

Summary
The intestinal tract is inhabited by a large diverse community of microbes collectively referred to as gut microbiota. While gut microbiota provide important benefits to its host, especially in metabolism and immune development, disturbance of the microbiota-host relationship is associated with numerous chronic inflammatory diseases, including inflammatory bowel disease (IBD) and the group of obesity-associated diseases collectively referred to as metabolic syndrome. A primary means by which the intestine is protected from its microbiota is via multilayered mucus structures that cover the intestinal surface thus allowing the vast majority of gut bacteria to be kept at a safe distance from epithelial cells that line the intestine 1. Thus, agents that disrupt mucus-bacterial interactions might have the potential to promote diseases associated with gut inflammation. Consequently, it has been hypothesized that emulsifiers, detergent-like molecules that are a ubiquitous component of processed foods and that can increase bacterial translocation across epithelia in vitro 2, might be promoting the post-mid 20th century increase in IBD 3. Herein, we observed that, in mice, relatively low concentrations of two commonly used emulsifiers, namely carboxymethylcellulose and polysorbate-80, induced low-grade inflammation and obesity/metabolic syndrome in WT hosts and promoted robust colitis in mice predisposed to this disorder. Emulsifier-induced metabolic syndrome was associated with microbiota encroachment, altered species composition, and increased pro-inflammatory potential. Use of germ-free mice and fecal transplants indicated that such changes in microbiota were necessary and sufficient for both low-grade inflammation and metabolic syndrome. These results support the emerging concept that perturbed host-microbiota interactions resulting in low-grade inflammation can promote adiposity and its associated metabolic effects. Moreover, they suggest that broad use of emulsifying agents might be contributing to increased societal incidence of obesity/metabolic syndrome and other chronic inflammatory diseases.
Keywords: Mucus, inflammation, innate immunity, flagellin, LPS, IL10, TLR5
Mice were administered the emulsifiers carboxymethylcellulose (CMC) or polysorbate-80 (P80), via drinking water (1.0% w/v or v/v, respectively) for 12 weeks. P80 has been studied for toxicity and carcinogenic potential 2,4,5 and is approved by the U.S. food and drug administration (FDA) for use in select foods at up to 1.0%. CMC has not been extensively studied but is deemed “generally regarded as safe (GRAS)” and used in various foods at up to 2.0% 3,6. We utilized wild-type (WT) C57Bl/6 mice and 2 engineered strains of mice, namely IL10−/− and TLR5−/−, that are prone to developing shifts in microbiota composition and inflammation 7,8. While observations that loss of TLR function alters microbiota composition and promotes inflammation was suggested to be an artifact of mouse husbandry practices 9, that epithelial cell specific deletion of TLR5 altered microbiota composition relative to WT siblings and resulted in low-grade inflammation/metabolic syndrome indicates that, at least for TLR5, this is not the case 10.
Confocal microscopy, using mucus-preserving Carnoy fixation 1,11 indicated that the closest bacteria resided, on average, about 25 μm from epithelial cells with no bacteria observed within 10 μm (Fig. 1A, D). In contrast, emulsifier-treated mice exhibited some bacteria in apparent contact with the epithelium while the average distance was reduced by more than 50% (Fig. 1, B-D). Such microbiota encroachment correlated with reduced mucus thickness; not correlated with altered expression of Muc2 (Fig. S1A). Both IL10−/− and TLR5−/− mice exhibited basal microbiota encroachment that was enhanced by both CMC and P80 (Fig. 1, E-H and S1B). (Fig. 1, E-H). Emulsifiers did not affect total levels of fecal bacteria in WT, IL10−/−, or TLR5−/− mice but resulted in more than a 2-fold increase in bacteria adherent to the colons of WT and IL10−/− mice (Fig. 1I, K and S1C). Moreover, interrogation of microbiota composition via 16S RNA sequencing, followed by phylogenic analysis, and use of the unweighted UniFrac algorithm to compare community structure 12, revealed that both CMC and P80 dramatically altered microbiota composition in both fecal and intestinal-adherent bacteria of WT, IL10−/−, and TLR5−/− mice (Fig. 1J, L and S2, Tables S1 and S2).
Figure 1. Dietary emulsifiers alter microbiota localization, composition, and pro-inflammatory potential.

WT and IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A-C, E-G) Confocal microscopy analysis of microbiota localization; Muc2 (green), actin (purple), bacteria (red), and DNA (Blue). Bar = 20μm. (D, H) Distances of closest bacteria to intestinal epithelial cells (IEC) per condition over 5 high-powered fields per mouse. Pictures are representatives of 20 biological replicates. (I, K) PCR-based quantification of bacterial load adhered to colonic mucosa. (J, L). Principal coordinates analysis (PCoA) of the UniFrac distance matrix of WT (J) and IL10−/− (L) mice. Black dashed ellipses indicate mice sharing a cage during treatment. (M-P) Bioactive levels of fecal flagellin and LPS assayed with TLR5 and TLR4 reporter cells. (Q-R) Intestinal permeability measured by levels of serum FITC-dextran (4kDa) following oral gavage. Data are the means +/- S.E.M. or geometric means with 95% confidence interval (for I and K) (n=20 for A-H and M-P; n=6 for I except n=8 for P80-treated group; n=5 for J except n=4 for P80-treated group; n=6 for K except n=7 for water-treated group; for L n=10, 4 and 5 for water, CMC and P80-treated groups, respectively; n=14 for Q, for R n=10, 11 and 15 for water, CMC and P80-treated groups, respectively). Points in I-L and Q-R are from individual mice. Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group.
