43, IQGAP, and the proteasome proteins PSMD4, PSMD8, PSMD10, while some other proteins were not tested (DCTN, NSF, SNX, STX17, VPS16, SEC22, etc.).
Figure EV1. Validation of the protein interacting with C9ORF72.
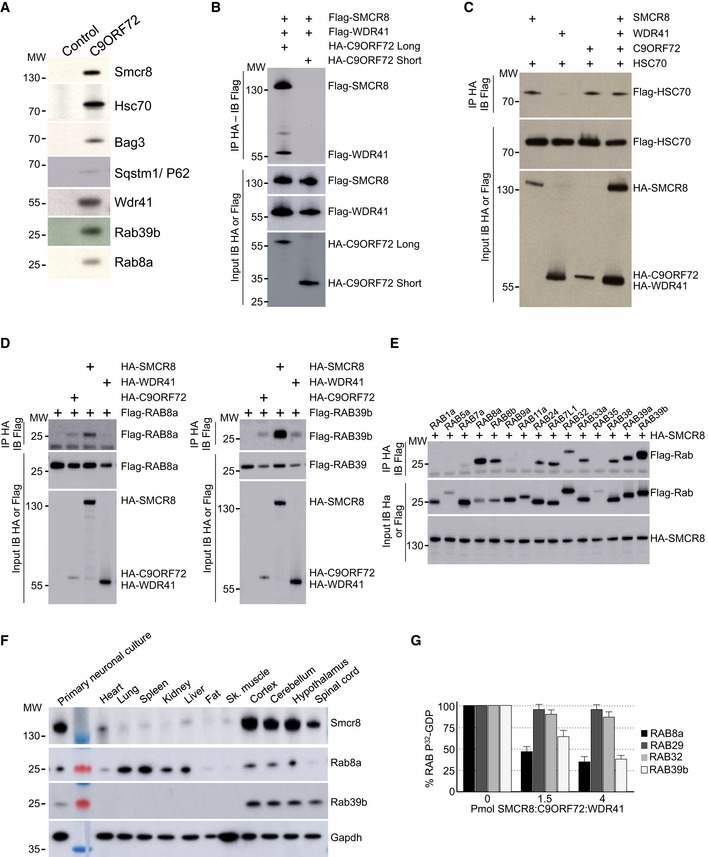
- Immunoblot analysis of endogenous proteins found associated by proteomic analysis with control or Flag‐HA‐tagged C9ORF72 expressed in N2A cells.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged long or short splicing variant of C9ORF72 with Flag‐tagged SMCR8 and Flag‐tagged WDR41.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged C9ORF72 and/or HA‐tagged SMCR8 and/or HA‐tagged WDR41 with Flag‐tagged HSC70.
- Left panel, immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing either HA‐tagged C9ORF72, HA‐tagged SMCR8, or HA‐tagged WDR41 with Flag‐tagged RAB8A. Right panel, immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing either HA‐tagged C9ORF72, HA‐tagged SMCR8, or HA‐tagged WDR41 with Flag‐tagged RAB39b.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged SMCR8 with various Flag‐tagged Rab GTPases.
- Immunoblot analysis of endogenous expression of Smcr8, Rab8a, Rab39b, and Gapdh in mouse adult tissues and primary culture of E18 cortical mouse neurons.
- 32P‐radiolabelled GDP release from GST‐tagged purified RAB8a, RAB39b, RAB29 (also known as RAB7L1), and RAB32 as a function of increased concentration of recombinant purified HIS‐C9ORF72:SMCR8:WDR41 complex purified from baculovirus‐infected insect cells. Error bars indicate SEM, N = 3.
Next, we confirmed by cell transfection and co‐immunoprecipitation experiments that HA‐tagged C9ORF72 interacts with Flag‐tagged SMCR8 and that the presence of both SMCR8 and C9ORF72 is required to recruit Flag‐tagged WDR41 (Fig 1B). The association of C9ORF72 with SMCR8 and WDR41 is direct since HIS‐tagged C9ORF72 was able to pull down both recombinant SMCR8 and WDR41 expressed in baculovirus‐infected insect cells (Fig 1C). Of technical interest, we noted that the interaction of C9ORF72 with SMCR8 stabilizes and increases expression of both C9ORF72 and SMCR8. C9ORF72 also encodes a putative shorter splicing form of ~30 kDa, but co‐immunoprecipitation assays demonstrated that this putative short C9ORF72 isoform does not interact with SMCR8 or WDR41 (Fig EV1B). Co‐immunoprecipitation experiments also indicated that C9ORF72 and SMCR8 bind to HSC70, but not to BAG3 (Fig EV1C), suggesting that BAG3 recruitment is not direct but mediated by HSC70. Whether C9ORF72 is a client or a regulator of the HSC70 and BAG3 pathway remains to be determined.
C9ORF72 in complex with SMCR8 and WDR41 interacts with RAB8a and RAB39b
Proteomic analysis revealed association of C9ORF72 with Rab8a and Rab39b (Fig EV1A). However, C9ORF72 was reported to interact with Rab1, Rab5, Rab7, and Rab11 (Farg et al, 2014). Since our proteomic analysis was performed using Neuro‐2A cell lysate that may incompletely represent the proteomic complexity of a tissue, we tested the association of C9ORF72 with various other Rab GTPases. Among the Rab proteins tested, we confirmed that the C9ORF72 complex interacts with RAB8a and weakly with RAB39b, as well as, but in a lesser extent, with their paralogs RAB8b and RAB39a (Fig 1D). Of interest, RAB8 is involved in a Drosophila model of FTD (West et al, 2015) and interacts with OPTN and TBK1 that are mutated in ALS (Maruyama et al, 2010; Cirulli et al, 2015; Freischmidt et al, 2015). Also, mutations in the X‐linked RAB39b gene lead to intellectual disability associated with autism, epilepsy, and early‐onset parkinsonism (Giannandrea et al, 2010; Wilson et al, 2014). Among other Rab tested, the C9ORF72 complex presents also some weak interaction with RAB6a, RAB12, RAB25, RAB33a, and RAB38 (Fig 1D). In contrast, C9ORF72 alone or in complex with SMCR8 present no or very little interaction with RAB1, RAB5, RAB7, and RAB11 (Fig 1D). Similarly, we found no interaction of RAGA/D or RALB GTPases with C9ORF72 alone or in complex with SMCR8. Of technical interest, co‐transfection of WDR41 abolished expression of various Rab cDNAs; thus, co‐immunoprecipitations were performed with the C9ORF72 and SMCR8 proteins (Fig 1D). Interactions between RAB8a or RAB39b and the C9ORF72, SMCR8, and WDR41 complex were not inhibited by mutations locking Rab proteins in a constitutive GDP‐bound inactive or a constitutively GTP‐bound active form. Further co‐immunoprecipitation assays indicated that the interaction between C9ORF72 complex and RAB8a or RAB39b is mainly dependent on the presence of SMCR8 since C9ORF72 or WDR41 alone immunoprecipitated little RAB8a or no RAB39b (Fig EV1D). In that aspect, SMCR8 expressed alone immunoprecipitated RAB8a and RAB39b but also some other Rab GTPases, including RAB24, RAB32, and RAB7L1, which is also known as RAB29 (Fig EV1E). These interactions were lost when SMCR8 was in complex with C9ORF72, suggesting that the specificity of SMCR8 toward Rab GTPase proteins may change according to the SMCR8 partners.
Since these experiments were performed in transfected cells, we next tested whether the endogenous C9ORF72 complex may immunoprecipitate endogenous RAB8a and RAB39b. Taking advantage of the recombinant HIS‐tagged C9ORF72, SMCR8, and WDR41 complex purified from baculovirus‐infected insect cells, we immunized mice, but failed to obtain antibodies specific to C9ORF72 despite many attempts. In contrast, we successfully developed a monoclonal antibody (1D2) against SMCR8. Immunoblotting indicated that Smcr8, Rab8a, and Rab39b are all expressed in mouse brain as well as in cultures of primary E18 cortical mouse neurons (Fig EV1F). We were not able to test expression of endogenous C9orf72 and Wdr41 due to the poor quality of the commercial antibodies tested, but expression of C9orf72 in mouse brain had been reported previously (Suzuki et al, 2013). Importantly, immunoprecipitation of endogenous Smcr8 from total mouse brain lysate successfully pulled down Smcr8, but also endogenous C9orf72 and Wdr41, as well as Rab8a and Rab39b (Fig 1E). In contrast, we did not detect endogenous Rab5 or Rab7 in Smcr8 immunoprecipitation (Fig 1E), thus confirming our transfection experiments.
C9ORF72 in complex with SMCR8 and WDR41 is a GEF for Rab GTPase
Both C9ORF72 and SMCR8 contain DENN domains characteristic of Rab‐guanine nucleotide exchange factors (Zhang et al, 2012; Levine et al, 2013). Thus, we tested whether the complex composed of C9ORF72, SMCR8, and WDR41 presents any GEF activity. Purified recombinant GST‐tagged RAB8a was preloaded with 32P‐labeled GDP, and nucleotide release was monitored in the presence of increasing amount of C9ORF72 recombinant complex purified from baculovirus‐infected insect cells. The complex formed by C9ORF72, SMCR8, and WDR41 stimulated GDP release since more than 90% of 40 pmol of RAB8a released its associated GDP in the presence of 25 pmol of C9ORF72, SMCR8, and WDR41 complex (Fig 1F). In contrast, addition of recombinant purified C9ORF72 alone had no or very little effect, suggesting that C9ORF72 is active only when in complex with SMCR8 (Fig 1F). As the C9ORF72 complex also interacts with RAB39b, we tested its GEF activity and found that, similar to RAB8a, the complex composed of C9ORF72, SMCR8, and WDR41 activated GDP release from recombinant purified GST‐RAB39b (Fig 1G). In contrast, addition of recombinant purified C9ORF72 alone had no or little effect (Fig 1G). As control, the complex formed by C9ORF72, SMCR8, and WDR41, which does not interact with RAB32 or RAB29, presented no or very little GEF activity toward these Rab GTPases (Fig EV1G). Overall, these results demonstrate that C9ORF72 forms a specific complex with SMCR8 and WDR41, which acts as a GEF effector for at least RAB8a and RAB39b (Fig 1H).
RAB8a and RAB39b interact with the autophagy receptors P62
Proteomic analysis identified that C9ORF72 potentially interacts with P62, which is encoded by the SQSTM1 gene and targets poly‐ubiquitinated proteins for autophagy degradation (Table EV1). Co‐immunoprecipitation experiments confirmed that the complex formed by C9ORF72, SMCR8, and WDR41 interacts weakly with P62, but also with OPTN that is alike P62, an autophagy receptor. Further co‐immunoprecipitation assays pinpointed that among the proteins of the C9ORF72 complex, P62 mainly interacts with WDR41 and SMCR8 (Fig EV2A), while OPTN mainly interacts with WDR41 (Fig EV2B). Of interest, mutations in SQSTM1 or OPTN cause ALS (Maruyama et al, 2010; Fecto et al, 2011), and OPTN is a potential modifier gene for FTD (Pottier et al, 2015). We noted that the quantities of P62 or OPTN immunoprecipitated by the C9ORF72 complex were low, suggesting a potential indirect association. Since OPTN interacts with RAB8 (Hattula & Peränen, 2000; Pilli et al, 2012), we tested whether interaction of the C9ORF72 complex with P62 would be mediated by intermediate proteins, namely RAB8 or RAB39. Indeed, both HA‐tagged RAB8a and RAB39b readily immunoprecipitated Flag‐tagged P62 (Fig EV2C).
Figure EV2. Reduced expression of C9ORF72 partly impairs autophagy.
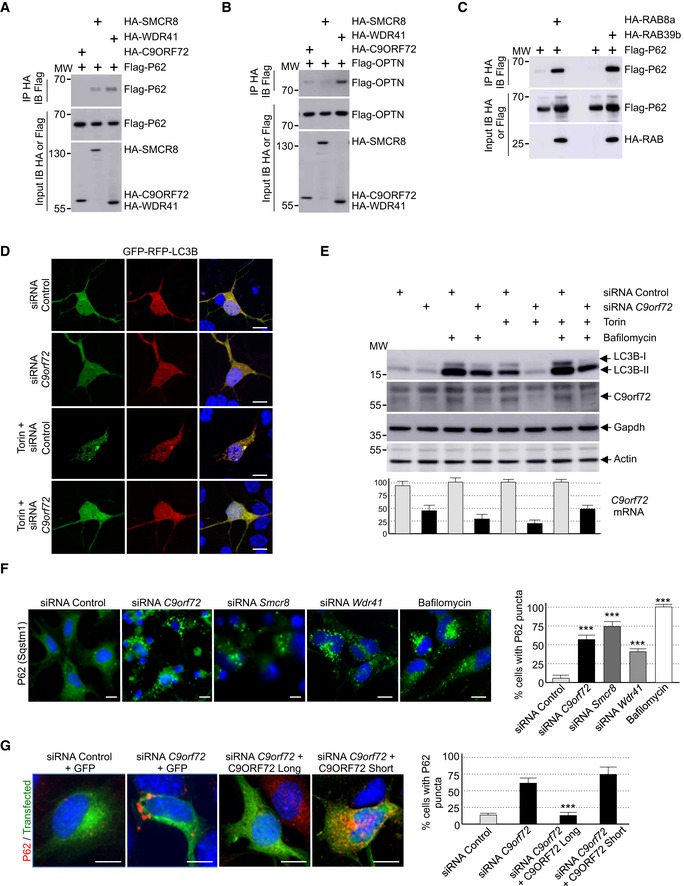
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing either HA‐tagged C9ORF72, HA‐tagged SMCR8, or HA‐tagged WDR41 with Flag‐tagged P62.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing either HA‐tagged C9ORF72, HA‐tagged SMCR8, or HA‐tagged WDR41 with Flag‐tagged OPTN.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing either HA‐tagged RAB8a or HA‐tagged RAB39b with Flag‐tagged P62.
