

Abstract
Human rhinovirus (HRV) infections target airway epithelium and are the leading cause of acute exacerbations of COPD. Cigarette smoke (CS) increases the severity of viral infections, but there is no effective therapy for HRV infection. We determined whether α1-antitrypsin (A1AT) reduces HRV-16 infection in CS-exposed primary human airway epithelial cells. Brushed bronchial epithelial cells from normal subjects and patients diagnosed with COPD were cultured at air–liquid interface to induce mucociliary differentiation. These cells were treated with A1AT or bovine serum albumin for 2 hours and then exposed to air or whole cigarette smoke (WCS) with or without HRV-16 (5×104 50% Tissue Culture Infective Dose [TCID50]/transwell) infection for 24 hours. WCS exposure significantly increased viral load by an average of fivefold and decreased the expression of antiviral genes interferon-λ1, OAS1, and MX1. When A1AT was added to WCS-exposed cells, viral load significantly decreased by an average of 29-fold. HRV-16 infection significantly increased HRV-16 receptor intercellular adhesion molecule-1 messenger RNA expression in air-exposed cells, which was decreased by A1AT. A1AT-mediated reduction of viral load was not accompanied by increased epithelial antiviral gene expression or by inhibiting the activity of 3C protease involved in viral replication or maturation. Our findings demonstrate that A1AT treatment prevents a WCS-induced increase in viral load and for the first time suggest a therapeutic effect of A1AT on HRV infection.
Keywords: α1-antitrypsin, rhinovirus, COPD, cigarette smoke, ICAM-1
Introduction
Airway epithelial cells represent the first line of defense against infections of the lungs through their wide variety of roles, from acting as a barrier to the outside environment to coordinating the innate and adaptive immune responses in the airway.1 Airway epithelial cells are also the primary target for tobacco cigarette smoke (CS). CS is known to be associated with various respiratory pathologic processes, including increased inflammation and viral infections.2,3 Long-term CS exposure is considered a risk factor in the development of COPD, the third leading cause of death in the US.1,4
A major cause of acute exacerbations of COPD and other respiratory diseases such as asthma are human rhinovirus (HRV) infections.5,6 HRV primarily acts on airway epithelial cells and causes roughly three-quarters of common colds.7 At this time, there are no approved treatments or vaccinations for HRV infections. Serotyped strains of HRV are categorized as either minor or major group viruses, depending on their entry receptor.8 Approximately 90% of identified serotypes are major group viruses (eg, HRV-16), which use intercellular adhesion molecule-1 (ICAM-1) as their receptor.8 ICAM-1 is a transmembrane glycoprotein receptor of the immunoglobulin superfamily and it functions in the uncoating of viral genetic material.9 Because ICAM-1 is the target for the entry of major group HRVs to the cell, any agent that targets ICAM-1 may potentially inhibit HRV infections.
α1-Antitrypsin (A1AT) is a 52 kDa serine protease inhibitor that is produced mainly in the liver and also in epithelial cells and macrophages. A1AT exhibits an anti-inflammatory function in bacteria-infected human airway epithelial cells.10 A1AT has also been shown to reduce replication of human immunodeficiency virus (HIV) in peripheral blood mononuclear cells of humans recently infected with HIV.11 While the antiviral properties of A1AT have been examined in the context of HIV, there have been no published reports about the possible effect of A1AT on HRV. We hypothesized that A1AT can be used as a therapeutic agent against viral (eg, HRV-16) infections, particularly in a smoking setting. Furthermore, we seek to determine the underlying mechanisms of action (eg, antiviral genes, HRV replication, and ICAM-1 expression) of A1AT on viral infections.