In order to verify that differences in microbiota composition were a consequence of emulsifier treatment rather than simply reflecting that mice shared cages during treatment, which results in microbiota composition clustering in coprophagic animals, multiple litters were split equally into groups that were to receive water, CMC or P80 (3 separate cages per condition as schematized in Fig S3). As expected, this experimental design avoided pre-treatment clustering. Yet, microbiota composition clustered strongly following treatment irrespective of cage clustering in both male and female mice in WT and TLR5−/− genotypes (Fig. S3), confirming that emulsifiers alter microbiota composition.
Alterations in microbiota composition included reduced levels of Bacteroidales, associated with health13-15, and increased levels several mucolytic OTU including Ruminococcus gnavus 16 (Fig. S2, S4, Tables S1 and S2). In IL10−/− mice, both CMC and P80 induced a marked reduction in microbial diversity, bloom in Verrucomicrobia phyla, especially Akkermansia muciniphila (Fig. S2D, Tables S1 and S2) 17, and enriched mucosa-associated inflammation-promoting Proteobacteria 18,19. Emulsifier-induced changes in microbiota composition were sufficiently uniform such that a low number of OTUs (<15) afforded reliable prediction of emulsifier treatment (Fig. S4, Tables S1 and S2). Such distinguishing OTUs were spread across the Bacteria domain and several were common to all 3 host genotypes.
While most experiments herein utilized young mice (4 weeks-old at start of experiment) based on the notion that microbiota are more prone to disturbance at an early age 18,20, administration of emulsifiers beginning at 4 months of age also destabilized and altered microbiota composition (Fig. S5A-B). Specifically, both CMC and P80 resulted in decreased alpha diversity and reduced stability, as evidenced by increased extent of week-to-week changes in principal coordinates, and greater extent of change over the course of the experiment that outweighed effects of cage clustering (Fig. S5A-B). Thus, emulsifiers can alter gut microbiota composition of hosts of a broad age range.
Microbiota composition influences ability of a microbiota to activate innate immune signaling 21. Thus, we measured the capacity of feces from control and emulsifier-treated mice to activate pro-inflammatory gene expression via the LPS and flagellin receptors TLR4 and 5, respectively. Exposure to emulsifiers increased fecal levels of bioactive LPS and flagellin in WT, IL10−/−, and TLR5−/− mice (Fig. 1M-P and S5C). Emulsifier treatment also increased gut permeability in WT and IL10−/− mice (Fig. 1Q-R), which correlated with increased levels of serum antibodies to flagellin and LPS (Fig. S5D) thought to reflect gut permeability 22. Thus, chronic exposure to dietary emulsifiers results in erosion of the protective function of the mucus, increased bacterial adherence, and a more pro-inflammatory microbiota.
The hallmark of active colitis is the presence of immune cell infiltrates, which is typically paralleled by changes in gross colon morphology, elevated levels of the leukocyte enzyme myeloperoxidase (MPO), and pro-inflammatory markers. Based on such criteria, emulsifiers promoted the extent and incidence of colitis in both IL10−/− and TLR5−/− mice (Fig. 2A-E, S6 and S7A). Emulsifiers did not induce overt colitis in WT mice but did result in subtle histopathologic and gross evidence of chronic intestinal inflammation including epithelial damage and shortened colons (Fig. 2F-I, and S6). Fecal lipocalin-2 (Lcn-2) is a sensitive and broadly dynamic marker of intestinal inflammation in mice 23. Emulsifier-treated WT mice exhibited modestly-elevated fecal Lcn-2 levels 4 weeks following initial exposure (Fig. 2F). In IL10−/− and TLR5−/− mice, basally elevated fecal Lcn-2 levels were further markedly elevated (about 10-fold) by 12 weeks of exposure to CMC and P80 (Fig. 2A and S7A). Such robust colitis in IL10−/−, but not TLR5−/−, correlated with Bilophila and Helicobacter enrichment, analogous to previous observations in IL10−/− mice (Tables S2 and S3) 24,25. The extent of inflammation inversely correlated with bacterial-epithelial distance in both WT and IL-10−/− mice (Fig. 2J and S6E). Thus, emulsifiers may promote robust colitis in susceptible hosts and induce low-grade inflammation in WT hosts.
Figure 2. Dietary emulsifiers promote colitis in susceptible mice and low-grade intestinal inflammation in WT mice.

(A-E) IL10−/− and (F-I) WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A, F) Fecal levels of the inflammatory marker Lcn2 over time, (B) colitis incidence over time, (C, G) MPO levels, (D, H) colon lengths and (E, I) histological score after 12 weeks of exposure. (J) Fecal levels of the inflammatory marker Lcn2 plotted vs. microbiota-epithelial distance obtained in figure 1D. Data are the means +/- S.E.M. or geometric means with 95% confidence interval (for A and F) (n=20). Points are from individual mice. Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group.
Metabolic syndrome is associated with, and may be promoted by, low-grade inflammation 26, Thus, we examined whether emulsifier-induced microbial dysbiosis and low-grade inflammation might promote this disorder in WT mice. Both CMC and P80 resulted in modest but significant gains in overall weight and a marked increase in adiposity as measured by fat mass (Fig. 3A-B). Such increased adiposity was associated with increased food consumption (Fig. 3C) that likely drove the increased adiposity. Emulsifier treatment also impaired glycemic control as assessed by fasting blood glucose concentration (Fig. 3D) and glucose/insulin tolerance testing (Fig. S7B).