- Representative images of mouse GT1‐7 neuronal cells co‐transfected with GFP‐RFP‐LC3B and either control siRNA or siRNA targeting endogenous C9orf72 mRNA and treated or not with Torin.
- Upper panel, immunoblot analysis of endogenous LC3B (Map1lc3b), C9orf72 and control Gapdh and actin of GT1‐7 neuronal cells transfected with either control siRNA or siRNA targeting C9orf72 mRNA and treated or not with Torin and/or bafilomycin A. Lower panel, real‐time RT–qPCR quantification of endogenous C9orf72 mRNA expression relative to Rplp0 mRNA.
- Left panel, representative images of immunofluorescence labeling of endogenous P62 (Sqstm1) on GT1‐7 neuronal cells transfected with either control siRNA or siRNA targeting either C9orf72, Smcr8, or Wdr41 mRNA. Right panel, quantification of P62 aggregates.
- Left panel, representative merged images of immunofluorescence labeling of transfected constructs (green) and endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with control siRNA or siRNA targeting C9orf72 and plasmids expressing control GFP or HA‐tagged long or short isoform of C9ORF72. Right panel, quantification of P62 aggregates.
Data information: Scale bars, 10 μm. Nuclei were counterstained with DAPI. Error bars indicate SEM. Student's t‐test, ***P < 0.001, n = 3.
Decreased expression of C9ORF72 alters autophagy
C9ORF72 is interacting with various proteins connected to autophagy (Table EV1; Behrends et al, 2010; Farg et al, 2014). Thus, we tested whether depletion of C9ORF72 expression modifies autophagy in neuronal cells. First, we evaluated the formation of vesicles containing LC3B, a protein encoded by the MAP1LC3B gene, which is specifically lipidated and localized to autophagic vesicles. Expression of double‐tagged GFP‐RFP‐LC3B in primary cultures of embryonic mouse cortical neurons shows diffuse cytoplasmic localization and rare punctuate structures. In contrast, treatment with torin, which inhibits the autophagic‐inhibitory mTOR pathway, induces formation of LC3B puncta (Fig 2A). Importantly, shRNA‐mediated depletion of C9orf72 abolishes this activation of autophagy (Fig 2A). GFP‐RFP‐LC3B expression in primary neuronal cultures revealed no differences in the GFP/RFP ratio. This ratio reflects the rate of formation of autophagosome relative to their fusion to lysosome, suggesting that C9ORF72 acts on the formation of autophagosome rather than on their degradation by the lysosome. Similar inhibitory effect of C9orf72 siRNA on autophagy activation was observed with primary neuronal cultures treated with rapamycin instead of torin, or when GT1‐7 cells, a transformed murine neuronal cell line expressing similar level of C9orf72 compared to primary neuronal cell cultures, were tested (Fig EV2D). Next, we confirmed these results by investigating the ratio of lipidated LC3B. Western blotting analysis revealed that the level of phosphatidylethanolamine‐linked LC3B‐II was slightly decreased in primary cultures of embryonic mouse cortical neurons transduced with lentivirus expressing shRNAs against C9orf72 in basal condition (Fig 2B). This inhibitory effect was much more evident in torin‐treated neurons since activation of the lipidation of LC3B was abolished by shRNA‐mediated depletion of C9orf72 (Fig 2B). Identical results were observed in neurons treated with rapamycin instead of torin, or in GT1‐7 neuronal cells (Fig EV2E). Depletion of at least 50% of C9orf72 expression was confirmed by Western blotting and, due to the poor quality of the commercial antibodies used, further confirmed by RT–qPCR (Figs 2B and EV2E). Also, we found that decreased expression of C9orf72 reduced but not totally abolished accumulation of LC3B‐II in neuronal cells treated with bafilomycin A1, an inhibitor of autophagosome degradation (Fig EV2E). Overall, these data suggest that depletion of C9ORF72 has a partial deleterious effect on autophagy.
Figure 2. Reduced expression of C9ORF72 partly impairs autophagy.
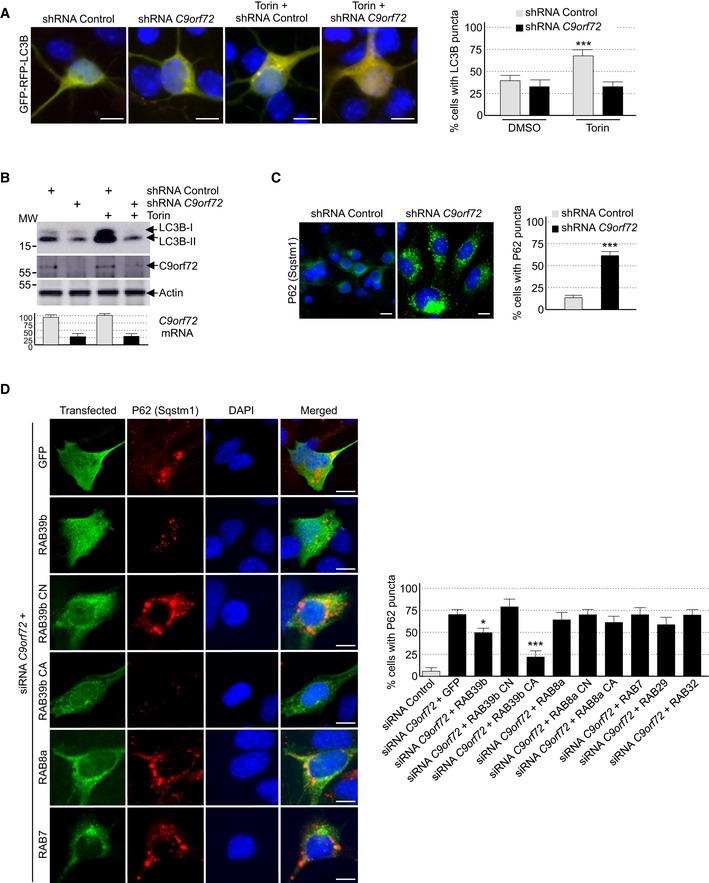
- Left panel, representative images of organotypic cultures of E18 mouse cortical neurons transfected with GFP‐RFP‐LC3B and transduced with lentivirus expressing either control shRNA or shRNA targeting C9orf72 mRNA and treated or not with Torin. Right panel, quantification of LC3B puncta.
- Upper panel, immunoblot analysis of endogenous LC3B (Map1lc3b), C9orf72, and control actin of E18 mouse cortical neurons transduced with lentivirus expressing either control shRNA or shRNA targeting C9orf72 mRNA and treated or not with Torin. Lower panel, real‐time RT–qPCR quantification of endogenous C9orf72 mRNA expression relative to Rplp0 mRNA.
- Left panel, representative images of immunofluorescence labeling of endogenous P62 (Sqstm1) on organotypic cultures of E18 mouse cortical neurons transduced with lentiviral particles expressing either control shRNA or shRNA targeting C9orf72. Right panel, quantification of P62 aggregates.
- Left panel, representative images of immunofluorescence labeling of transfected constructs (green) and endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with siRNA targeting C9orf72 and plasmids expressing control GFP, and HA‐tagged wild‐type or mutant RAB39b (CA, Q68L or CN, S22N), RAB8a, or RAB7. Right panel, quantification of P62 aggregates.
Autophagy is a crucial mechanism to clear misfolded or aggregated proteins. Thus, we tested whether depletion of C9ORF72 had any effect on the degradation of protein aggregate. The P62 protein, which is encoded by the SQSTM1 gene, bridges aggregates of poly‐ubiquitinated proteins to LC3B, thereby targeting these proteins toward autophagy. Furthermore, P62‐positive aggregates are a histological hallmark of ALS‐FTD patients with expansion of GGGGCC repeats in C9ORF72 (Al‐Sarraj et al, 2011). Immunofluorescence labeling of P62 indicated an accumulation of unresolved P62 aggregates upon depletion of C9orf72 in primary cultures of embryonic mouse cortical neurons (Fig 2C). Identical results were obtained for GT1‐7 neuronal cells or when partners of C9orf72, namely Smcr8 and Wdr41, were siRNA‐depleted (Fig EV2F). As control, expression of optimized HA‐tagged C9ORF72, which has a nucleotide sequence resistant to the siRNAs targeting endogenous mouse C9orf72 mRNA, fully rescued autophagy dysfunction caused by siRNA‐mediated depletion of C9orf72 (Fig EV2G). Of interest, while the long isoform of C9ORF72 rescued autophagy dysfunction, the short form of C9ORF72 was inactive in that respect (Fig EV2G). These results suggest that the short isoform of C9ORF72 is either a null variant or possesses a cellular function unrelated to autophagy.
RAB39b corrects autophagy dysfunction caused by depletion of C9ORF72
Depletion of C9ORF72 alters autophagy, and C9ORF72 in complex with SMCR8 interacts and promotes GDP/GTP exchange of RAB8a and RAB39b, which are two Rab GTPases involved in autophagy (Pilli et al, 2012; Seto et al, 2013). This questions the relevance of the interaction of C9ORF72 with RAB8a or RAB39b, notably to control autophagy. Importantly, expression of a constitutively active (Q68L, mutant CA) form of RAB39b, which is locked in its GTP conformation and consequently does not require any GEF activity, fully corrected autophagy alteration caused by a decreased expression of C9orf72 (Fig 2D). Expression of wild‐type HA‐tagged RAB39b had only a partial effect on autophagy rescue, while a GDP‐locked constitutively negative (S22N, mutant CN) RAB39b did not rescue autophagy alterations caused by siRNA‐mediated depletion of C9orf72. As control of RAB39b specificity, expression of RAB7, RAB29, RAB32, or wild‐type, constitutively active or inactive RAB8A did not rescue autophagy alteration caused by C9orf72 depletion (Fig 2D). These results suggest that the GEF activity of the C9ORF72 complex toward RAB39b is important to control autophagy in neuronal cells.
TBK1 phosphorylates SMCR8
Tandem‐tag purification of C9ORF72 identified a putative weak interaction with TANK‐Binding Kinase 1 (TBK1) (Table EV1). Of interest, mutations in TBK1 lead to ALS (Cirulli et al, 2015; Freischmidt et al, 2015), and RAB8, which interacts with the C9ORF72 complex, is involved in the autophagic elimination of intracellular pathogens through the kinase TBK1 (Pilli et al, 2012). Thus, we tested whether the C9ORF72 complex would interact with the TBK1 kinase complex. Co‐immunoprecipitation experiments indicated that the complex formed by C9ORF72 and SMCR8 did not bind to TBK1 directly, but interacted with all three TBK1 adaptor proteins (Fig 3A), namely TANK, SINTBAD (TBKBP1), and NAP1 (AZI2). These adaptor proteins are essential to direct TBK1 toward specific cellular comportments and functions, notably autophagy (Goncalves et al, 2011). These interactions question whether TBK1 phosphorylates C9ORF72. In vitro kinase assay demonstrated that TBK1 phosphorylates SMCR8, but not C9ORF72 or WDR41 (Fig 3B). We repeated that experiment using purified TBK1 overexpressed from HEK293 cells and recombinant HIS‐C9ORF72, SMCR8, and WDR41 complex purified from baculovirus‐infected insect cells. Mass spectrometry analysis identified SMCR8 serine 402 and threonine 796 as TBK1 phosphorylation sites (Fig 3C). Consistent with the consensus motif identified in other substrates of TBK1 (Ma et al, 2012), both SMCR8 serine 402 and threonine 796 are followed by a leucine.
Figure 3. SMCR8 is phosphorylated by TBK1.