Methods
Air–liquid interface culture of human brushed bronchial epithelial cells
Brushed bronchial epithelial cells from normal human subjects (n=4) and patients diagnosed with COPD (n=4) were processed as previously described12 and stored in liquid nitrogen until ready for use. The Institutional Review Board of National Jewish Health approved the collection and use of these cells. All patients provided written informed consent. The characteristics of human subjects are shown in Table 1. There was no significant difference between the ages and smoking history (pack-years) of the two groups. As expected, the forced expiratory volume in 1 second (FEV1%) and FEV1/forced vital capacity (FVC)% were significantly lower in patients diagnosed with COPD than in normal subjects. Airway epithelial cells at passage 1 were cultured and expanded at 37°C, 5% CO2, until 90% confluence in collagen-coated 60 mm tissue culture dishes containing bronchial epithelial cell growth medium with supplements (Lonza, Walkersville, MD, USA). Epithelial cells at passage 2 were then seeded into collagen-coated 12-well transwell plates (Transwell 2460; Corning Incorporated, Corning, NY, USA) at 4×104 cells/well and cultured as previously described.13 After 7 days of submerged culture, cells were shifted to air–liquid interface (ALI) for the next 10 days to induce mucociliary differentiation. On day 10 of ALI, cells were pretreated with A1AT (1 mg/mL; Grifols Inc., Research Triangle Park, NC, USA) or bovine serum albumin (BSA, 1 mg/mL, control; Sigma-Aldrich Co., St Louis, MO, USA) 2 hours prior to CS exposure. The 1 mg/mL dose of A1AT has been optimized in cultured human bronchial epithelial cells and mouse alveolar macrophages in previous studies.14,15 Furthermore, we performed a dose–response study using 0.1 mg/mL and 1 mg/mL doses of A1AT in lipopolysaccharide-treated lung epithelial cells (NCI-H292 cell line), where the 1 mg/mL dose significantly reduced interleukin-8 production by lipopolysaccharide. A1AT stock was prepared in sterile H2O at 100 mg/mL and then diluted directly in cell culture medium on the day of use. The BSA (control) stock was prepared in sterile phosphate-buffered saline at 100 mg/mL and diluted directly in cell culture medium on the day of use. In our preliminary studies, we did not observe any differences regarding the effect of H2O versus phosphate-buffered saline on antiviral responses in airway epithelial cells.
Table 1.
Characteristics of human study subjects
| Age (years) | Sex (M/F) | Smoking (pack-years) | FEV1, % predicted | FVC, % predicted | FEV1/FVC% | |
|---|---|---|---|---|---|---|
| Normal (n=4) | 53.0±3.2 | 1/3 | 1 (0) 3 (34.3±16.7) |
93.7±12.8 | 88.0±11.6 | 83.0±2.5 |
| COPD (n=4) | 61.5±8.1 | 1/3 | 4 (55.3±21.3) | 41.0±13.4a | 62.5±21.9 | 41.7±6.2a |
Notes: Data are presented as mean ± SEM.
Indicates that P<0.05 compared to normal subjects.
Abbreviations: M, male; F, female; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; SEM, standard error of the mean.
Whole cigarette smoke exposure in cultured human brushed bronchial epithelial cells
On day 10 of ALI culture, cells were exposed to air (control) or whole cigarette smoke (WCS) from research cigarette 2R4F (University of Kentucky, Lexington, KY, USA) for 10 minutes using a mainstream smoking chamber specifically designed for cell culture experiments (British American Tobacco Science, Southampton, UK) as previously described.16,17 Immediately after smoking, the BSA control or A1AT-treated cells were infected apically with HRV-16 in ALI culture medium at 5×104 TCID50/transwell. Cells were harvested after 24 hours of viral infection with no wash or removal of virus from the apical surface.
HRV preparation
HRV-16 (American Type Culture Collection, Manassas, VA, USA) was propagated in HeLa cells (CRL-1958; American Type Culture Collection), purified, and titrated as described previously.18
Quantitative real-time reverse-transcription polymerase chain reaction
TaqMan gene expression assays for HRV and human interferon-λ1 (IFN-λ1) were custom made from Integrated DNA Technologies (Coralville, IA, USA). The specific primers and probes were HRV (forward: 5′-CCT CCG GCC CCT GAA T-3′; reverse: 5′-GGT CCC ATC CCG CAA TT-3′, probe: 5′-CTA ACC TTA AAC CTG CAG CCA-3′) and IFN-λ1 (forward: 5′-GGG AAC CTG TGT CTG AGA ACG T-3′; reverse: 5′-GAG TAG GGC TCA GCG CAT AAA TA-3′; probe: 5′-CTG AGT CCA CCT GAC ACC CCA CAC C-3′). TaqMan gene expression assays for OAS1 (Hs00973637_m1), MX1 (Hs00895608_m1), and ICAM-1 (Hs00164932_m1) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). The housekeeping gene GAPDH (4352665; Thermo Fisher Scientific) was evaluated as an internal positive control. We focused primarily on IFN-λ1 (a type III interferon) as it is considered to be the primary gene (vs type I interferons IFN-α and IFN-β) involved in the epithelial cell response to HRV infections.19,20 Nonetheless, we measured MX1 (also known as MxA) and OAS1, which are considered as the inducible genes by both IFN-α and IFN-β.21 Quantitative real-time polymerase chain reaction was performed on the CFX96™ real-time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA). The comparative cycle of threshold (ΔΔCt) method was used to demonstrate the relative levels of target genes relative to GAPDH.