Figure 3. Dietary emulsifiers promote metabolic syndrome.

(A-D) WT Mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A) Body weight over time, (B) fatpad mass, (C) food intake, and (D) 15-hours fasting blood glucose concentration. (E-F) WT and (G-H) TLR5−/− mice were given mouse chow containing CMC or P80 (1.0%) for 12 weeks. (E, G) Body weights over time, (F, H) fat-pad mass. WT mice were exposed to drinking water containing 0.1-1.0% CMC (I-J) or P80 (K-L) for 12 weeks. (I, K) Body weight over time, (J, L) fat-pad mass. Data are the means +/- S.E.M. (n=20 for A-D, n=5 for E-L). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05) or 2-way group ANOVA corrected for multiple comparisons with Bonferroni test (# indicates p<0.05) compared to control group.
Human exposure to dietary emulsifiers occurs in liquid and solid foods. Hence, we next examined whether incorporating emulsifiers into mouse chow would drive similar phenotypes. Supplementation of chow with 1.0% CMC or P80 fully mimicked the pro-inflammatory effects and metabolic changes induced by emulsifiers in drinking water (Fig. 3E-F and S7C). While human emulsifier consumption is not a widely tracked parameter, their pervasive use in many foods exceed the 1.0% level (in food or water) used herein 6. Nonetheless, we next sought to better define the minimum doses of emulsifiers that would produce evidence of low-grade inflammation/metabolic syndrome. As little as 0.1% CMC resulted in modest increases in body weight and fasting glucose while 0.5% resulted in clear evidence of low-grade inflammation (shortened colon, enlarged spleen) and increased adiposity (Fig. 3I-J and S7D). For P80, as little as 0.1% resulted in evidence of low-grade inflammation and increased adiposity, while 0.5% resulted in mild dysglycemia (Fig. 3K-L and S7D). Emulsifier-induced metabolic syndrome was also observed in older mice (Fig. S5A) and persisted for at least 6 weeks after stopping emulsifier consumption (Fig. S8A). Promotion of metabolic syndrome was not seen upon exposure to sodium sulfite, which is a common food additive but not an emulsifier (Fig. S8B). TLR5−/− mice are prone to developing metabolic syndrome 27, which results from poor microbiota management 18. Emulsifiers markedly promoted multiple parameters of metabolic syndrome in TLR5−/− mice (Fig. S7B and S8C) including hyperphagia, increased adiposity, and glucose dysregulation, and was observed upon supplementation of chow or drinking water with as little as 0.1% P80 (Fig. 3G-H and S8D,E). A trend toward development of metabolic syndrome in emulsifier-treated non-colitic IL10−/− mice was also observed, particularly evident upon exclusion of colitic mice, which exhibited weight loss (Fig. S9A). Emulsifier-induced metabolic syndrome was observed in mice from multiple vivaria and strains, including Swiss-Webster, which, in contrast to C57BL/6 mice, are considered obesity-resistant 28 (Fig. 4A-D and S9B, D).
Figure 4. Altered microbiota is necessary and sufficient for emulsifier-induced metabolic syndrome.

(A-D) Conventionally-housed and germ-free (E-H) Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A, E) Body weight over time, (B, F) fat-pad mass, (C, G) food intake, (D, H) 15-hours fasting blood glucose concentration. (I-P) Germ-free Swiss-Webster mice were conventionalized via microbiota transplant from mice that received standard drinking water or drinking water containing CMC or P80 (1.0%). (I-K) Confocal microscopy analysis of microbiota localization; Muc2 (green), actin (purple), bacteria (red), and DNA (Blue). Bar = 20μm. (L) Distances of closest bacteria to intestinal epithelial cells per condition over 5 high-powered fields per mouse. Pictures are representatives of 10 biological replicates. (M) Body weight over time, (N) fat-pad mass, and (O) 15-hours fasting blood glucose concentration. Data are the means +/- S.E.M. (n=5 for A-L, n=3 for M-P). Points are from individual mice. Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05) or 2-way group ANOVA corrected for multiple comparisons with Bonferroni test (# indicates p<0.05) compared to control group.
Emulsifier-induced low grade inflammation and metabolic syndrome required the presence of a microbiota in that administration of emulsifiers to germ-free mice resulted in neither low-grade inflammation, as assessed by fecal Lcn-2, colon length and splenomegaly, nor all measured parameters of metabolic syndrome including body mass, fat mass, food intake and fasting glucose levels (Fig. 4E-H and S9B). This result could reflect direct action of CMC and P80 on gut bacteria composition or metabolism. In accord with both of these possibilities, emulsifiers altered fecal levels of short-chain fatty acids (Fig. S9C), including decreased levels of butyrate, which is thought to play a key role in dampening inflammation 29. Emulsifiers also altered bile acid levels (Fig. S9E), which influence microbiota composition 24. However, in germ free mice, emulsifiers did not alter bile acids, reduce mucus thickness, nor alter penetrance of 0.5 μm beads into the mucus (Fig. S9E and S10A-B) suggesting these changes are not purely a direct effect of emulsifiers on mucus structures. Collectively, these data suggest alterations in mucus in emulsifier-treated mice might result, at least in part, from altered gut microbiota composition but do not exclude direct effects on the host.