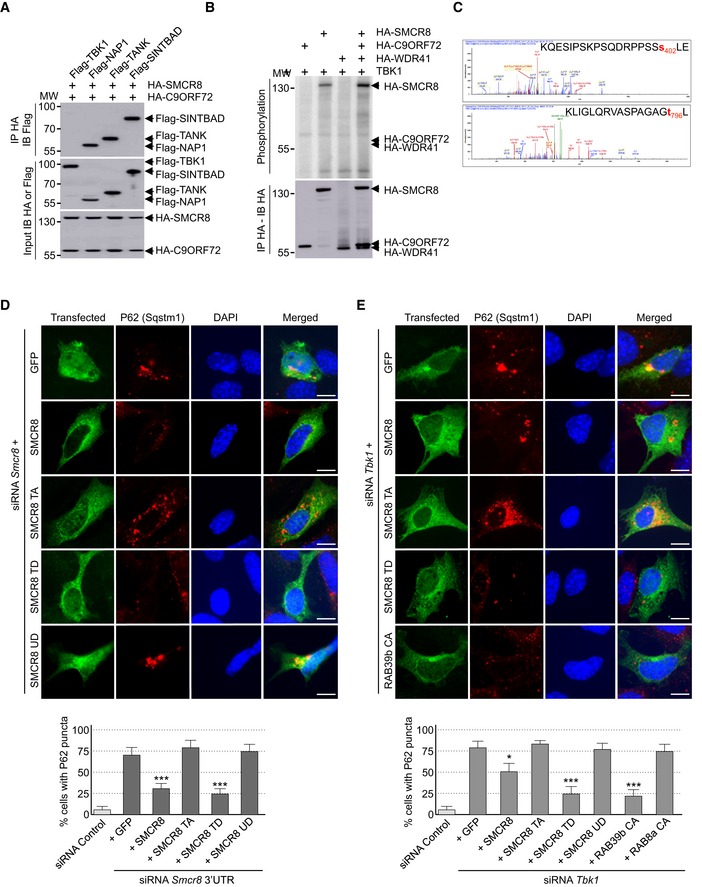
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged C9ORF72 and HA‐tagged SMCR8 with Flag‐tagged TBK1, NAP1, TANK, or SINTBAD.
- Immunoprecipitated HA‐tagged C9ORF72, HA‐tagged SMCR8, and HA‐tagged WDR41 expressed in HEK293 were subjected to in vitro TBK1 kinase assay in the presence of γ‐32P‐radiolabelled ATP. Proteins were separated by SDS–PAGE migration and phosphorylation was detected by autoradiography (upper panel), while expression was detected by Western blotting (lower panel).
- Mass spectrometry identification of SMCR8 phosphorylation sites by in vitro TBK1 kinase assay of HIS‐tagged C9ORF72:SMCR8:WDR41 complex purified from baculovirus‐infected insect cells.
- Upper panel, representative images of immunofluorescence labeling of transfected constructs (green) and endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with siRNA targeting the 3′UTR of Smcr8 mRNA and plasmids expressing control GFP and HA‐tagged wild‐type or mutant SMCR8 (TA, S402A and T796A; TD, S402D and T796D; UD, S400D, S492D, S562D, and T666D). Lower panel, quantification of P62 aggregates.
- Upper panel, representative images of immunofluorescence labeling of transfected constructs (green) and endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with siRNA targeting Tbk1 and plasmids expressing control GFP and HA‐tagged wild‐type or mutant SMCR8 or RAB39b. Lower panel, quantification of P62 aggregates.
TBK1 phosphorylation of SMCR8 is important for autophagy
We constructed TBK1 phospho‐dead (S402A, T796A) and phosphomimetic (S402D, T796D) mutants of SMCR8 and noted that these mutants were normally localized and expressed in transfected neuronal cells. Also, phospho‐dead and phosphomimetic mutants of SMCR8 immunoprecipitated C9ORF72 and WDR41 as well as wild‐type SMCR8, suggesting that mutation of SMCR8 S402 and T796 did not alter expression, stability, structure, and ability of SMCR8 to form a complex. Next, we tested whether SMCR8 phosphorylation was important for its cellular function. Depletion of Smcr8 altered autophagy as illustrated by accumulation of P62 aggregates in neuronal GT1‐7 cells transfected with a siRNA targeting the 3′UTR of Smcr8 mRNA (Fig 3D). Depletion of endogenous Smcr8 expression was confirmed by immunoblotting (Fig EV3A). As control, co‐transfection of a siRNA‐resistant HA‐tagged SMCR8 cDNA corrected autophagy dysfunction due to reduced expression of Smcr8 (Fig 3D). Importantly, expression of HA‐tagged S402D and T796D double mutant of SMCR8 (mutant TD), which mimics a constitutive TBK1 phosphorylation of SMCR8, also corrected autophagy dysfunction caused by siRNA‐mediated depletion of Smcr8 (Fig 3D). In contrast, a phospho‐dead (S402A, T796A; mutant TA) SMCR8 was unable to rescue autophagy alteration (Fig 3D), demonstrating importance of SMCR8 phosphorylation for its function. As a further control, a phosphomimetic mutant of SMCR8 unrelated to TBK1 (SMCR8 S400D, S492D, S562D, T666D; mutant UD) did not correct autophagy alteration caused by siRNA‐mediated depletion of Smcr8 (Fig 3D).
Figure EV3. SMCR8 is phosphorylated by ULK1.
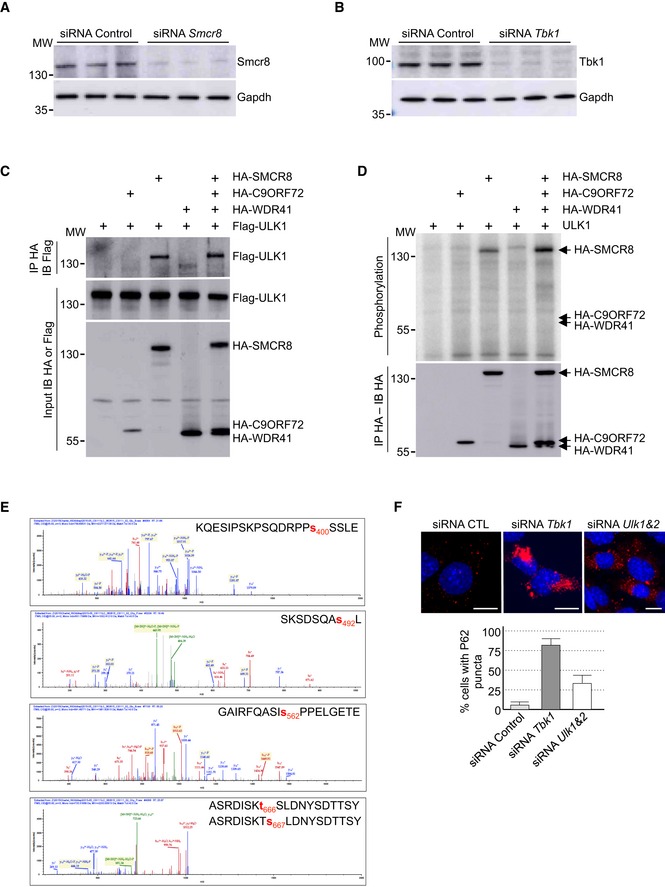
- Immunoblot analysis of endogenous Smcr8 or Gapdh in GT1‐7 cells transfected with control siRNA or a siRNA targeting the 3′UTR of Smcr8.
- Immunoblot analysis of endogenous Tbk1 or Gapdh in GT1‐7 cells transfected with control siRNA or a siRNA targeting Tbk1.
- Immunoblot analysis of HA‐immunoprecipitated proteins and lysate of HEK293 cells co‐expressing HA‐tagged C9ORF72 and/or HA‐tagged SMCR8 and/or HA‐tagged WDR41 with Flag‐tagged ULK1.
- HA‐tagged C9ORF72, HA‐tagged SMCR8, and HA‐tagged WDR41 were co‐expressed in HEK293, immunoprecipitated, and subjected to in vitro ULK1 kinase assay in the presence of γ‐32P‐radiolabelled ATP. Proteins were separated by migration on SDS–PAGE gel and phosphorylation was detected by autoradiography (upper panel), while expression was detected by Western blotting (lower panel).
- Recombinant HIS‐tagged C9ORF72:SMCR8:WDR41 complex purified from baculovirus‐infected insect cells was phosphorylated in vitro by ULK1 and SMCR8 phosphorylation sites were identified by mass spectrometry.
- Upper panel, representative images of immunofluorescence labeling of endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with control siRNA or siRNA targeting Tbk1 or Ulk1 and Ulk2. Lower panel, quantification of P62 aggregates.
Data information: Error bars indicate SEM, n = 3. Scale bars, 10 μm. Nuclei were counterstained with DAPI.
We then investigated whether SMCR8 phosphorylation was important for TBK1 cellular function. siRNA‐mediated depletion of Tbk1 promoted accumulation of P62 aggregates in neuronal GT1‐7 cells (Fig 3E). Decreased expression of Tbk1 was confirmed by immunoblotting (Fig EV3B). Of interest, expression of wild‐type HA‐tagged SMCR8 rescued only partially the dysfunction of autophagy caused by Tbk1 depletion (Fig 3E). In contrast, transfection of the S402D and T796D mutants of SMCR8 (mutant TD), which mimics a constitutive TBK1 phosphorylation, fully corrected autophagy alteration caused by Tbk1 depletion (Fig 3E). As controls, a phospho‐dead (S402A, T796A; mutant TA) as well as a phosphomimetic mutant of SMCR8 unrelated to TBK1 (SMCR8 S400D, S492D, S562D, T666D; mutant UD) was unable to rescue autophagy alteration caused by siRNA‐mediated depletion of Tbk1 (Fig 3E). Importantly, depletion of Tbk1 was also corrected by expression of constitutively active GTP‐locked HA‐tagged RAB39b, but not by constitutively active RAB8A (Fig 3E). Overall, these results suggest that phosphorylation of SMCR8 by TBK1 is important to control autophagy, but also that TBK1, C9ORF72 complex, and RAB39b belong to a common pathway regulating autophagy in neuronal cells.
ULK1 phosphorylates SMCR8
We noted that SMCR8 was initially identified as an interactant of the ULK1 kinase complex, which initiates autophagy (Behrends et al, 2010). Similarly, our tandem‐tag purification of C9ORF72 identified a potential weak interaction with a component of the ULK1 kinase complex, namely Fip200, encoded by the Rb1cc1 gene (Table EV1). Co‐immunoprecipitation experiments confirmed that the complex formed by C9ORF72, WDR41, and SMCR8 interacts weakly with ULK1 and that this interaction is mostly dependent on the presence of SMCR8 (Fig EV3C). Similar to TBK1, in vitro kinase assay demonstrated that ULK1 phosphorylates SMCR8 alone or in complex, but not C9ORF72 or WDR41 (Fig EV3D). Mass spectrometry analysis identified SMCR8 serine 400, serine 492, serine 562, and threonine 666 or serine 667 as ULK1 phosphorylation sites (Fig EV3E). However, depletion of Ulk1 or of both Ulk1 and Ulk2 using siRNA in neuronal GT1‐7 cells leads to only partial accumulation of P62 aggregates compared to siRNA‐mediated depletion of Tbk1 (Fig EV3F). Furthermore, a phosphomimetic mutant of SMCR8 (S400D, S492D, S562D, T666D; mutant UD) simulating constitutive phosphorylation of SMCR8 by the ULK1 kinase did not correct autophagy alteration caused by depletion of Smcr8 (Fig 3D). Overall, these results suggest that in GT1‐7 neuronal cells and in the time frame of our study, phosphorylation of SMCR8 by the TBK1 kinase plays a crucial role to regulate autophagy compared to ULK1.
Decreased expression of C9ORF72 promotes aggregation of TDP‐43
Brain sections of ALS‐FTD patients are characterized by the presence of neuronal cytoplasmic inclusions containing the TAR DNA‐binding protein 43 (TDP‐43) that is abnormally ubiquitinated, phosphorylated, and truncated (Arai et al, 2006; Neumann et al, 2006). Since aggregates of TDP‐43 are resolved by autophagy (Filimonenko et al, 2007; Ju et al, 2009; Urushitani et al, 2010; Wang et al, 2012; Barmada et al, 2014; Scotter et al, 2014), we tested whether decreased expression of C9orf72 had any effect on TDP‐43. Importantly, Western blotting assays demonstrated that shRNA‐mediated depletion of C9orf72 in mouse embryonic cortical neurons induced the accumulation of a ~30 kDa truncated fragment of Tdp‐43 (Fig 4A). Similarly, immunofluorescence analysis against phosphorylated serine 409/410 of Tdp‐43 confirmed accumulation of cytoplasmic aggregates of Tdp‐43 in neurons depleted of C9orf72 (Fig 4B). Note that the accumulation of Tdp‐43 aggregates was evident in primary neuronal cultures upon 7 days of transduction with C9orf72 shRNA lentivirus, while we did not detect accumulation of Tdp‐43 aggregates in GT1‐7 neuronal cells treated with C9orf72 siRNA for 24 h, suggesting that accumulation of Tdp‐43 may be cell or time dependent. Finally, mutations in TARDBP, encoding TDP‐43, lead to aggregate of TDP‐43 proteins and cause ALS‐FTD (Gitcho et al, 2008; Kabashi et al, 2008; Rutherford et al, 2008; Sreedharan et al, 2008; Van Deerlin et al, 2008; Yokoseki et al, 2008). We found that overexpression of C9ORF72 reduced the aggregation of the D169G mutant of TDP‐43 (Fig 4C). These results are consistent with previous works showing that enhancing autophagy corrects aggregation and toxicity of mutant TDP‐43 (Wang et al, 2012; Barmada et al, 2014).