A1AT effects on the activity of HRV-3C protease
To determine whether A1AT inhibits the replication of HRV, we focused on HRV-3C protease, a key protease involved in the replication and maturation of HRV. Incubating A1AT with 3C protease and its substrate, GST-Syk fusion protein, indicates if A1AT may inhibit viral replication. Human A1AT (1 mg/mL) or BSA (1 mg/mL) was incubated overnight at 4°C with GST-Syk protein (0.1 mg/mL), HRV-3C protease (40 units/mL), and HRV-3C buffer (control) from the HRV-3C protease kit (Thermo Fisher Scientific, Cat#88946). Samples were electrophoresed on a 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel and stained with Coomassie blue dye. The gel was destained with 10% acetic acid and 50% ethanol until bands were resolved.
Statistical analysis
When the data were not normally distributed, one-way nonparametric analysis was used for group comparisons by applying the Wilcoxon test (two-group comparison) or the Kruskal–Wallis test (multiple-group comparison). When the data were normally distributed, one-way analysis of variance was used for multiple comparisons, followed by a paired t-test for two-group comparisons. P<0.05 was considered statistically significant.
Results
WCS decreases antiviral gene expression and increases viral load in human brushed bronchial epithelial cells
Previous studies on the effects of CS in host defense against rhinovirus infection were performed in cell lines or primary cells that were exposed to CS extract.1,2,22 This study, to the best of our knowledge, is the first to investigate the impact of WCS on antiviral responses of primary brushed airway epithelial cells from both normal subjects and patients diagnosed with COPD. As the antiviral response was similar for both normal and COPD cells, we chose to combine the data from both groups for statistical analysis. As expected, in air-exposed cells, antiviral gene expression significantly increased in response to viral infection: IFN-λ1 by 90-fold, OAS1 by 12-fold, and MX1 by 49-fold (Figure 1; the normal and COPD groups are distinguished using different colors). WCS suppressed the antiviral response to HRV-16 infection as it significantly decreased expression of IFN-λ1 by 38-fold, OAS1 by eightfold, and MX1 by 21-fold (Figure 2A). There was no significant difference in the reduction of antiviral genes by WCS between normal cells and COPD cells. In line with the impaired antiviral gene expression, WCS increased viral load by an average of fivefold in normal and COPD cells (Figure 2B).
Figure 1.
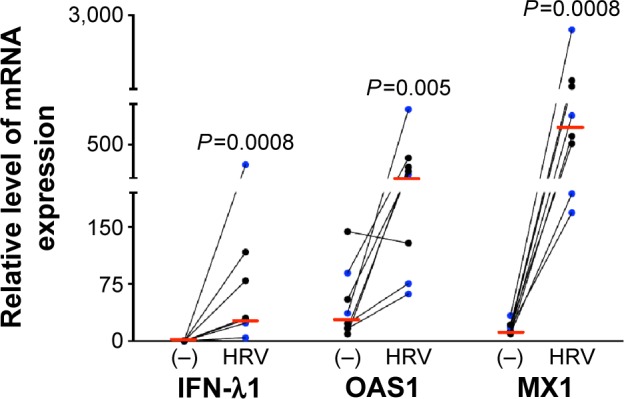
Rhinovirus infection increased expression of antiviral genes in human bronchial epithelial cells.
Notes: Brushed bronchial epithelial cells cultured in ALI from normal subjects (n=4) and patients diagnosed with COPD (n=4) were exposed to air for 10 minutes and then infected with HRV-16 for 24 hours or treated with cell culture medium (−) as a control. HRV-16 significantly increased the mRNA expression of antiviral genes IFN-λ1, OAS1, and MX1. Relative levels of the three genes were normalized to the housekeeping gene GAPDH. The red horizontal bars represent the medians of the eight subjects. The Wilcoxon test was used to compare the difference between (−) and HRV-infected groups. Blue represents normal subjects, while black represents patients diagnosed with COPD.