Transfer of microbiota from emulsifier-treated mice to germ-free mice (not exposed to emulsifiers) transferred microbiota encroachment, low-grade inflammation (Fig. S10D), increased adiposity, and dysglycemia (Fig. 4M-O). Microbiota transplant from CMC-treated resulted in a more lasting increase in adiposity, paralleling that adiposity was less rapidly reversible following stoppage of CMC exposure (Fig. S8A). Such transplanted phenotypes correlated with acquisition of elevated fecal LPS and flagellin (Fig. S10C) and altered microbiota composition (Fig. S10E). Thus, emulsifier-induced changes in microbiota play a role in driving the inflammation and metabolic changes promoted by these food additives.
The last half-century has witnessed a steady increase in consumption of food additives, many of which have not been carefully tested as they were given GRAS status at the time government entities charged with regulating food safety were created and/or expanded. Moreover, the testing of food additives that has been performed has generally utilized animal models designed to detect acute toxicity and/or promotion of cancer. Our data herein suggests that such testing may be inadequate - a notion supported by the recent observation that artificial sweeteners induce dysglycemia in humans 30. More specifically, our data suggests that one ubiquitous class of food additives, namely emulsifiers, can disturb the host-microbiota relationship resulting in a microbiota with enhanced mucolytic and pro-inflammatory activity that promotes intestinal inflammation. Such chronic gut inflammation can manifest as colitis or metabolic syndrome. While additional studies will be needed to determine if CMC, P80, and/or other emulsifiers impact human health, our observations in mice suggest the possibility that dietary emulsifiers may have contributed to the post-mid 20th century increased incidence of IBD, metabolic syndrome, and perhaps other chronic inflammatory diseases. Importantly, this hypothesis does not dispute the commonly held assumption that excess caloric consumption is a predominant factor driving the metabolic syndrome epidemic. Rather, it suggests such hyperphagia may be driven, in part, by food additives and other factors that might alter gut microbiota and promote low-grade intestinal inflammation.
METHODS
Mice
WT C57BL/6 and IL10−/− mice were purchased from Jackson Laboratories. Swiss-Webster mice were purchased from Taconic, Inc. TLR5−/− were originally generated by Dr. Shizuo Akira (Osaka University, Japan) 31 and backcrossed/maintained as previously described 27. All mice were then bred and housed at Georgia State University, Atlanta, Georgia, USA under institutionally-approved protocols (IACUC # A14033). For WT mice, experiments were also performed on mice purchased directly from the above-mentioned providers and used within 3 days of receipt. Mice were fed Purina rodent chow # 5001, which is commonly used in many vivaria. Mice were weaned at 3 weeks of age and put into new cages (randomized to littermates) such that each experimental group contained mice from at least 2 different litters, and that each litter were used for at least 3 experimental groups. Experiments used either male or female mice (comparisons within a gender) except for figures 1M-P and 2 that utilized both male and female mice. For littermate control experiments (Fig. S7 and S8), all the mice from 10 (S7) and 8 (S8) different litters (from distinct mothers) were used and placed into cages such that each litter was split equally amongst groups that were to receive water, CMC or P80 (3 (S7) or 2 (S8) cages per sex per condition). For experiments using older mice (Fig. S13 and S14), mice were weaned at 3 weeks of age, put into new cages, and subjected to emulsifier treatment starting 3 months later.
Materials
Sodium carboxymethylcellulose (CMC, average MW~250,000), Polysorbate-80 (P80) and Sodium Sulfite were purchased from Sigma (Sigma, St. Louis, MO).
Emulsifier agent treatment
Mice were exposed to CMC, P80 or sodium sulfite diluted in the drinking water (1.0% or indicated concentration) (not blinded). The same water (reverse-osmosis treated Atlanta city water) was used for the water-treated (control) group. These solutions were changed every week. Emulsifier solutions were autoclaved for experiments presented in figure 4A-H. When required, emulsifier solutions were replaced by water after 8 weeks of treatment for an additional 6 weeks. Body weights were measured every week and are expressed as % compare to the initial body weight (day 0) defined as 100%. Mice developing colitis (Lcn2 level ≥ 500 ng/g of feces) were excluded for metabolic syndrome parameters analysis. Fresh feces were collected every week for downstream analysis. After 3 months of emulsifier treatment, mice were fasted for 5-h at which time blood was collected by retrobulbar intraorbital capillary plexus. Hemolysis-free serum was generated by centrifugation of blood using serum separator tubes (Becton Dickinson, Franklin Lakes, NJ). Mice were then euthanized, and colon length, colon weight, spleen weight and adipose weight were measure. Organs were collected for downstream analysis. Incorporation of CMC and P80 in the diet was performed by Research Diets Inc. (New Brunswick, NJ, USA) using Purina Rodent Chow diet #5001. These diets are now referenced by Research Diets as C13050701 and C13050702, respectively.
Food intake measurement
Groups of mice were placed in a clean cage with a known amount of food. Twenty-four hours later, amount of remaining food was measured with the difference viewed as food intake per 24h. Error bars reflect S.E.M. of 3 measurements made one week apart.
Overnight fasting blood glucose measurement
Mice were placed in a clean cage and fasted for 15h. Blood glucose concentration was then determined using a Nova Max Plus Glucose Meter and expressed in mg/dL.