Figure 4. Reduced expression of C9ORF72 promotes aggregation of TDP‐43.
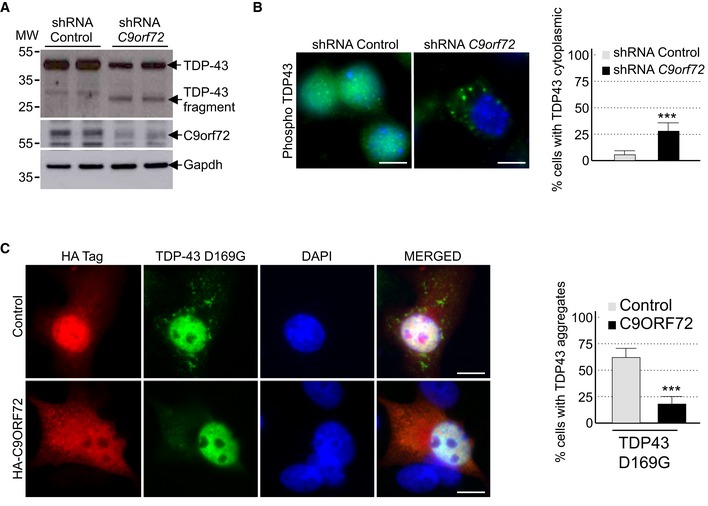
- Immunoblot analysis of endogenous Tdp‐43, C9orf72, and control Gapdh of E18 mouse cortical neurons transduced with lentivirus expressing either control shRNA or shRNA targeting C9orf72 mRNA.
- Left panel, representative images of immunofluorescence labeling of endogenous phosphorylated Ser409/410‐Tdp‐43 on primary cultures of E18 mouse cortical neurons transduced with lentiviral particles expressing either control shRNA or shRNA targeting C9orf72. Right panel, quantification of cytoplasmic Tdp‐43 aggregates.
- Left panel, representative images of immunofluorescence labeling of D169G mutant GFP‐tagged TDP‐43 on primary cultures of E18 mouse cortical neurons transfected with either HA‐tagged control or HA‐C9ORF72 plasmid. Right panel, quantification of cells with cytoplasmic aggregates of TDP‐43.
Phosphomimetic mutants of SMCR8 and constitutively active GTP‐locked RAB39b correct autophagy alteration due to depletion of Smcr8, C9orf72, or Tbk1, suggesting that these proteins belong to the same pathway. Since TDP‐43 regulates indirectly autophagy (Bose et al, 2011; Xia et al, 2016), we tested whether C9ORF72 or RAB39b can rescue autophagy misregulation caused by reduced expression of Tardbp. Depletion of Tdp‐43 partly altered autophagy as illustrated by accumulation of P62 aggregates in neuronal GT1‐7 cells transfected with a siRNA targeting Tardbp (Fig EV4). However, we found no correction of autophagy upon transfection of C9ORF72, SMCR8, or wild‐type or constitutively active RAB8A or RAB39b (Fig EV4). These negative results highlight the specificity of RAB39b toward TBK1 and C9ORF72, but also indicate that while TDP‐43 regulates autophagy, this regulation is independent or downstream of the TBK1, C9ORF72, and RAB39b pathway. This is consistent with the recent report that loss of TDP‐43 downregulates Dynactin 1 mRNA, which impairs autophagy at the late step of fusion of autophagosomes to lysosomes (Xia et al, 2016).
Figure EV4. Reduced expression of C9ORF72 promotes aggregation of TDP‐43.
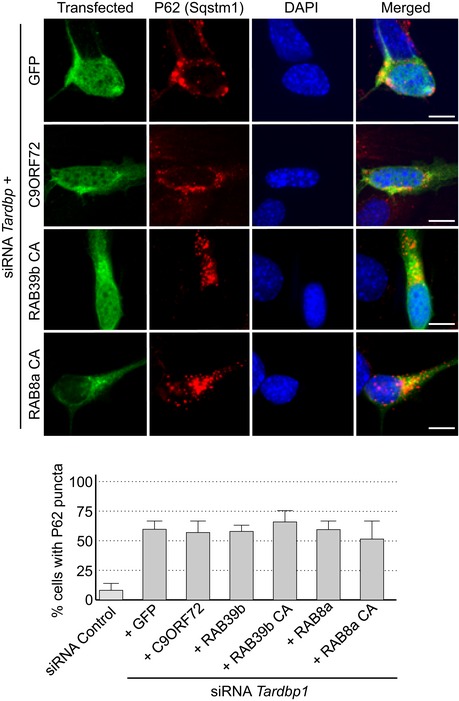
Upper panel, representative images of immunofluorescence labeling of transfected constructs (green) and endogenous P62 (Sqstm1, red) on GT1‐7 neuronal cells transfected with siRNA targeting Tardbp and plasmids expressing control GFP and HA‐tagged C9ORF72 or wild‐type or mutant RAB8a or RAB39b. Lower panel, quantification of P62 aggregates. Error bars indicate SEM, n = 3.
Decreased expression of C9ORF72 synergizes Ataxin‐2 Q30x toxicity
Since decreased expression of C9ORF72 inhibits autophagy and promotes accumulation of aggregates of TDP‐43, we searched for other proteins prone to aggregation that may accumulate upon depletion of C9ORF72. Ataxin‐2 (encoded by the gene ATXN2) is a cytoplasmic RNA‐binding protein that interacts with the poly(A)‐binding protein (PABP) and regulates mRNA stability (Kozlov et al, 2001; Yokoshi et al, 2014). Abnormal expansion over 34 glutamines in ATXN2 leads to spinocerebellar ataxia type 2 (SCA2) (Imbert et al, 1996; Pulst et al, 1996; Sanpei et al, 1996), while intermediate expansion of polyQ (27–33 repeats) is an increased risk of ALS‐FTD (Elden et al, 2010; Daoud et al, 2011; Ross et al, 2011; Van Damme et al, 2011; Lattante et al, 2014). We found that both HA‐tagged Ataxin‐2 with control length (Q22x) and intermediate size (Q30x) of polyQ localized diffusely into the cytoplasm of primary cultures of embryonic mouse cortical neurons (Fig 5A). Importantly, shRNA‐mediated decreased expression of C9orf72 promoted accumulation of aggregates of Ataxin‐2 with intermediate polyQ length in primary cultures of neurons (Fig 5A). Similar results were observed in GT1‐7 neuronal cells or when partners of C9orf72, namely Smcr8 and Wdr41, were siRNA‐depleted (Fig EV5A). As a control, treatment of neuronal cells with bafilomycin A1, a drug that blocks autophagy, also promoted accumulation of Ataxin‐2 Q30x aggregates (Fig EV5A). In contrast, depletion of C9orf72, Smcr8, or Wdr41 or bafilomycin treatment had no effect on the diffuse localization of Ataxin‐2 with control (Q22x) polyQ size (Figs 5A and EV5A). Immunoblotting confirmed identical expression of HA‐tagged Ataxin‐2 Q22x upon siRNA‐mediated depletion of C9orf72, Smcr8, or Wdr41 or bafilomycin treatment compared to control siRNA (Fig EV5B). Similarly, expression of HA‐tagged Ataxin‐2 Q30x is identical between control and siRNA‐treated C9orf72, Smcr8, or Wdr41, but slightly increased upon bafilomycin A1 treatment (Fig EV5B). Consistent with a previous observation of an increased stability of Ataxin‐2 with intermediate size of polyQ (Elden et al, 2010), we noted that HA‐tagged Ataxin‐2 Q30x is expressed at twofold to threefold higher levels than HA‐tagged Ataxin‐2 Q22x (Fig EV5B and C). As further controls, the effect of C9orf72 loss on aggregation of Ataxin‐2 Q30x was specific since we did not observe increased aggregation of mutant SOD1, mutant FUS, huntingtin, or Ataxin‐3 with polyQ expansion upon siRNA‐mediated depletion of C9orf72 expression, at least in transfected cells and in the time frame of our study (Fig EV5D).
Figure 5. Reduced expression of C9ORF72 synergizes Ataxin‐2 toxicity.
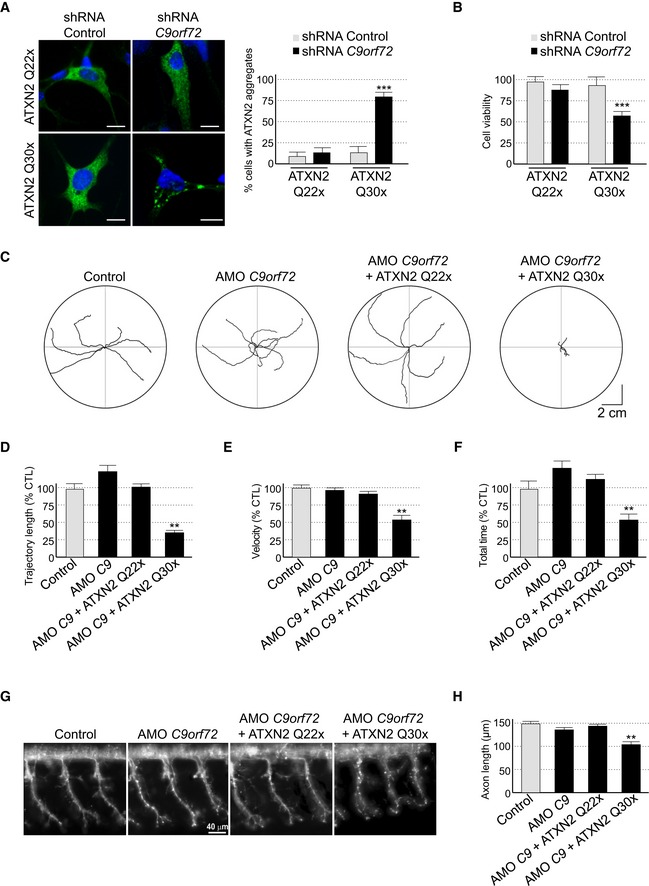
-
ALeft panel, representative images of organotypic cultures of E18 mouse cortical neurons co‐transfected with HA‐tagged ATXN2 with control (Q22x) or intermediate (Q30x) polyQ size and transduced with lentivirus expressing either control shRNA or shRNA targeting C9orf72 mRNA. Right panel, quantification of Ataxin‐2 aggregates. Scale bars, 10 μm. Nuclei were counterstained with DAPI.
-
BCell viability (tetrazolium assay) of GT1‐7 neuronal cells co‐transfected with HA‐tagged ATXN2 with control (Q22x) or intermediate (Q30x) polyQ size and control siRNA or siRNA targeting C9orf72 mRNA. Error bars indicate SEM, n = 3.
-
CTracing of the swimming trajectories of 48 h post‐fertilization zebrafish larvae following light touch.
-
D–FQuantification of the touch‐evoked swimming distance (D), average velocity (E), and maximum velocity attained (F) shows significant functional impairment of the zebrafish injected with HA‐tagged ATXN2 with intermediate length of polyQ (Q30x) and antisense morpholino oligonucleotides (AMOs) against C9orf72 compared to control HA‐tagged ATXN2 (Q22x) or to the sole injection of AMO against C9orf72.
-
GRepresentative images of motor neurons visualized with anti‐Sv2 immunohistochemistry show severe axonopathy in fish injected with both ATXN2 Q30x and AMO against C9orf72 compared to zebrafish injected with control ATXN2 Q22x or with AMO against C9orf72 alone.
-
HQuantification of the motor neuron axonal length demonstrates a significant decrease in axonal length in 48 h post‐fertilization zebrafish larvae injected with both ATXN2 Q30x and AMO against C9orf72 compared to controls.
Figure EV5. Reduced expression of C9ORF72 synergizes Ataxin‐2 toxicity.
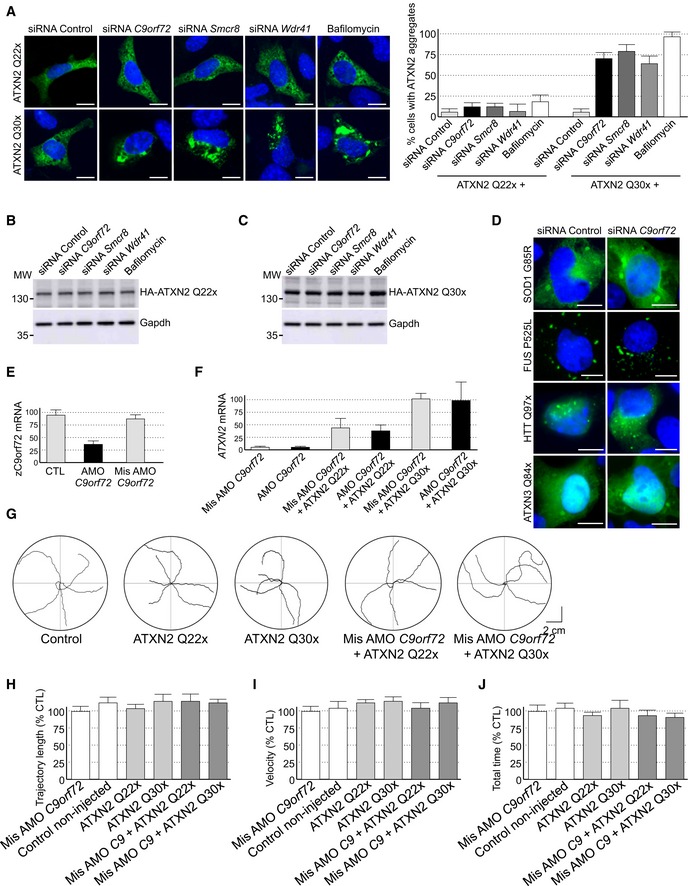
-
ARepresentative images of GT1‐7 neuronal cells co‐transfected with HA‐tagged ATXN2 with control (Q22x) or intermediate (Q30x) polyQ size and either control siRNA or siRNA targeting endogenous C9orf72 mRNA.