Abbreviations: ALI, air–liquid interface; HRV, human rhinovirus; mRNA, messenger RNA; IFN-λ1, interferon-λ1.
Figure 2.

WCS increased viral load and decreased antiviral gene expression in rhinovirus-infected human bronchial epithelial cells.
Notes: Brushed bronchial epithelial cells cultured in ALI from normal subjects (n=4) and patients diagnosed with COPD (n=4) were exposed to air for 10 minutes and then infected with HRV-16 for 24 hours or treated with cell culture medium as a control. WCS significantly increased viral load (A) but reduced the mRNA expression of antiviral genes (B). Relative levels of the three genes were normalized to the housekeeping gene GAPDH. The red horizontal bars represent the medians of the eight subjects. The Wilcoxon test was used to compare the difference between air and WCS groups. Blue represents normal subjects, while black represents patients diagnosed with COPD.
Abbreviations: WCS, whole cigarette smoke; ALI, air–liquid interface; HRV, human rhinovirus; mRNA, messenger RNA; IFN-λ1, interferon-λ1.
A1AT decreases viral load in WCS-exposed human brushed bronchial epithelial cells
How to reduce viral infection in CS-exposed airways remains a challenge in COPD health care. Here, we found that A1AT treatment significantly decreased viral load in WCS-exposed cells by an average of 29-fold (Figure 3A). However, in air-exposed cells, A1AT did not significantly reduce viral load (<10% reduction, P>0.05).
Figure 3.

A1AT decreased viral load in WCS-exposed human bronchial epithelial cells.
Notes: Brushed bronchial epithelial cells cultured in ALI from normal subjects (n=4) and patients diagnosed with COPD (n=4) were exposed to air or WCS for 10 minutes. Cells were treated with BSA as a control or A1AT (1 mg/mL) in the presence of HRV-16 for 24 hours. A1AT decreased viral load in cells exposed to WCS (A), and expression of the antiviral genes IFN-λ1, OAS1, or MX1 in cells exposed to WCS (B) or air (C). Relative levels of the three genes were normalized to the housekeeping gene GAPDH. The red horizontal bars represent the medians of the eight subjects. The Wilcoxon test was used to compare the difference between BSA and AIAT groups. Blue represents normal subjects, while black represents patients diagnosed with COPD.
Abbreviations: A1AT, α1-antitrypsin; WCS, whole cigarette smoke; ALI, air–liquid interface; BSA, bovine serum albumin; HRV, human rhinovirus; IFN-λ1, interferon-λ1.
Mechanisms underlying anti-HRV effect of A1AT
Effects of A1AT on antiviral gene expression
We measured antiviral gene expression to determine if A1AT decreases viral load by enhancing the expression of antiviral genes. In contrast to our hypothesis, A1AT-mediated reduction of viral load in WCS-exposed cells is accompanied by a significant reduction of IFN-λ1 messenger RNA (mRNA) expression (Figure 3B). A1AT treatment tended to reduce both OAS1 and MX1 gene expression in cells exposed to WCS (Figure 3B). Our data suggest that reduction of viral load by A1AT in WCS-exposed cells may lead to less antiviral gene induction, and A1AT does not enhance the antiviral gene expression. In air-exposed cells (Figure 3C), antiviral genes were also reduced by A1AT.
Effects of A1AT on viral replication and maturation
We further explored the possibility that A1AT may interfere with HRV replication and maturation. One of the critical mechanisms involved in HRV replication and maturation is the role of HRV nonstructural components such as the 3C protease, which is essential to the formation of mature capsid proteins and the completion of viral replication cycle. We hypothesized that A1AT may inactivate 3C protease, thus inhibiting HRV replication. When A1AT was directly added to HRV-3C protease (Figure 4), it did not reduce the degradation of GST-Syk fusion protein, a 3C protease substrate, suggesting that A1AT does not inhibit the activity of 3C protease.
Figure 4.

A1AT did not affect HRV-3C protease activity.