Glucose and insulin tolerance test
WT and TLR5−/− mice were treated with CMC or P80 diluted to 1% drinking water for 8 weeks. Followed a 5h fasting, baseline blood glucose levels were measured using a Nova Max Plus Glucose Meter and expressed in mg/dL. Mice were then injected intraperitoneally with 2 g glucose/kg body weight in sterile PBS or with 0.5 U insulin/kg body weight (Sigma, St. Louis, MO), and blood glucose levels were measured 30, 60, 90, and 120 min after injection, as previously described 27.
In vivo epithelial barrier permeability
In vivo assay of intestinal barrier function was performed using an FITC-labeled dextran method, as previously described 32. Mice were deprived of food and water for 4 hours, and were then gavaged with 15mg of permeability tracer FITC–labeled dextran 4 kDa (FD4, Sigma, St. Louis, MO). Blood was collected retro-orbitally after 3 hours, and fluorescence intensity was measured in the serum (excitation, 490 nm; emission, 520 nm; BIOTEK Fluorescence Spectrophotometer). FITC-dextran concentrations were determined using a standard curve generated by serial dilution of FITC-dextran in mice serum.
Germ-free experiments
Germ-free Swiss Webster mice were kept under germ-free conditions in a Park Bioservices isolator in our germ-free facility. CMC and P80 were diluted to 1% in water and then autoclaved for germ-free purpose. Same water was used for the water-treated (control) group. After 3 months of emulsifier agent treatment, mice were fasted for 5-h and then removed from isolator to be euthanized. Samples were collected as previously described. For the analysis of mucus layer integrity, germ-free C57BL/6 mice were kept under germ-free conditions and treated with CMC or P80 as described above. After 2 months of emulsifier agent treatment, mice were removed from isolator, and inoculated with 0.5 μm green fluorescent beads (Polysciences, Warrington, PA). Seven hours post inoculation, mice were euthanized, colonic tissue were collected and placed in methanol-Carnoy's fixative solution, followed by immunostaining of mucins, as described below.
Microbiota transplantation
Cecal contents from detergent treated mice were suspended in 30% glycerol diluted in PBS (1.0 ml) and stocked at −80°C until analysis. Germ-free Swiss Webster mice (4 weeks old) were removed from isolator and were orally administered with 200μL of fecal suspension made using glycerol stocks. Transplanted mice were then monitored as previously described.
Colonic myeloperoxidase (MPO) assay
Neutrophil influx in tissue was analyzed by assaying the enzymatic activity of MPO, a marker for neutrophils. Briefly, tissue (50 mg/mL) were thoroughly washed in PBS and homogenized in 0.5% hexadecyltrimethylammonium bromide (Sigma, St. Louis, MO) in 50 mM PBS, (pH 6.0), freeze-thawed 3 times, sonicated and centrifuged. MPO was assayed in the clear supernatant by adding 1 mg/mL of dianisidine dihydrochloride (Sigma, St. Louis, MO) and 5×10−4% H2O2 and the change in optical density measured at 450 nm. Human neutrophil MPO (Sigma, St. Louis, MO) was used as standard. One unit of MPO activity was defined as the amount that degraded 1.0 μmol of peroxide/min at 25°C 33.
H&E staining and histopathologic analysis
Following euthanasia, mouse colons and small intestines were fixed in 10% buffered formalin for 24 hours at room temperature and then embedded in paraffin. Tissues were sectioned at 5-μm thickness and stained with hematoxylin & eosin (H&E) using standard protocols. H&E stained slides were scored as follows. Each colon was assigned four scores based on the degree of epithelial damage and inflammatory infiltrate in the mucosa, submucosa and muscularis/serosa, as previously described 34. A slight modification was made to this scoring system 23; each of the four scores was multiplied by 1 if the change was focal, 2 if it was patchy and 3 if it was diffuse. The 4 individual scores per colon were added, resulting in a total scoring range of 0–36 per mouse.
Periodic Acid Schiff Staining
Following euthanasia, mouse colons were fixed in 10% buffered formalin for 24 hours at room temperature and then embedded in paraffin. Tissues were sectioned at 5-μm thickness, deparaffinized and oxidized for 5 minutes in 0.5% periodic acid solution (Sigma, St. Louis, MO). After rinsing, tissues were placed in Schiff reagent (Sigma, St. Louis, MO) for 15 minutes, washed in warm water, and counterstained using hematoxylin (Sigma, St. Louis, MO) for one minute.
Immunostaining of mucins and localization of bacteria by FISH
Mucus immunostaining was paired with fluorescent in situ hybridization (FISH), as previously described 11, in order to analyze bacteria localization at the surface of the intestinal mucosa. Briefly, colonic tissues (proximal colon, 2nd cm from the cecum) containing fecal material were placed in methanol-Carnoy's fixative solution (60% methanol, 30% chloroform, 10% glacial acetic acid) for a minimum of 3 h at room temperature. Tissue were then washed in methanol 2 × 30 min, ethanol 2 × 15 min, ethanol/xylene (1:1) 15 min and xylene 2 × 15 min, followed by embedding in Paraffin with a vertical orientation. Five μm sections were performed and dewax by preheating at 60°C for 10 min, followed by xylene 60°C for 10 min, xylene for 10 min and 99.5% ethanol for 10 minutes. Hybridization step was performed at 50°C overnight with EUB338 probe (5’-GCTGCCTCCCGTAGGAGT-3’, with a 5' labeling using Alexa 647) diluted to a final concentration of 10 μg/mL in hybridization buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl, 0.1% SDS, 20% formamide). After washing 10 min in wash buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl) and 3 × 10 min in PBS, PAP pen (Sigma, St. Louis, MO) was used to mark around the section and block solution (5% fetal bovine serum in PBS) was added for 30 min at 4°C. Mucin-2 primary antibody (rabbit H-300, Santa Cruz Biotechnology) was diluted 1:1500 in block solution and apply overnight at 4°C. After washing 3 × 10 min in PBS, block solution containing anti-rabbit Alexa 488 secondary antibody diluted 1:1500, Phalloidin-Tetramethylrhodamine B isothiocyanate (Sigma, St. Louis, MO) at 1μg/mL and Hoechst 33258 (Sigma, St. Louis, MO) at 10μg/mL was applied to the section for 2h. After washing 3 × 10 min in PBS slides were mounted using Prolong anti-fade mounting media (Life Technologies). Observations were performed with a Zeiss LSM 700 confocal microscope with software Zen 2011 version 7.1. This software was used to determine the distance between bacteria and epithelial cell monolayer, as well as the mucus thickness.