-
B, CImmunoblot analysis of endogenous Gapdh or transfected HA‐tagged Ataxin‐2 with (B) control (Q22x) length or (C) intermediate (Q30x) length of polyQ in GT1‐7 cells.
-
DRepresentative images of GT1‐7 neuronal cells co‐transfected with either GFP‐tagged mutant SOD1, FUS, HTT, or Ataxin‐3 and either control siRNA or siRNA targeting endogenous C9orf72 mRNA.
-
ERT–qPCR quantification of endogenous C9orf72 expression relative to Gapdh mRNA in mismatched or C9orf72 antisense morpholino oligonucleotide (AMO)‐injected zebrafish.
-
FRT–qPCR quantification of exogenous HA‐tagged ATXN2 with normal (Q22x) or intermediate (Q30x) length of polyQ relative to endogenous Gapdh mRNA in zebrafish injected with control or HA‐tagged ATXN2 constructs and with mismatched AMOs or AMOs against C9orf72.
-
GTracing of the swimming trajectories of 48 h post‐fertilization zebrafish larvae following light touch.
-
H–JQuantification of the touch‐evoked swimming distance (H), average velocity (I), and maximum velocity attained (J) shows no impairment upon the sole injection of HA‐tagged ATXN2 with control (Q22x) or intermediate length of polyQ (Q30x). Similarly, injection of mismatched antisense morpholino oligonucleotides (AMOs) against C9orf72 alone or with HA‐tagged ATXN2 Q22x or Q30x shows no functional alterations.
Data information: Scale bars, 10 μm. Nuclei were counterstained with DAPI. Error bars indicate SEM, n = 3.
Next, we investigated whether decrease expression of C9ORF72 leads to any reduction in neuronal viability. Consistent with a previous report (Wen et al, 2014), neither siRNA‐ nor shRNA‐mediated decreased expression of C9orf72 in neuronal primary cultures induced significant cell death, at least in the conditions and time frame of our study (Fig 5B). Similarly, expression of Ataxin‐2 with control (Q22x) or intermediate (Q30x) size of polyQ had little effect on neuronal viability (Fig 5B). In contrast, decreased expression of C9orf72 and simultaneous expression of Ataxin‐2 with intermediate size of polyQ (Q30x) induced neuronal cell death (Fig 5B). As a control, siRNA‐mediated depletion of C9orf72 with concomitant expression of control Ataxin‐2 (Q22x) did not reduce neuronal cell viability (Fig 5B).
To confirm these results in vivo, we developed zebrafish models with decreased expression of C9orf72 and which express Ataxin‐2 with either normal or intermediate size of polyQ. Of technical interest, we used a reduced quantity (50%) of antisense morpholino oligonucleotides (AMOs) known to block translation of the zebrafish ortholog of C9ORF72 compared to a previous study (Ciura et al, 2013). In these conditions, we observed little toxicity and no abnormal motor phenotype associated with a reduction in C9orf72 (Fig 5C). Quantification of endogenous zebrafish C9orf72 mRNA by RT–qPCR confirmed a partial (50–60%) decreased expression of C9orf72 upon antisense AMO injection compared to control conditions (Fig EV5E). We also controlled by RT–qPCR the equal expression of HA‐tagged Ataxin‐2 Q22x or Q30x in injected zebrafish (Fig EV5F). Consistent with previous results (Elden et al, 2010), we noted a higher expression of Ataxin‐2 Q30x compared to Ataxin‐2 Q22x (Fig EV5F). Importantly, we observed no toxicity associated with the sole expression of Ataxin‐2 with normal (Q22x) or intermediate (Q30x) length of polyQ (Fig EV5G). In contrast, the decreased expression of C9orf72 associated with the expression of Ataxin‐2 with intermediate size of polyQ (Q30x) resulted in an abnormal motor behavior, specifically a reduced touch‐evoked escape response (Fig 5C). Indeed, after stimulation by light touch to the fish tail, swimming distance, average velocity, and maximum velocity were significantly reduced in zebrafish embryos knocked down for C9orf72 and expressing Ataxin‐2 with thirty glutamines (Fig 5D–F). As a control, decreased expression of C9orf72 with concomitant expression of Ataxin‐2 with control length of polyQ (Q22x) had no significant pathogenic effect on swimming episodes following escape response (Fig 5D–F). Furthermore, the specificity of the phenotype was confirmed by injection of the same concentration of a control mismatch morpholino, which did not cause any locomotor phenotype (Fig EV5G–J). As further controls, expression of Ataxin‐2 with either control or intermediate length of polyQ alone or with a mismatch antisense morpholino oligonucleotide also did not cause any locomotor phenotypes (Fig EV5G–J).
Finally, we analyzed the morphology of the axonal projections from spinal motor neurons by immunofluorescence against the synaptic vesicle marker Sv2. Similar to results presented above, knockdown of C9orf72 alone or the sole expression of Ataxin‐2 (Q22x or Q30x) had no effect compared to control non‐injected or mismatch AMO‐injected fish (Fig 5G). Also, knockdown of C9orf72 with concomitant expression of Ataxin‐2 with control size (Q22x) of polyQ had no toxic effect (Fig 5G and H). In contrast, the decreased expression of C9orf72 with simultaneous expression of Ataxin‐2 with intermediate size of polyQ (Q30x) resulted in disrupted arborization and shortening of the motor neuron axons (Fig 5G and H). Overall, these results indicate that the partial knockdown of C9ORF72 does not induce major neuronal cell death but synergizes the toxicity of Ataxin‐2 with intermediate length of polyQ, resulting in alterations of the motor neuron and of the locomotor phenotype in vivo.
Discussion
In conclusion, we found that C9ORF72 belongs to a complex containing the SMCR8 and WDR41 proteins. This complex is phosphorylated by TBK1 and ULK1 kinases, is a GDP/GTP exchange factor for RAB8a and RAB39b GTPases, and regulates autophagy (Fig 6). These results are reminiscent of the complex formed by folliculin (FLCN) and the folliculin‐interacting protein‐1 and folliculin‐interacting protein‐2 (FNIP1 and FNIP2), which contain divergent DENN modules presenting some homology with SMCR8 and C9ORF72, respectively (Zhang et al, 2012; Levine et al, 2013). FLCN is a tumor suppressor protein disrupted in various cancers and the Birt–Hogg–Dubé syndrome, which presents GTPase‐activating activity for RagC/D regulating mTORC1 activity (Petit et al, 2013; Tsun et al, 2013). Also and similar to the C9ORF72 complex, FLCN is a GEF for Rab GTPases, is phosphorylated by ULK1, and is involved in the control of autophagy (Nookala et al, 2012; Dunlop et al, 2014).
Figure 6. Tentative model of C9ORF72 function.
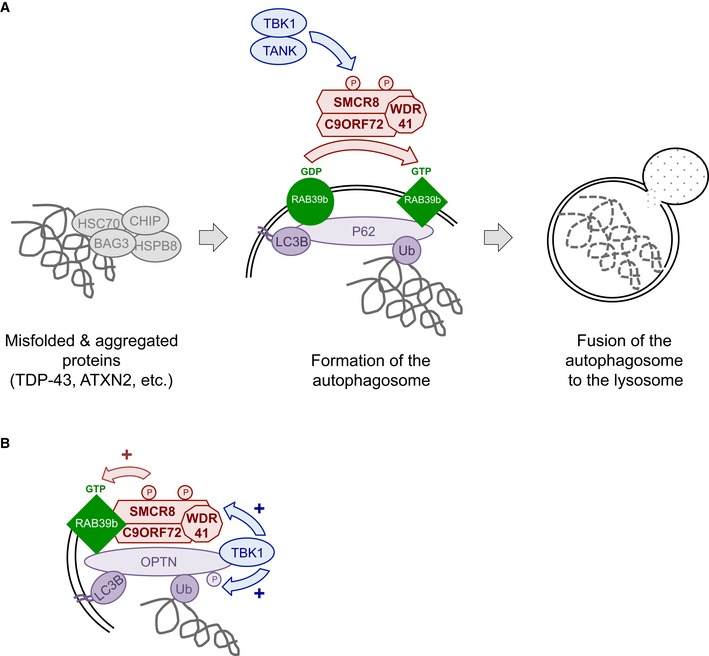
- C9ORF72 in complex with SMCR8 and WDR41 acts as a GDP/GTP exchange factor for RAB39b GTPase, which interacts with the P62 autophagy receptor. Phosphorylation of SMCR8 by TBK1 potentially promotes C9ORF72 GEF activity and enhances autophagy turnover of proteins such as TDP‐43 or Ataxin‐2 with intermediate polyQ size. In the absence of C9ORF72, autophagy clearance of these proteins is reduced and TDP‐43, P62, or Ataxin‐2 with intermediate length of polyQ accumulates into cytoplasmic aggregates.
- Autophagy receptors such as P62 or OPTN act as hubs to gather Rab GTPases with their GEF effectors and kinase regulators to initiate autophagy precisely at the site of damaged organelles, protein aggregates, or intracellular pathogens.
However, our results indicating that C9ORF72 in complex with SMCR8 interacts with RAB8a and RAB39b are different from a previous report where C9ORF72 was found to interact with RAB1, RAB5, RAB7, and RAB11 (Farg et al, 2014). These discrepancies are inherent to the different approaches used in these two studies. Notably, we tested C9ORF72 in complex, while Farg and colleagues studied C9ORF72 in isolation. Similarly, composition of the immunoprecipitation washing buffers is different with 50 mM Tris in Farg and colleagues versus 50 mM Tris with 150 mM NaCl in our study. Moreover, we found that commercial antibodies against C9ORF72 were of poor specificity to carry immunoprecipitation of endogenous proteins. A second divergence between these two studies is that Farg and colleagues observed that siRNA‐mediated depletion of C9orf72 increases LC3B‐II levels in basal condition. In contrast, we observed little effect of siRNA‐ or shRNA‐mediated depletion of C9ORF72 on LC3B in basal condition, but observed a significant inhibition of LC3B lipidation when autophagy flux was investigated. These differences may originate from the different degree of siRNA‐mediated depletion of C9ORF72 and/or from the different cells employed (primary E18 cortical neurons and GT1‐7 cells in our study versus SH‐SY5Y cells in Farg and colleagues) since basal autophagy and LC3B levels are known to vary according to the cell types.
Importantly, a role of C9ORF72 in autophagy is consistent with the increased accumulation of P62 and susceptibility to inhibition of autophagy observed in human neurons derived from C9ORF72 iPS cells (Almeida et al, 2013). Similarly, accumulation of TDP‐43‐ and P62‐positive protein aggregates upon reduction in C9ORF72 expression is reminiscent of a dysfunction of autophagy and reproduces key histopathological features of ALS‐FTD patients (Neumann et al, 2006; Al‐Sarraj et al, 2011). In that aspect, it is also striking to note that various mutations causing ALS‐FTD are found in genes involved in protein clearance pathways, including UBQLN2, CHMP2B, VCP, OPTN, SQSTM1, and TBK1 (Skibinski et al, 2005; Johnson et al, 2010; Maruyama et al, 2010; Deng et al, 2011; Fecto et al, 2011; Cirulli et al, 2015; Freischmidt et al, 2015). Thus, our work linking decreased expression of C9ORF72 to partial deficient autophagy provides further support to compromised protein clearance mechanisms in ALS‐FTD. However, it is important to note that our siRNA approach resulted in loss of 75% or more of C9orf72 expression, while C9ORF72 levels are reduced by 50% or less in brain of individuals with ALS‐FTD. Similarly, mutations in TBK1 cause disease through a haploinsufficiency mechanism, while our siRNA reduced Tbk1 expression by nearly 80%. Thus, we can only speculate that partial reduction in C9ORF72 or TBK1 activity in patients may result in a suboptimal autophagy pathway, which in turn may contribute to disease pathogenesis.