Notes: A1AT (1 mg/mL) was added to HRV-3C protease and its substrate, GST-Syk fusion protein, incubated overnight, electrophoresed on an SDS-PAGE gel, and then stained with Coomassie blue. The image is from one of the three independent experiments, all showing similar results. (−) indicates no HRV-3C.
Abbreviations: A1AT, α1-antitrypsin; HRV, human rhinovirus; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; BSA, bovine serum albumin.
Effect of A1AT on the expression of ICAM-1 in bronchial epithelial cells
As previously reported,23,24 HRV infection itself significantly increased airway epithelial ICAM-1 mRNA expression (Figure 5A). A1AT treatment in air-exposed and HRV-infected cells significantly reduced ICAM-1 mRNA expression (Figure 5A). In WCS-exposed cells, A1AT tended to reduce ICAM-1 expression, but this did not reach statistical significance (Figure 5B).
Figure 5.

A1AT treatment decreased ICAM-1 mRNA expression in human bronchial epithelial cells.
Notes: Brushed bronchial epithelial cells cultured in ALI from normal subjects (n=4) and patients diagnosed with COPD (n=4) were exposed to air or WCS for 10 minutes. Cells were treated with BSA as a control or A1AT (1 mg/mL) in the presence of HRV-16 for 24 hours. In air-exposed and HRV-16-infected cells, A1AT significantly reduced ICAM-1 mRNA (A). In WCS-exposed and HRV-16-infected cells, A1AT tended to decrease ICAM-1 mRNA (B). Relative ICAM-1 mRNA levels were normalized to the housekeeping gene GAPDH. An ANOVA was used to compare all the groups, followed by a paired t-test to compare the two groups. Data are presented as mean ± SEM.
Abbreviations: A1AT, α1-antitrypsin; ICAM-1, intercellular adhesion molecule-1; mRNA, messenger RNA; ALI, air–liquid interface; WCS, whole cigarette smoke; BSA, bovine serum albumin; HRV, human rhinovirus; ANOVA, analysis of variance; SEM, standard error of the mean.
Discussion
Our study is the first to demonstrate a therapeutic effect of A1AT on airway epithelial HRV infection, particularly in the context of CS. First, we extended previous studies regarding the detrimental effects of CS on the innate antiviral response in human airway epithelial cells. Exposing airway epithelial cells to WCS for only 10 minutes caused a fivefold increase in viral load. Application of A1AT significantly reduced the viral load.
When airway epithelial cells are exposed to HRV infection, they react with an increase in antiviral responses to combat the infection. Our results showed a significant increase of antiviral genes IFN-λ1, MX1, and OAS1. Interestingly, when cells were exposed to WCS, this defense mechanism was impaired as WCS decreased their expression. Although in this study, we focused on these three genes, other interferons such as IFN-λ2/3 may need to be measured to fully understand the effect of WCS on antiviral genes. How WCS dampened the upregulation of antiviral genes remains unclear. Although unraveling this mystery is not the focus of this study, previous studies have suggested several potential mechanisms. For example, CS exposure was shown to diminish the RIG-1 and JAK/STAT pathway, leading to a dampened type 1 interferon response to HRV infections.2,25 As SOCS1, an immune negative regulator, was shown to suppress induction of interferons by rhinovirus,26 it remains to be determined if WCS upregulates SOCS1 and subsequently inhibits the induction of antiviral genes following rhinovirus infection. Regardless of the underlying mechanisms whereby CS suppresses airway epithelial antiviral mechanisms, our data suggest that smoke-induced increase in viral load may contribute to acute exacerbations of lung diseases such as COPD.
A1AT has mainly been used as an augmentation therapy for patients with primary A1AT deficiency, but its effect in COPD patients remains to be determined. Because of its anti-inflammatory property, A1AT has been used to treat lung diseases such as cystic fibrosis.27–30 As pathogen (eg, viral) infection contributes to the disease process, it is imperative to reveal whether A1AT affects viral infections in a disease setting (eg, CS exposure). The data presented here are the first to suggest A1AT as a therapeutic agent against HRV infections in CS-exposed airway epithelial cells. The fact that A1AT reduced viral load in cells acutely exposed to CS suggests that it may be used in current smokers, but whether it is effective in ex-smokers remains to be determined.