Quantification of fecal Lcn-2 by ELISA and determination of colitis incidence
For quantification of fecal Lcn-2 by ELISA, frozen fecal samples were reconstituted in PBS containing 0.1% Tween 20 to a final concentration of 100 mg/mL and vortexed for 20 min to get a homogenous fecal suspension 23. These samples were then centrifuged for 10 min at 14 000 g and 4°C. Clear supernatants were collected and stored at −20°C until analysis. Lcn-2 levels were estimated in the supernatants using Duoset murine Lcn-2 ELISA kit (R&D Systems, Minneapolis, Minnesota, USA) using the colorimetric peroxidase substrate tetramethylbenzidine, and optical density (OD) was read at 450 nm (Versamax microplate reader). For determination of colitis incidence, a fecal Lcn2 level ≥ 500 ng/g of feces was use to determine colitic mice.
Fecal flagellin and lipopolysaccharide load quantification
We quantified flagellin and lipopolysaccharide (LPS) as previously described 21 using human embryonic kidney (HEK)-Blue-mTLR5 and HEK-BluemTLR4 cells, respectively (Invivogen, San Diego, California, USA). We resuspended fecal material in PBS to a final concentration of 100 mg/mL and homogenized for 10 s using a Mini-Beadbeater-24 without the addition of beads to avoid bacteria disruption. We then centrifuged the samples at 8000 g for 2 min and serially diluted the resulting supernatant and applied to mammalian cells. Purified E. coli flagellin and LPS (Sigma, St Louis, Missouri, USA) were used for standard curve determination using HEK-Blue-mTLR5 and HEK-Blue-mTLR4 cells, respectively. After 24 h of stimulation, we applied cell culture supernatant to QUANTI-Blue medium (Invivogen, San Diego, California, USA) and measured alkaline phosphatase activity at 620 nm after 30 min.
Serum flagellin and LPS-specific immunoglobulins
Flagellin- and LPS-specific IgA and IgG levels were quantified by ELISA, as previously reported 22,35. Microtiter plates were coated overnight with purified E. coli flagellin (100 ng/well) or LPS (1 μg/well). Serum samples diluted 1:500 were then applied. After incubation and washing, wells were incubated either with anti-mouse IgA or anti-mouse IgG. Quantification was performed using the colorimetric peroxidase substrate tetramethylbenzidine, as described above. Data are reported as OD corrected by subtracting background (determined by readings in samples lacking serum).
Gene expression analysis by q-RT-PCR
Total RNAs were isolated from colonic tissues using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Quantitative RT-PCR was performed using the iScriptTM One-Step RT-PCR Kit with SYBR Green (Bio-Rad, Hercules, CA) in a CFX96 apparatus (Bio-Rad, Hercules, CA) with specific mouse oligonucleotides. The sense and antisense oligonucleotides used were, respectively, the following: 36B4 5’-TCCAGGCTTTGGGCATCA-3’ and 5’-CTTTATTCAGCTGCACATCACTCAGA-3’, Tff3 5’-CCTGGTTGCTGGGTCCTCTGG-3’ and 5’-GTCTCCTGCAGAGGTTTGAAGC-3’, Klf4F 5’-TGTGACTATGCAGGCTGTGG-3’ and 5’-AGTGCCTGGTCAGTTCATCG-3’. Results were normalized to the housekeeping 36B4 gene.
Bacterial quantification by q-PCR
For quantification of total fecal bacterial load, total bacterial DNA was isolated from weighted feces using QIAamp DNA Stool Mini Kit (Qiagen). DNA was then subjected to quantitative PCR using QuantiFast SYBR Green PCR kit (Biorad) with universal 16S rRNA primers 8F: 5’-AGAGTTTGATCCTGGCTCAG-3’ and 338R: 5’-CTGCTGCCTCCCGTAGGAGT-3’ to measure total bacteria 36. Results are expressed as bacteria number per mg of stool, using a standard curve. For quantification of mucosa-associated bacteria, total DNA was isolated from PBS-washed and weighted colonic tissue using DNeasy Blood & Tissue Kit (Qiagen). DNA was then subjected to quantitative PCR as described above, and results are expressed as bacteria number per mg of colonic tissue, using a standard curve.