A second significant conclusion of this study is that constitutively active GTP‐locked RAB39b corrects autophagy alteration caused by either loss of TBK1 or C9ORF72. Thus, we propose that TBK1, C9ORF72 complex, and RAB39b belong to a common pathway regulating autophagy in neuronal cells (Fig 6A). This model is similar to the ULK1‐mediated phosphorylation of the guanine nucleotide exchange factor DENND3 that activates Rab12 and promotes autophagy (Xu et al, 2015), suggesting that phosphorylation of GDP/GTP exchange factors may be a widespread and novel pathway to activate Rab GTPase in autophagy. Since RAB39b and RAB8a interact with P62 and OPTN, our results also support a model where autophagy receptors, such as P62 or OPTN, function as essential hubs to gather autophagy substrates with LC3B but also with specific Rab GTPases and their GEF effectors and kinase regulators in order to initiate autophagy precisely at the site of protein aggregates, dysfunctional organelles, or intracellular pathogens (Fig 6B). This model is supported by the interaction and phosphorylation of P62 or OPTN by TBK1 in xenophagy (Wild et al, 2011; Pilli et al, 2012), but also by the recent reports of OPTN and TBK1 importance for the PINK1‐Parkin mitophagy pathway, which is altered in Parkinson's disease (Heo et al, 2015; Lazarou et al, 2015; Matsumoto et al, 2015). In that aspect, mutations in the X‐linked RAB39b gene lead to early‐onset Parkinson's disease (Wilson et al, 2014), while atypical parkinsonism has been observed in rare cases of individuals with GGGGCC expansion in C9ORF72 (Wilke et al, 2016).
This work also raises several questions. First, we noted that the C9ORF72 complex acts as a GEF toward RAB8a and RAB39b; however, we did not test all existing Rab proteins and the C9ORF72 complex may potentially regulate various other Rab GTPases. Similarly, it is unclear which Rab proteins initiate and regulate autophagy in which tissue, developmental time, or conditions. In that aspect, the precise molecular functions of RAB8 and RAB39 in autophagy remain also to be elucidated. Binding of RAB8a and RAB39b to P62 suggest that these Rab GTPases may act on formation of the autophagosome. However, whether these Rab GTPases can also regulate the transport or fusion of the autophagosome to multivesicular body or lysosome is to be determined. Also, we found that phosphorylation of SMCR8 by TBK1 is important to control autophagy in neuronal cells. However, it remains to test the importance of SMCR8 phosphorylation on the GDP/GTP exchange activity of the C9ORF72 complex. Likewise, the signaling pathways activating TBK1 is yet unclear. Indeed, TBK1 activation may require an upstream kinase (Heo et al, 2015), while a second and non‐exclusive model proposes that local concentration of TBK1 through its recruitment via OPTN may auto‐activate TBK1 through trans‐autophosphorylation (Matsumoto et al, 2015). Furthermore, SMCR8 is also interacting with proteins of the ULK1, mTOR, and AMPK kinase complexes (Table EV1) and SMCR8 is reported phosphorylated by mTOR (Hsu et al, 2011) and AMPK (Hoffman et al, 2015; Schaffer et al, 2015), but with unknown consequences.
Also intriguing is the accumulation of TDP‐43 aggregates upon depletion of C9ORF72 in primary culture of E18 cortical mouse neurons but not in GT1‐7 cells. This discrepancy may originate from the reduced time frame (24–48 h) of analysis in GT1‐7 cells compared to primary neuronal culture transduced with shRNA lentivirus for 7 days. In support of our work, a causal link between altered autophagy and accumulation of cytoplasmic aggregates of TDP‐43 is long established (Filimonenko et al, 2007; Ju et al, 2009; Urushitani et al, 2010; Wang et al, 2012; Barmada et al, 2014; Scotter et al, 2014) and is consistent with the TDP‐43‐positive neuronal inclusions observed in ALS‐FTD patients with mutation in genes involved in autophagy or in protein clearance pathway, including UBQLN2, TBK1, OPTN, SQSTM1, VCP, or GRN (Hu et al, 2010; Brady et al, 2013; review in Majcher et al, 2015 and in Taylor, 2015). However, this questions whether the aggregates of TDP‐43 observed in the vast majority of sporadic ALS‐FTD cases are also caused by some deficiency in autophagy and/or protein clearance mechanisms, and if so, what would be the underlying pathogenic mechanisms. In that aspect, recent evidences of accumulation of TDP‐43 aggregates in aging human brain (Uchino et al, 2015) might be related to the known decline of autophagy with age (Sun et al, 2015).
Finally, we found that partial loss of C9ORF72 promoted accumulation of P62 in aggregates, but had only mild effect on LC3B levels. Similarly, reduced expression of C9orf72 had little effect on neuronal cell viability. This is consistent with the absence of neurodegenerative phenotypes observed in mouse depleted of C9orf72 expression (Lagier‐Tourenne et al, 2013; Koppers et al, 2015). These results may suggest that either the activity of C9ORF72 is redundant with other proteins or that C9ORF72 is not crucial for basal autophagy but may play a more restricted role in a specific autophagy subpathway. In support of that latter hypothesis, we found that decreased expression of C9ORF72 promotes specifically the aggregation of Ataxin‐2 with intermediate length of polyQ. These results support the co‐incidence of expanded GGGGCC repeats in C9ORF72 with intermediate length of polyQ repeats in Ataxin‐2 in ALS‐FTD patients (Elden et al, 2010; Daoud et al, 2011; Ross et al, 2011; Van Damme et al, 2011; Lattante et al, 2014). However, why the decreased expression of C9ORF72 promotes the aggregation and toxicity of Ataxin‐2 with polyQ expansion, but not the aggregation of Ataxin‐3 or Huntingtin with polyQ expansion, is intriguing and the cause of this specificity remains to be explored. In that aspect, both TDP‐43 and Ataxin‐2 are RNA‐binding proteins, thus questioning whether a selective autophagy of RNA‐protein granules (RNAphagy) is controlled by C9ORF72 and altered specifically in ALS‐FTD (Buchan et al, 2013; Fujiwara et al, 2013). It is also possible that we missed the deleterious effect of C9ORF72 loss on other polyQ proteins due to the reduced time frame of our study or to other inherent limitations of cell cultures. Similarly, it is possible that Ataxin‐2 with a control length (22Q) of polyglutamine might form microaggregates that would be toxic on a longer time period of analysis. This hypothesis is sustained by the observation that Ataxin‐2 with both normal and intermediate length of polyQ synergizes the toxicity of TDP‐43 in fly, however with normal polyQ size being less toxic than intermediate polyQ length (Kim et al, 2014). Thus, murine models would be instrumental to test the pathological consequences of loss of C9ORF72 in the presence of Ataxin‐2 or other proteins with polyglutamine expansion. It also remains to test whether decreased expression of C9ORF72 may synergize toxicity of other stress, notably some that are inherent to the expanded GGGGCC repeats such as accumulation of GGGGCC RNA foci and/or RAN‐translated DPRs. This hypothesis is particularly appealing in light of the association of C9ORF72 mRNA expression with patient survival (van Blitterswijk et al, 2015) and the recent reports of no overt neurodegenerative phenotype in BAC transgenic mouse models with normal expression of C9orf72 but overexpression of expanded GGGGCC repeats. These mice present RNA foci and aggregates of dipeptide repeat proteins but develop only subtle behavioral phenotypes (O'Rourke et al, 2015; Peters et al, 2015). Similarly, mice depleted of C9orf72 expression in brain or in neurons present no overt neurodegenerative phenotypes (Lagier‐Tourenne et al, 2013; Koppers et al, 2015). These results suggest that the sole loss of C9ORF72 or the expression of GGGGCC RNA and DPR in isolation is not sufficient to be pathogenic, but it remains to test whether a reduced expression of C9ORF72 would synergize the toxicity of GGGGCC RNA or DPRs. This synergic model is consistent with the absence of ALS/FTD patients with null alleles or missense mutations in C9ORF72, as well as by increasing genetic evidences of oligogenicity in ALS‐FTD (Ferrari et al, 2012; Van Blitterswijk et al, 2012; van Blitterswijk et al, 2013; Cady et al, 2015; Lattante et al, 2015a; Pottier et al, 2015).
In conclusion, our results support a double‐hit mechanism in ALS‐FTD, where the sole decreased expression of C9ORF72 does not explain alone the pathogenicity of the expanded GGGGCC repeats but may synergize other stress such as Ataxin‐2 with intermediate polyQ length, while association with other stress such as GGGGCC RNA foci or Ran‐translated DPRs remains to be formally tested. Finally, if loss of C9ORF72 leads to partial dysfunction of autophagy, one may hope that pharmacological compounds activating autophagy may contribute to alleviate some pathological features of ALS‐FTD.
Materials and Methods
Constructions
PCMV6 containing C‐terminally Flag‐tagged human cDNAs of SQSTM1 (P62), OPTN, SMCR8, WDR41, ULK1, NAP1, SINTBAD, TANK, and RAB GTPases were purchased from OriGene. Optimized cDNAs for human N‐terminally HA‐tagged C9ORF72 and Flag‐tagged TBK1 cloned into pcDNA3 were purchased from GenScript. Human cDNAs of ATXN2 with 22 or 30 glutamines were fused to an N‐terminal HA tag and cloned into pcDNA3. Constitutively active Q68L and negative S22N mutants of N‐terminally HA‐tagged RAB39b were constructed by inverse PCR. Similarly, mutations of C‐terminally HA‐tagged SMCR8 phosphorylation sites (S400, S402, S492, S562, T666, and T796D) in alanine or aspartate were constructed by inverse PCR.
HA‐Flag tandem affinity purification
12 × 106 Neuro‐2A cells were transfected with 48 μg of either control‐ or C9ORF72‐Flag‐Ha plasmid using Fugene HD (Promega) for 24 h, and proteins were purified by Ha‐Flag tandem purification kit according to the manufacturer's instruction (Sigma‐Aldrich). The bound proteins were visualized by silver staining (SilverQuest, Invitrogen) after separation on a 4–12% bis‐Tris Gel (NuPAGE), and interactant proteins were identified using NanoESI_Ion Trap (Thermo Fisher).
Immunofluorescence
Coverslips were incubated for 10 min in PBS with 4% paraformaldehyde, washed with PBS, and incubated in PBS plus 0.5% Triton X‐100 for 10 min. The cells were washed three times with PBS, and the coverslips were incubated for 1 h with primary antibody against the HA tag (26183, Pierce). P62/Sqstm1 (ab56416, Abcam) or anti‐phospho‐TDP‐43 (pS409/410, Cosmo Bio). After washing with PBS, the coverslips were incubated with goat anti‐mouse secondary antibody conjugated with Alexa 488 (Interchim SA) for 1 h, washed twice with PBS, and incubated for 2 min in PBS/DAPI (1/10,000 dilution). Coverslips were rinsed twice before mounting in Pro‐Long media (Molecular Probes) and were examined using confocal microscope.
Immunoprecipitation
6.25 × 105 HEK293 cells (1 well of a 6‐well plate) were co‐transfected for 24 h with 1 μg of plasmids expressing HA‐tagged cDNA and 1 μg of plasmids expressing Flag‐tagged cDNA using Fugene HD (Promega). Cells were scraped into RIPA buffer (50 mM Tris–HCI pH 7.6, 150 mM NaCl, 1% NP‐40) and centrifuged for 15 min at 18,000 g at 4°C; 20 μl of pre‐washed HA magnetic beads (Dynabeads) was added, and immunoprecipitation was carried for 1 h at 4°C with constant rotation. After three washes with 50 mM Tris–HCI pH 7.6, 150 mM NaCl, 0.05% Tween, bound proteins were eluted in SDS–PAGE loading buffer and analyzed by Western blot using antibodies against the Flag tag (PA1‐984B, Pierce) or the HA tag (26183, Pierce). For endogenous immunoprecipitation, SMCR8 1D2 mouse monoclonal antibody was incubated with mouse brain extract overnight in RIPA buffer. Pre‐cleared A/G magnetic beads (Life Technologies) were added, and immunoprecipitation was carried out for 1 h at 4°C with constant rotation. The beads were washed three times with 50 mM Tris–HCI, 150 mM NaCl, 0.05% Tween and then boiled at 95°C for 5 min in SDS–PAGE loading buffer. Bound proteins were analyzed by Western blot using antibodies against SMCR8 (1D2), C9ORF72 (22637‐1‐AP, Proteintech), WDR41 (NBP1‐83812, Novus Biological), Rab8a (11792‐1‐AP, Proteintech), Rab39b (12162‐1‐AP, Proteintech), Rab5 (11671‐1‐AP, Proteintech), Rab7 (Ab137029, Abcam).
In vitro GDP/GTP exchange assay
About 40 pmol (~2 μg) of purified recombinant GST‐RAB protein was loaded with 1 μCurie of α32P‐labeled GDP (Hartmann Analytic) in 10 μl of buffer assay (150 mM NaCl, 50 mM Hepes, 1 mg/ml BSA, 2.5 mM EDTA, pH 7.5) for 30 min at 30°C. Increased quantities of recombinant purified C9ORF72 or C9ORF72/SMCR8/WDR41 complex were added and incubated for further 30 min at 30°C. Twenty microliters of pre‐washed GST magnetic beads (Dynabeads) was added, and pull‐down was carried out for 30 min at 4°C. After three washes in reaction buffer, radioactivity was measured by using a scintillation counter (Beckman Coulter).