A major challenge in this study is discerning the mechanisms by which A1AT reduces HRV-16 load. First, we determined whether A1AT increases antiviral gene expression. We found that A1AT-mediated reduction of viral load is accompanied by reduced antiviral gene expression. We speculate that the reduction of antiviral genes following A1AT treatment is the result of reduced viral load and that A1AT does not induce antiviral gene expression. Whether A1AT has a direct role in regulating signaling pathways involved in antiviral gene expression deserves further investigation.
Because viral replication is a critical step in determining the viral level, we examined the direct effect of A1AT on HRV-3C protease, a protein critical to HRV replication and maturation. If A1AT acted on HRV by inhibiting activity of HRV-3C protease, there should be less degradation of its substrate, such as the GST-Syk fusion protein we examined in this study. However, A1AT did not inhibit the activity of HRV-3C protease, suggesting that it is unlikely that A1AT blocks viral replication or maturation. Next, we determined if A1AT reduces the expression of ICAM-1, a receptor for HRV-16. We found that HRV infection increases ICAM-1 mRNA expression, which was significantly inhibited by A1AT in air-exposed, but not WCS-exposed cells. Our findings are in line with the study by Churg et al31 who reported that A1AT prevents increases in ICAM-1 at 24 hours in dust-exposed mice. However, the insignificant decrease of ICAM-1 in WCS-exposed and HRV-infected airway epithelial cells suggests that other mechanisms still exist and warrant further investigation. Together, our data on antiviral genes and protease 3C do not indicate the role of A1AT in inhibiting viral replication. However, reduction of ICAM-1 by A1AT during HRV infection may in part explain its antiviral function although A1AT did not completely reduce (normalize) ICAM-1 expression following HRV infection.
Since A1AT has multiple functions in inflammation and immunity, it is likely that it may exert the antiviral function through additional mechanisms such as inhibition of the inflammasome pathway. Inflammasomes regulate (either suppress or promote) viral infection/replication.32,33 The effect of A1AT on caspase-1, a component of inflammasomes, is controversial as some studies demonstrate the inhibitory effect of A1AT on caspase-1 activity,34 while others did not find any effects.35
There are several limitations to this study. First, as secondary A1AT deficiency occurs in COPD patients and viral infection remains to be a major contributor of acute exacerbations of COPD and other lung diseases, future studies in humans and animal models should be pursued to determine if A1AT may serve as a broad-spectrum intervention or therapy in COPD and other lung diseases associated with rhinovirus infections. Second, our sample size of eight (four normal subjects and four COPD patients) is relatively small. Although we did not see a significant difference of antiviral response between normal and COPD cells, we realize that this needs to be confirmed in future studies with a larger sample size. The COPD patients included in our study had moderate-to-severe COPD (GOLD stages II–IV) with an average age of 61.5 years (Table 1). With a relatively small COPD sample size, it is difficult to dissect the effect of the COPD GOLD stage or age on our results. In the future, we hope to investigate the effects of GOLD stage and age on airway epithelial responses to A1AT in the context of CS exposure and viral infection in a large COPD cohort.
Research findings from our study on the antiviral effects of A1AT are of particular importance because rhinoviruses are the most common respiratory viruses affecting millions of people in the world. A1AT augmentation therapy has been used as an approach to restore A1AT levels in COPD patients with A1AT deficiency as it may reduce disease severity and the frequency of COPD exacerbations.36–41 Our report demonstrates the ability of A1AT in reducing viral infections in CS-exposed airway epithelial cells, thus providing a novel mechanism underlying the therapeutic effects of A1AT in COPD.