Fecal microbiota analysis by 16S rRNA gene sequencing using Illumina technology
16S rRNA gene amplification and sequencing were done using the Illumina MiSeq technology following the protocol of Earth Microbiome Project with their modifications to the MOBIO PowerSoil DNA Isolation Kit procedure for extracting DNA (www.earthmicrobiome.org/emp-standard-protocols) 37,38. Bulk DNA were extracted from frozen extruded feces using a PowerSoil-htp kit from MoBio Laboratories (Carlsbad, California, USA) with mechanical disruption (bead-beating). The 16S rRNA genes, region V4, were PCR amplified from each sample using a composite forward primer and a reverse primer containing a unique 12-base barcode, designed using the Golay error-correcting scheme, which was used to tag PCR products from respective samples 38. We used the forward primer 515F 5’-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGT AA-3’: the italicized sequence is the 5’ Illumina adapter B, the bold sequence is the primer pad, the italicized and bold sequence is the primer linker and the underlined sequence is the conserved bacterial primer 515F. The reverse primer 806R used was 5’-CAAGCAGAAGACGGCATACGAGAT XXXXXXXXXXXX AGTCAGTCAG CCGGACTACHVGGGTWTCTAAT-3’: the italicized sequence is the 3’ reverse complement sequence of Illumina adapter, the 12 X sequence is the golay barcode, the bold sequence is the primer pad, the italicized and bold sequence is the primer linker and the underlined sequence is the conserved bacterial primer 806R. PCR reactions consisted of Hot Master PCR mix (Five Prime), 0.2 μM of each primer, 10-100 ng template, and reaction conditions were 3 min at 95°C, followed by 30 cycles of 45 s at 95°C, 60s at 50°C and 90 s at 72°C on a Biorad thermocycler. Four independent PCRs were performed for each sample, combined, purified with Ampure magnetic purification beads (Agencourt), and products were visualized by gel electrophoresis. Products were then quantified (BIOTEK Fluorescence Spectrophotometer) using Quant-iT PicoGreen dsDNA assay. A master DNA pool was generated from the purified products in equimolar ratios. The pooled products were quantified using Quant-iT PicoGreen dsDNA assay and then sequenced using an Illumina MiSeq sequencer (paired-end reads, 2 × 250 bp) at Cornell University, Ithaca.
16S rRNA gene sequence analysis
The sequences were demultiplexed, quality filtered using Quantitative Insights Into Microbial Ecology (QIIME, version 1.8.0) software package 39, and forward and reverse Illumina reads were joined using the fastq-join method 40,41. We used QIIME default parameters for quality filtering (reads truncated at first low-quality base and excluded if: (1) there were more than three consecutive low quality base calls (2), less than 75% of read length was consecutive high quality base calls (3), at least one uncalled base was present (4), more than 1.5 errors were present in the bar code (5), any Phred qualities were below 20, or (6) the length was less than 75 bases). Sequences were assigned to operational taxonomic units (OTUs) using UCLUST algorithm 42 with a 97% threshold of pairwise identity (without the creation of new clusters with sequences that do not match the reference sequences), and classified taxonomically using the Greengenes reference database 43. A single representative sequence for each OTU was aligned and a phylogenetic tree was built using FastTree 44. The phylogenetic tree was used for computing the unweighted UniFrac distances between samples 12,45, rarefaction were performed (3,500 – 20,000 sequences per sample) and used to compare abundances of OTUs across samples. Principal coordinates analysis (PCoA) plots were used to assess the variation between experimental group (beta diversity) and Jackknifed beta diversity was used to estimate the uncertainty in PCoA plots. In addition to using PCoA, samples were clustered using UPGMA (Unweighted Pair Group Method with Arithmetic mean). Alpha diversity curves were determined for all samples using the determination of the number of observed species, and Shannon diversity index was used to characterize species diversity in a community. Our full 16S rRNA gene sequence data are deposited under Study ID XXXX and YYYY in the QIIME-DB database (http://www.microbio.me/qiime/).
Short-chain fatty acids (SCFA) and bile acids composition analysis
Fecal samples were used to analyze SCFA and bile acid composition at Metabolomics Core of the University of Michigan (supported by grant U24 DK097153 of NIH Common Funds Project to the University of Michigan).
Statistical analysis
All replicates in this study were biological; i.e. repeat experiments with additional mice. D'Agostino-Pearson omnibus test was used to verify that all data were normally distributed. Significance was determined using t-test, one-way ANOVA corrected for multiple comparisons with Sidak test, two-way ANOVA corrected for multiple comparisons with Bonferroni test, or two-way group ANOVA (GraphPad Prism software, version 6.01). Differences were noted as significant *p≤0.05. A ‘nearest-shrunken centroid’ classification approach was performed to detect the OTUs that were particularly representative of each experimental group 46. The amount of shrinkage was set to minimize the misclassification error. When OTUs were used to classify samples, accuracy of the classification was perfect (overall error rate=0). These analyses allowed the identification of the 15 OTUs whose abundances were the most significantly different between experimental groups, which were then used for heatmap generation and sample clustering. These analyses were performed using the Prediction Analysis for Microarrays (PAM) package within R software 46.
Extended Data
Extended data figure 1.