In vitro phosphorylation
Eight micrograms of recombinant purified C9ORF72/SMCR8/WDR41 complex was incubated for 30 min at 30°C with 10 μCurie of γ32P‐labeled ATP in 20 μl of kinase buffer assay (150 mM NaCl, 20 mM HEPES, 2 mM MgCl2, 25 mM β‐glycerophosphate, 100 μM orthovanadate, pH 7.5) with or without 2 μg of recombinant purified ULK1 or TBK1 protein (OriGene). The reaction was stopped by the addition of SDS–PAGE loading buffer, boiled for 3 min at 90°C, and run on 4–12% bis‐Tris Gel (NuPAGE). The gel was then either used for Western blotting assay or dried and imaged (Typhoon scanner, GE Healthcare).
Neuronal cell cultures, transfection, and treatments
Primary cortical neurons were prepared from C57Bl/6 E18 embryos and grown on poly‐L‐lysine‐coated 24‐well plates in neurobasal medium (NBM) supplemented with 1× B27, 0.5 mM L‐glutamine, and 100 IU/ml penicillin/streptomycin at 37°C with 5% CO2. Neurons were transduced at Day 3 with recombinant lentivirus expressing either control shRNA or shRNA against C9orf72 (SK02‐040236‐00‐10 SMARTvector 2.0 hEF1a Lentiviral Mouse 3110043O21Rik shRNA, Thermos). After overnight incubation, lentivirus was removed and fresh media were added. After 5–7 days, neurons were analyzed by immunofluorescence or by Western blot analysis. GT1‐7 cells were grown in 10% fetal bovine serum, gentamicin, and penicillin at 37°C in 5% CO2, plated in DMEM and 0.1% fetal bovine serum, and transfected for 24–48 h using Lipofectamine 2000 (Fisher Scientific) and/or RNAimax (Fisher Scientific) with siRNA control or targeting C9orf72, Smcr8 3′UTR, or Tbk1 (ON‐TARGETplus, Dharmacon). Neurons were treated with either 10 μM of rapamycin (Millipore) for 15 h or 100 nM of bafilomycin (Sigma) for 15 h or 250 nM of Torin‐1 (Tocris) for 2 h before analysis.
Recombinant protein production and purification
For RAB GTPases, E. coli BL21(RIL) pRARE competent cells (Invitrogen) were transformed with pet28‐GST‐RAB GTPase vectors, grown at 37°C in 400 ml of LB medium supplemented with kanamycin until OD600 = 0.5, 0.5 mM IPTG was added and the culture was further incubated for 4 h at 30°C. Harvested cells were sonicated in 300 mM NaCl, 50 mM Tris–Cl pH 7.5, 1 mM DTT, 5 mM EDTA and centrifuged for 20 min at 20,000 g and recombinant GST‐RAB GTPase proteins were purified using the GST purification Kit (Novagen), quantified, dialyzed, and stored in 150 mM NaCl, 20 mM HEPES, 2 mM MgCl2, 20% glycerol. Concerning C9ORF72 complex, 2 × 106 of SF9 (Spodoptera frugiperda) cells (one T25 flask) were co‐transfected with 500 ng of Bsu36I‐linearized BAC10:KO1629 DNA and 2 μg of pMF‐Dual vectors containing either HIS‐C9ORF72, SMCR8, or WDR41 and incubated at 27°C for 6 days. Harvested baculovirus were then tested, amplified and used to infect 2 l of SF9 cell culture for protein production and purification using the HIS purification kit (Novagen) with sonication and washing in 500 mM NaCl, 50 mM Tris–Cl pH 7.5, 50 mM imidazole, elution with 150 mM NaCl, 50 mM Tris–Cl pH 7.5, 1 mM DTT, 5 mM EDTA, 200 mM imidazole, dialysis, and storage in 150 mM NaCl, 20 mM HEPES, 2 mM MgCl2, 20% glycerol.
Monoclonal antibody production
To generate anti‐SMCR8 monoclonal antibodies, 8‐week‐old female BALB/c mice were injected intraperitoneally with 100 μg of recombinant purified SMCR8:C9ORF72 complex and 200 μg of poly(I/C) as adjuvant. Three injections were performed at 2‐week intervals, and 4 days prior to hybridoma fusion, mice with positively reacting sera were re‐injected. Spleen cells were fused with Sp2/0.Agl4 myeloma cells as described by De StGroth and Scheidegger (1980). Hybridoma culture supernatants were tested at Day 10 by ELISA for cross‐reaction with recombinant purified C9ORF72:SMCR8 complex. Positive supernatants were then tested by immunofluorescence and Western blot on HA‐tagged SMCR8 and HA‐tagged C9ORF72 transfected COS‐1 cells. Specific cultures were cloned twice on soft agar. Specific hybridomas were established, and ascites fluid was prepared by injection of 2 × 106 hybridoma cells into Freund adjuvant‐primed BALB/c mice. All animal experimental procedures were performed according to the European authority guidelines.
Western blotting
Proteins were denatured for 3 min at 95°C, separated on 4–12% bis‐Tris Gel (NuPAGE), transferred on nitrocellulose membranes (Whatman Protran), blocked with 5% non‐fat dry milk in Tris‐buffered saline (TBS) buffer, incubated with anti‐Flag (PA1‐984B, Pierce), HA (26183, Pierce), C9ORF72 (22637‐1‐AP, Proteintech), LC3B (ab51520, Abcam), GAPDH (ab125247, Abcam), TDP‐43 (3449S, Cell Signaling), SMCR8 (1D2, 1/200) in TBS plus 5% non‐fat dry milk, washed three times, and incubated with anti‐rabbit or anti‐mouse peroxidase antibody (1:3,000, Cell Signaling) for 1 h in TBS, followed by washing and ECL chemoluminescence revelation (Amersham ECL Prime).
Zebrafish studies
Adult and larval zebrafish (Danio rerio) were maintained at the ICM fish facility and bred according to the National and European Guidelines for Animal Welfare. Experiments were performed on wild‐type embryos from AB and TL strains. Hb9:GFP Tg(Mnx1:GFP) transgenic lines were used to label motor neurons and their axonal projections. All procedures for zebrafish experimentation were approved by the Institutional Ethics Committee at the Research Center of the ICM and by French and European legislation. The ATXN2 constructs were used in all the experiments described here at the final DNA concentration of 75 ng/μl. Embryos were maintained at 28°C and manually dechorionated using fine forceps at 24 hpf. Only zebrafish without developmental abnormalities (qualified as malformed) and the swimming trajectories at 48 h post‐fertilization were selected and their swimming trajectories appropriately traced. For the percentage analysis, behavioral analysis, and axonal projections, more than three independent experiments were performed for each of the conditions described here. Antisense morpholino oligonucleotides (AMOs) were designed complementary to bind to an upstream ATG that would block both transcripts of the zebrafish C9orf72 (C9orf72) and synthesized from GeneTools. The C9orf72 AMO sequence was ATTGTGGAGGACAGGCTGAAGACAT and known to bind to the following sequence in the C9orf72 mRNA: [(ATG)TCTTCAGCCTGTCCTCCACAAT]. A control AMO (mismatch), containing five mismatch nucleotides with the C9orf72 AMO sequence and not binding anywhere in the zebrafish genome, was used to assess the specificity of the observed phenotype (ATTcTcGAGcACAGcCTcAAGACAT). For the genetic interaction experiments, microinjections were performed at 0.2 mM for C9orf72‐AMO and 0.2 mM for C9orf72‐mis. At these concentrations, C9orf72 and mismatch AMOs do not lead to phenotypic features associated with deficient swimming or morphological abnormalities (Ciura et al, 2013; Lattante et al, 2015a,b). For the percentage analysis, behavioral analysis, and axonal projections, more than three independent experiments were performed for each of the conditions including co‐injections of ATXN2 Q22x and ATXN2 Q30x alongside C9orf72 and mismatch AMOs. Zebrafish embryos at 48 hpf were analyzed for any morphological abnormalities and touched lightly at the level of the tail with a pipette tip with their locomotor behavior scored. Thus, for each injection set, larvae and embryos were separated into the following groups: dead, curly, and monster groups (developmentally aberrant fish). TEER episodes were performed only in zebrafish that appeared morphologically normal (normal TEER observed) and were recorded for each of the conditions with a Grasshopper 2 Camera (Point Grey Research) at 30 Hz. The videos were then analyzed using the manual tracking plugin of ImageJ 1.45r software, and the swim duration, swim distance, and maximum swim velocity of the fish were calculated as previously described (Ciura et al, 2013; Lattante et al, 2015b). To correlate gene expression with cell morphology, the axonal projections of motor neurons in selected HB9 zebrafish embryos GFP‐positive at 48 hpf. In addition, axonal projections were labeled using the synaptic vesicle marker, SV2 as previously described (Kabashi et al, 2010). Fluorescent images of fixed embryos were taken using the fluorescence Automated Inverted Microscope System Olympus IX83 equipped with a Hamamatsu ORCA‐flash 2.8 digital camera. Image acquisition was performed with the Olympus cellSens software. Axonal projections from primary motor neurons at a defined location in the intersomitic segments were determined. Analysis of Z‐stacks by fluorescence microscopy was performed in three to four axonal projections per animal. The axonal length to the first branching was determined by tracing the labeled axon from the spinal cord to the point where it branches using ImageJ. These values were averaged for each of the animals analyzed (minimum 15 zebrafish per condition) for the various conditions. Primers for RT–qPCR quantification of HA‐tagged ATXN2 injected in zebrafish are F‐TGGTTCTCCAGCTCCTGTCT and R‐TGACCACTGATGACCACGTT. ATXN2 levels were normalized to endogenous Gapdh (QuantiTect Primer Assay, Dr_gapdh_1_SG).
Statistical analysis
All cell experiments are represented as average ± standard error of mean (SEM) with significance determined using Student's t‐test. All data values for the zebrafish experiments are represented as average ± standard error of mean (SEM) with significance determined using one‐way ANOVAs.
Author contributions
Experiments were performed by CS, MLC, AG, IKC, CJC, MOA, FR and SC. Data were collected and analyzed by CS, AP, SC, EK, and NCB. The study was designed, coordinated, and written by CS, SC, EK, and NCB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Pamela Mellon (UCSD, USA) for the gift of the GT1‐7 cells; Terje Johansen (University of Tromsø, Norway) for the gift of the P62, OPTN, and LC3B plasmids; Jochen Weishaupt (Ulm University, Germany) for the gift of the TBK1 vector; and Aaron Gitler (Stanford University School of Medicine, USA) and Daisuke Ito (Keio University, Tokyo, Japan) for the gift of the Ataxin‐2 plasmids. This work was supported by Fondation de France Thierry Latran #57486 “Model‐ALS”, AFM grant #18605 “Role of C9ORF72 in ALS‐FTD”, ERC‐2012‐StG #310659 “RNA DISEASES”, ANR‐10‐LABX‐0030‐INRT, and ANR‐10‐IDEX‐0002‐02 (NCB); Atip/Avenir from Inserm, Career Integration Grant (Marie Curie Actions), Robert Packard Foundation, E‐rare ERA‐NET program, AFM, ARSLA, France‐Alzheimer Association, and the program “Investissements d'avenir” ANR‐10‐IAIHU‐06 (EK). SC is supported by an AFM postdoctoral fellowship and M‐LC by a postdoctoral fellowship from the Fondation Cognacq‐Jay.