Acknowledgments
This work was supported by the Flight Attendant Medical Research Institute (FAMRI) CIA 130041 and by the National Institutes of Health grants R01HL122321 and R01AI106287.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Logan J, Chen L, Gangell C, Sly PD, Fantino E, Liu K. Brief exposure to cigarette smoke impairs airway epithelial cell innate anti-viral defence. Toxicol In Vitro. 2014;28(8):1430–1435. doi: 10.1016/j.tiv.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Proud D, Hudy MH, Wiehler S, et al. Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS One. 2012;7(7):e40762. doi: 10.1371/journal.pone.0040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hudy MH, Traves SL, Wiehler S, Proud D. Cigarette smoke modulates rhinovirus-induced airway epithelial cell chemokine production. Eur Respir J. 2010;35(6):1256–1263. doi: 10.1183/09031936.00128809. [DOI] [PubMed] [Google Scholar]
- 4.Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol. 2014;50(3):471–482. doi: 10.1165/rcmb.2013-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zlateva KT, de Vries JJ, Coenjaerts FE, et al. GRACE Study Group Prolonged shedding of rhinovirus and re-infection in adults with respiratory tract illness. Eur Respir J. 2014;44(1):169–177. doi: 10.1183/09031936.00172113. [DOI] [PubMed] [Google Scholar]
- 6.George SN, Garcha DS, Mackay AJ, et al. Human rhinovirus infection during naturally occurring COPD exacerbations. Eur Respir J. 2014;44(1):87–96. doi: 10.1183/09031936.00223113. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of rhinovirus infection. Curr Opin Virol. 2012;2(3):287–293. doi: 10.1016/j.coviro.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–7426. doi: 10.1128/JVI.02290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukhopadhyay S, Malik P, Arora SK, Mukherjee TK. Intercellular adhesion molecule-1 as a drug target in asthma and rhinitis. Respirology. 2014;19(4):508–513. doi: 10.1111/resp.12285. [DOI] [PubMed] [Google Scholar]
- 10.Jiang D, Persinger R, Wu Q, Gross A, Chu HW. alpha1-Antitrypsin promotes SPLUNC1-mediated lung defense against Pseudomonas aeruginosa infection in mice. Respir Res. 2013;14:122. doi: 10.1186/1465-9921-14-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro L, Pott GB, Ralston AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001;15(1):115–122. doi: 10.1096/fj.00-0311com. [DOI] [PubMed] [Google Scholar]
- 12.Chu HW, Balzar S, Seedorf GJ, et al. Transforming growth factor-beta2 induces bronchial epithelial mucin expression in asthma. Am J Pathol. 2004;165(4):1097–1106. doi: 10.1016/s0002-9440(10)63371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross CA, Bowler RP, Green RM, Weinberger AR, Schnell C, Chu HW. beta2-Agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm Med. 2010;10:30. doi: 10.1186/1471-2466-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao W, Zhao J, Kim H, et al. alpha1-Antitrypsin inhibits ischemia reperfusion-induced lung injury by reducing inflammatory response and cell death. J Heart Lung Transplant. 2014;33(3):309–315. doi: 10.1016/j.healun.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 15.Churg A, Wang X, Wang RD, Meixner SC, Pryzdial EL, Wright JL. Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144–151. doi: 10.1165/rcmb.2006-0345OC. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Case S, Bowler RP, Martin RJ, Jiang D, Chu HW. Cigarette smoke modulates PGE(2) and host defence against Moraxella catarrhalis infection in human airway epithelial cells. Respirology. 2011;16(3):508–516. doi: 10.1111/j.1440-1843.2010.01920.x. [DOI] [PubMed] [Google Scholar]
- 17.Wu Q, Jiang D, Chu HW. Cigarette smoke induces growth differentiation factor 15 production in human lung epithelial cells: implication in mucin over-expression. Innate Immun. 2012;18(4):617–626. doi: 10.1177/1753425911429837. [DOI] [PubMed] [Google Scholar]
- 18.Wu Q, van Dyk LF, Jiang D, et al. Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology. 2013;446(1–2):199–206. doi: 10.1016/j.virol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okabayashi T, Kojima T, Masaki T, et al. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011;160(1–2):360–366. doi: 10.1016/j.virusres.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Khaitov MR, Laza-Stanca V, Edwards MR, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64(3):375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 21.Haller O, Kochs G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res. 2011;31(1):79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- 22.Hudy MH, Proud D. Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respir Res. 2013;14:88. doi: 10.1186/1465-9921-14-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papi A, Papadopoulos NG, Stanciu LA, et al. Reducing agents inhibit rhinovirus-induced up-regulation of the rhinovirus receptor intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells. FASEB J. 2002;16(14):1934–1936. doi: 10.1096/fj.02-0118fje. [DOI] [PubMed] [Google Scholar]