The EMBO Journal (2016) 35: 1276–1297
See also: S Almeida & F‐B Gao (June 2016)
References
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet‐Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol 126: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al‐Chalabi A, Hortobágyi T, Shaw CE (2011) P62 positive, TDP‐43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72‐linked FTLD and MND/ALS. Acta Neuropathol 122: 691–702 [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611 [DOI] [PubMed] [Google Scholar]
- Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, Hoch M, Höhfeld J (2010) Chaperone‐assisted selective autophagy is essential for muscle maintenance. Curr Biol 20: 143–148 [DOI] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, van Blitterswijk MM, Jansen‐West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, Shaw C, Chandran S, Finkbeiner S (2014) Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10: 677–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW (2010) Network organization of the human autophagy system. Nature 466: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Baker MC, DeJesus‐Hernandez M, Ghidoni R, Benussi L, Finger E, Hsiung GY, Kelley BJ, Murray ME, Rutherford NJ, Brown PE, Ravenscroft T, Mullen B, Ash PE, Bieniek KF, Hatanpaa KJ, Karydas A, Wood EM, Coppola G, Bigio EH et al (2013) C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology 81: 1332–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Gendron TF, Baker MC, DeJesus‐Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, Lagier‐Tourenne C, Edbauer D, Cleveland DW, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Petrucelli L, Boeve BF, Graff‐Radford NR et al (2015) Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol 130: 863–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose JK, Huang CC, Shen CK (2011) Regulation of autophagy by neuropathological protein TDP‐43. J Biol Chem 286: 44441–44448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 22: 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153: 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, Mitra RD, Ravits J, Harms MB, Baloh RH (2015) Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 77: 100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Kir S, Tooze SA (2007) siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 282: 25464–25474 [DOI] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes‐Casey M, Lee CW, Jansen‐West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 348: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day‐Williams AG et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347: 1436–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol 74: 180–187 [DOI] [PubMed] [Google Scholar]
- Cooper‐Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, Shaw PJ (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137 : 2040–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud H, Belzil V, Martins S, Sabbagh M, Provencher P, Lacomblez L, Meininger V, Camu W, Dupré N, Dion PA, Rouleau GA (2011) Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol 68: 739–742 [DOI] [PubMed] [Google Scholar]
- De StGroth SF, Scheidegger D (1980) Production of monoclonal antibodies: strategy and tactics. J Immunol Methods 35: 1–21 [DOI] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Miller BL, Dickson DW, Boylan KB, Graff‐Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL et al (2011) Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 477: 211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Blackshaw S, Sattler R, Rothstein JD (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop EA, Seifan S, Claessens T, Behrends C, Kamps MA, Rozycka E, Kemp AJ, Nookala RK, Blenis J, Coull BJ, Murray JT, van Steensel MA, Wilkinson S, Tee AR (2014) FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy 10: 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen‐Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay‐Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM et al (2010) Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23: 3579–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud‐Driss S, Sufit RL, Heller SL, Deng HX, Siddique T (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68: 1440–1446 [DOI] [PubMed] [Google Scholar]
- Ferrari R, Mok K, Moreno JH, Cosentino S, Goldman J, Pietrini P, Mayeux R, Tierney MC, Kapogiannis D, Jicha GA, Murrell JR, Ghetti B, Wassermann EM, Grafman J, Hardy J, Huey ED, Momeni P (2012) Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging 33: 1850.e1–1850.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179: 485–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani MH, Dorst J, Graf E, Nordström U et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 18: 631–636 [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Furuta A, Kikuchi H, Aizawa S, Hatanaka Y, Konya C, Uchida K, Yoshimura A, Tamai Y, Wada K, Kabuta T (2013) Discovery of a novel type of autophagy targeting RNA. Autophagy 9: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J 28: 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang YJ, Jansen‐West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes‐Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, D'Elia E, Vecellio M, Russo S, Cogliati F, Larizza L, Ropers HH, Tzschach A, Kalscheuer V, Oehl‐Jaschkowitz B, Skinner C, Schwartz CE, Gecz J, Van Esch H, Raynaud M, Chelly J et al (2010) Mutations in the small GTPase gene RAB39B are responsible for X‐linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet 86: 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ et al (2012) A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11: 54–65 [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL 3rd, Bigio EH, Caselli R, Baker M, Al‐Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ (2008) TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 63: 535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A, Bürckstümmer T, Dixit E, Scheicher R, Górna MW, Karayel E, Sugar C, Stukalov A, Berg T, Kralovics R, Planyavsky M, Bennett KL, Colinge J, Superti‐Furga G (2011) Functional dissection of the TBK1 molecular network. PLoS ONE 6: e23971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattula K, Peränen J (2000) FIP‐2, a coiled‐coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol 10: 1603–1606. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher‐Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, Stöckli J, Burchfield JG, Jensen TE, Jothi R, Kiens B, Wojtaszewski JF, Richter EA, James DE (2015) Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise‐Regulated Kinases and AMPK Substrates. Cell Metab 22: 922–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM (2011) The mTOR‐regulated phosphoproteome reveals a mechanism of mTORC1‐mediated inhibition of growth factor signaling. Science 332: 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM (2010) Sortilin‐mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68: 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, Dürr A, Didierjean O, Stevanin G, Agid Y, Brice A (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14: 285–291 [DOI] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez‐Lage M, Falcone D, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Galassi G et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68: 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica‐Worms D, Baloh RH, Weihl CC (2009) Valosin‐containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40: 572–574 [DOI] [PubMed] [Google Scholar]
- Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P (2010) Gain and loss of function of ALS‐related mutations of TARDBP (TDP‐43) cause motor deficits in vivo. Hum Mol Genet 19: 671–683 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM (2014) Therapeutic modulation of eIF2α phosphorylation rescues TDP‐43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46: 152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884 [DOI] [PubMed] [Google Scholar]
- Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Jeroen Pasterkamp R (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78: 426–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov G, Trempe JF, Khaleghpour K, Kahvejian A, Ekiel I, Gehring K (2001) Structure and function of the C‐terminal PABC domain of human poly(A)‐binding protein. Proc Natl Acad Sci U S A 98: 4409–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345: 1139–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis‐Downes M, Markmiller S, Hutt KR et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 110: E4530–E4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Millecamps S, Stevanin G, Rivaud‐Péchoux S, Moigneu C, Camuzat A, Da BS, Mundwiller E, Couarch P, Salachas F, Hannequin D, Meininger V, Pasquier F, Seilhean D, Couratier P, Danel‐Brunaud V, Bonnet AM, Tranchant C, LeGuern E, Brice A et al (2014) Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 83: 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Ciura S, Rouleau GA, Kabashi E (2015a) Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet 31: 263–273 [DOI] [PubMed] [Google Scholar]
- Lattante S, de Calbiac H, Le Ber I, Brice A, Ciura S, Kabashi E (2015b) Sqstm1 knock‐down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum Mol Genet 24: 1682–1690 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez‐Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE (2013) Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5: 1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab‐GEFs. Bioinformatics 29: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen‐Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59: 1077–1079 [DOI] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC (2012) Molecular basis of Tank‐binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A 109: 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majcher V, Goode A, James V, Layfield R (2015) Autophagy receptor defects and ALS‐FTLD. Mol Cell Neurosci 66 : 43–52. [DOI] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon‐Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 11: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226 [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through P62/SQSTM1 phosphorylation. Hum Mol Genet 24: 4429–4442 [DOI] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf‐Strathmann J, Mann M, Meissner F, Edbauer D (2014) C9orf72 FTLD/ALS‐associated Gly‐Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128: 485–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126: 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering‐Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EM, Partridge L, Isaacs AM (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345: 1192–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Lammich S, Mackenzie IR, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C (2013a) hnRNP A3 binds to GGGGCC repeats and is a constituent of P62‐positive/TDP43‐negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125: 413–423 [DOI] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013b) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339: 1335–1338 [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997 [DOI] [PubMed] [Google Scholar]
- Nookala RK, Langemeyer L, Pacitto A, Ochoa‐Montaño B, Donaldson JC, Blaszczyk BK, Chirgadze DY, Barr FA, Bazan JF, Blundell TL (2012) Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol 2: 120071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L et al (2015) C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 88: 892–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, Sun H, Boylan KB, Dickson D, Kennedy Z, Lin Z, Zhang YJ, Daughrity L, Jung C, Gao FB, Sapp PC et al (2015) Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 88: 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CS, Roczniak‐Ferguson A, Ferguson SM (2013) Recruitment of folliculin to lysosomes supports the amino acid‐dependent activation of Rag GTPases. J Cell Biol 202: 1107–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V (2012) TBK‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, DeTure M, Knopman DS, Josephs KA, Parisi JE, Petersen RC, Boylan KB, Boeve BF, Graff‐Radford NR, Veltman JA, Gilissen C et al (2015) Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130: 77–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes‐Cendes I, Pearlman S, Starkman S, Orozco‐Diaz G, Lunkes A, DeJong P, Rouleau GA, Auburger G, Korenberg JR, Figueroa C, Sahba S (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14: 269–276 [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65: 586–590 [DOI] [PubMed] [Google Scholar]
- Ross OA, Rutherford NJ, Baker M, Soto‐Ortolaza AI, Carrasquillo MM, DeJesus‐Hernandez M, Adamson J, Li M, Volkening K, Finger E, Seeley WW, Hatanpaa KJ, Lomen‐Hoerth C, Kertesz A, Bigio EH, Lippa C, Woodruff BK, Knopman DS, White CL 3rd, Van Gerpen JA et al (2011) Ataxin‐2 repeat‐length variation and neurodegeneration. Hum Mol Genet 20: 3207–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR et al (2008) Novel mutations in TARDBP (TDP‐43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4: e1000193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, Koide R, Saito M, Sato A, Tanaka T, Hanyu S, Takiyama Y, Nishizawa M, Shimizu N, Nomura Y, Segawa M et al (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14: 277–284 [DOI] [PubMed] [Google Scholar]
- Sato T, Iwano T, Kunii M, Matsuda S, Mizuguchi R, Jung Y, Hagiwara H, Yoshihara Y, Yuzaki M, Harada R, Harada A (2014) Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J Cell Sci 127: 422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, Benayoun BA, Banko MR, Shaw RJ, Shokat KM, Brunet A (2015) Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large‐Scale Substrate Prediction. Cell Metab 22: 907–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC, Shaw CE (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP‐43 species. J Cell Sci 127: 1263–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto S, Sugaya K, Tsujimura K, Nagata T, Horii T, Koide Y (2013) Rab39a interacts with phosphatidylinositol 3‐kinase and negatively regulates autophagy induced by lipopolysaccharide stimulation in macrophages. PLoS ONE 8: e83324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G1, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Johannsen P, Sørensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37: 806–808. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al‐Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319: 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmström KM, Fergusson MM, Yoo YH, Combs CA, Finkel T (2015) Measuring In Vivo Mitophagy. Mol Cell 60: 685–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis‐Dusenbery BN, Eggan K (2013) The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci 16: 1725–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation‐induced cytotoxicity. Hum Mol Genet 24: 2426–2441 [DOI] [PubMed] [Google Scholar]
- Taylor JP (2015) Multisystem proteinopathy: Intersecting genetics in muscle, bone, and brain degeneration. Neurology 85: 658–660 [DOI] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat Immunol 10: 1215–1221 [DOI] [PubMed] [Google Scholar]
- Tsun ZY, Bar‐Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 52: 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino A, Takao M, Hatsuta H, Sumikura H, Nakano Y, Nogami A, Saito Y, Arai T, Nishiyama K, Murayama S (2015) Incidence and extent of TDP‐43 accumulation in aging human brain. Acta Neuropathol Commun. 20: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M, Sato T, Bamba H, Hisa Y, Tooyama I (2010) Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP‐43. J Neurosci Res 88: 784–797 [DOI] [PubMed] [Google Scholar]
- Van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, Bourque PR, Schelhaas HJ, van der Kooi AJ, de Visser M, de Bakker PI, Veldink JH, van den Berg LH (2012) Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 21: 3776–3784 [DOI] [PubMed] [Google Scholar]
- Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W (2011) Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 76: 2066–2072 [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen‐Plotkin AS, Martinez‐Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7: 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35:1779.e5–1779.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA‐binding protein 43. Proc Natl Acad Sci U S A 109: 15024–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, Pasinelli P, Ichida JK, Trotti D (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84: 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RJ, Lu Y, Marie B, Gao FB, Sweeney ST (2015) Rab8, POSH, and TAK1 regulate synaptic growth in a Drosophila model of frontotemporal dementia. J Cell Biol 208: 931–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke C, Pomper JK, Biskup S, Puskás C, Berg D, Synofzik M (2016) Atypical parkinsonism in C9orf72 expansions: a case report and systematic review of 45 cases from the literature. J Neurol 263: 558–574 [DOI] [PubMed] [Google Scholar]
- Wilson GR, Sim JC, McLean C, Giannandrea M, Galea CA, Riseley JR, Stephenson SE, Fitzpatrick E, Haas SA, Pope K, Hogan KJ, Gregg RG, Bromhead CJ, Wargowski DS, Lawrence CH, James PA, Churchyard A, Gao Y, Phelan DG, Gillies G et al (2014) Mutations in RAB39B cause X‐linked intellectual disability and early‐onset Parkinson disease with α‐synuclein pathology. Am J Hum Genet 95: 729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13: 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, Wang G (2016) TDP‐43 loss of function increases TFEB activity and blocks autophagosome‐lysosome fusion. EMBO J 35: 121–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Fotouhi M, McPherson PS (2015) Phosphorylation of the exchange factor DENND3 by ULK in response to starvation activates Rab12 and induces autophagy. EMBO Rep 16: 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O (2008) TDP‐43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63: 538–542 [DOI] [PubMed] [Google Scholar]
- Yokoshi M, Li Q, Yamamoto M, Okada H, Suzuki Y, Kawahara Y (2014) Direct binding of Ataxin‐2 to distinct elements in 3′ UTRs promotes mRNA stability and protein expression. Mol Cell 55: 186–198 [DOI] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, He F, Aravind L (2012) Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet 13: 283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Jansen‐West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, Chew J, Belzil VV, Prudencio M, Stankowski JN, Castanedes‐Casey M, Whitelaw E, Ash PE, DeTure M, Rademakers R, Boylan KB et al (2014) Aggregation‐prone c9FTD/ALS poly(GA) RAN‐translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128: 505–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Bañez‐Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110: E4968–E4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4