- 24.Whiteman SC, Bianco A, Knight RA, Spiteri MA. Human rhinovirus selectively modulates membranous and soluble forms of its intercellular adhesion molecule-1 (ICAM-1) receptor to promote epithelial cell infectivity. J Biol Chem. 2003;278(14):11954–11961. doi: 10.1074/jbc.M205329200. [DOI] [PubMed] [Google Scholar]
- 25.Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol. 2011;44(1):118–126. doi: 10.1165/rcmb.2009-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gielen V, Sykes A, Zhu J, et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol. 2015;136(1):177–188.e11. doi: 10.1016/j.jaci.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siekmeier R. Lung deposition of inhaled alpha-1-proteinase inhibitor (alpha 1-PI) – problems and experience of alpha1-PI inhalation therapy in patients with hereditary alpha1-PI deficiency and cystic fibrosis. Eur J Med Res. 2010;15(Suppl 2):164–174. doi: 10.1186/2047-783X-15-S2-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griese M, Latzin P, Kappler M, et al. alpha1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29(2):240–250. doi: 10.1183/09031936.00047306. [DOI] [PubMed] [Google Scholar]
- 29.Sorrells S, Camprubi S, Griffin R, Chen J, Ayguasanosa J. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med. 2015;109(4):490–499. doi: 10.1016/j.rmed.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Geraghty P, Eden E, Pillai M, Campos M, McElvaney NG, Foronjy RF. alpha1-Antitrypsin activates protein phosphatase 2A to counter lung inflammatory responses. Am J Respir Crit Care Med. 2014;190(11):1229–1242. doi: 10.1164/rccm.201405-0872OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Churg A, Dai J, Zay K, et al. Alpha-1-antitrypsin and a broad spectrum metalloprotease inhibitor, RS113456, have similar acute anti-inflammatory effects. Lab Invest. 2001;81(8):1119–1131. doi: 10.1038/labinvest.3780324. [DOI] [PubMed] [Google Scholar]
- 32.Lupfer C, Malik A, Kanneganti TD. Inflammasome control of viral infection. Curr Opin Virol. 2015;12:38–46. doi: 10.1016/j.coviro.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gastaldello S, Chen X, Callegari S, Masucci MG. Caspase-1 promotes Epstein-Barr virus replication by targeting the large tegument protein deneddylase to the nucleus of productively infected cells. PLoS Pathog. 2013;9(10):e1003664. doi: 10.1371/journal.ppat.1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, He Y, Abraham B, et al. Cytosolic, autocrine alpha-1 proteinase inhibitor (A1PI) inhibits caspase-1 and blocks IL-1beta dependent cytokine release in monocytes. PLoS One. 2012;7(11):e51078. doi: 10.1371/journal.pone.0051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman MA, Mitra S, Sarkar A, Wewers MD. Alpha 1-antitrypsin does not inhibit human monocyte caspase-1. PLoS One. 2015;10(2):e0117330. doi: 10.1371/journal.pone.0117330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118(5):1480–1485. doi: 10.1378/chest.118.5.1480. [DOI] [PubMed] [Google Scholar]
- 37.Campos MA, Kueppers F, Stocks JM, et al. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK) COPD. 2013;10(6):687–695. doi: 10.3109/15412555.2013.800852. [DOI] [PubMed] [Google Scholar]
- 38.Stoller JK, Fallat R, Schluchter MD, et al. Augmentation therapy with alpha1-antitrypsin: patterns of use and adverse events. Chest. 2003;123(5):1425–1434. doi: 10.1378/chest.123.5.1425. [DOI] [PubMed] [Google Scholar]
- 39.Kueppers F. The role of augmentation therapy in alpha-1 antitrypsin deficiency. Curr Med Res Opin. 2011;27(3):579–588. doi: 10.1185/03007995.2010.548750. [DOI] [PubMed] [Google Scholar]
- 40.Marciniuk DD, Hernandez P, Balter M, et al. Canadian Thoracic Society COPD Clinical Assembly Alpha-1 Antitrypsin Deficiency Expert Working Group Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Can Respir J. 2012;19(2):109–116. doi: 10.1155/2012/920918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stockley RA. Emerging drugs for alpha-1-antitrypsin deficiency. Expert Opin Emerg Drugs. 2010;15(4):685–694. doi: 10.1517/14728214.2010.512287. [DOI] [PubMed] [Google Scholar]