(A-D) Dietary emulsifiers did not affect mucus and mucus-related genes expression in WT mice. WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A-C) mRNA expression analysis by q-RT-PCR of Muc2 (A), Tff3 (B) and Klf4 (C) genes in the colonic mucosa. Points are from individual mice, bar represent the mean +/- S.E.M, (n=9). (D) Colons were stained using Periodic Acid Schiff. Bar = 200μm.Pictures are representatives of 10 biological replicates. (E-J) Dietary emulsifiers alter microbiota localization, composition, and pro-inflammatory potential in TLR5−/− mice. TLR5 −/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (E-G) Confocal microscopy analysis of microbiota localization; Muc2 (green), actin (purple), bacteria (red), and DNA (Blue). Bar = 20μm. (H) Distances of closest bacteria to intestinal epithelial cells per condition over 5 high-powered fields per mouse. Pictures are representatives of 5 biological replicates. (I-J) PCR-based quantification of total bacterial load (I) and bacterial load adhered to colonic mucosa (J). Data are the means +/- S.E.M., (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group. (K-L) Dietary emulsifiers do not modify total bacterial load in WT and IL10−/− mice. WT (K) and IL10−/− (L) mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (K) Total bacterial load in stool of WT and (L) IL10−/− mice. Points are from individual mice. Data are geometric means with 95% confidence interval (n=5 for K except n=4 for CMC and P80-treated groups; for L, n=8, 4 and 6 for water, CMC and P80-treated groups, respectively).
Extended data figure 2.

(A-H) Dietary emulsifiers induce profound alterations in gut microbiota composition in WT and IL10−/− mice. WT and IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A, E) Day 0 and (B, F) day 93 microbiota richness and diversity in WT (A, B) and IL10−/− (E, F). (C-D, G-H) Principal coordinates analysis (PCoA) of the unweighted UniFrac distance matrix of fecal microbiota (C, G) and mucosa-associated bacteria (D, H) in WT (C-D) and IL10−/− (G-H) mice. Treatment of each mouse is indicated by point color and matching colored circles represent clustering by treatment (blue = water; orange = CMC; purple = P80). Black dashed circles represent mice sharing a cage. Data are the means +/- S.E.M. (n=5, except n=4 for WT mice P80-treated, n=4 for IL10−/− mice CMC-treated and n=9 for IL10−/− mice water-treated). Significance was determined using 2-way group ANOVA (# indicates p<0.05) compared to water-treated group. (I-J) Phylum characterization of emulsifier-induced alteration of gut microbiota composition in WT and IL10−/− mice. WT and IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Relative abundance of phyla are represented for fecal microbiota at day 93 (I) and for colonic mucosa associated bacteria (J). Data are the means +/- S.E.M. (n=5). Significance was determined using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group. (K-O)Dietary emulsifiers induce profound alterations in gut microbiota composition in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (K) Day 0 and (L) day 93 microbiota richness and diversity (n=5). (M-O) PCoA of the unweighted UniFrac distance matrix of fecal microbiota at day 0 (M), day 93 (N) and of mucosa-associated bacteria (O). Data are the means +/- S.E.M. (for M, n=4, 5 and 5 for water, CMC and P80-treated groups, respectively; for N, n=4, 5 and 3 for water, CMC and P80-treated groups, respectively; for O, n=4). Significance was determined using 2-way group ANOVA (# indicates p<0.05) compared to water-treated group. (P-T) Prevalence analysis of OTUs related to mucolytic bacteria in IL10−/− mice treated with dietary emulsifier. IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. OTUs (P) Prok_MSA # 52947 (related to Clostridium perfringens), (Q) 264696 (related to Akkermansia muciniphila), (R) 315982 (related to Clostridium perfringens), (S) 363731 (related to Akkermansia muciniphila), and (T) 178331(related to Akkermansia muciniphila) were analyzed. Data are expressed as % of the total sequences analyzed and are the means +/- S.E.M. (n=6, except n=9 for IL10−/− mice water-treated). Significance was determined using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group.
Extended data figure 3.

(A-G) Dietary emulsifiers alter gut microbiota composition in WT mice relative to littermate controls. All the female (n=33, A-B) and male (n=33, C-D) mice from 10 different litters were placed into cages in a manner such that each litter was split equally amongst the 3 experimental groups that were to receive water, CMC or P80 (3 cages per sex per condition). Mice were exposed to drinking water containing CMC or P80 (1.0%) for 8 weeks. (A, C) Day 0 and (B, D) day 63 PCoA of the unweighted UniFrac distance matrix of fecal microbiota in WT mice. Treatment of each mouse is indicated by point color (blue = water; orange = CMC; purple = P80). Mother of each mouse is indicated by point color. Cage of each mouse is indicated by point color. (E-F) Mice were clustered using UPGMA (Unweighted Pair Group Method with Arithmetic mean) method. Treatment of each mouse is indicated by line color (blue = water; orange = CMC; purple = P80). (G) Schematic representation of the above experimental design. (H-N) Dietary emulsifiers alter gut microbiota composition in TLR5−/− mice relative to littermate controls. All the female (n=25, H-I) and male (n=23, J-K) mice from 8 different litters were placed into cages in a manner such that each litter was split equally amongst the 3 experimental groups that were to receive water, CMC or P80 (2 cages per sex per condition). Mice were exposed to drinking water containing CMC or P80 (1.0%) for 8 weeks. (H, J) Day 0 and (I, K) day 63 PCoA of the unweighted UniFrac distance matrix of fecal microbiota in TLR5−/− mice. Treatment of each mouse is indicated by point color (blue = water; orange = CMC; purple = P80). Mother of each mouse is indicated by point color. Cage of each mouse is indicated by point color. (L-M) Mice were clustered using UPGMA (Unweighted Pair Group Method with Arithmetic mean) method. Treatment of each mouse is indicated by line color (blue = water; orange = CMC; purple = P80). (N) Schematic representation of the above experimental design.
Extended data figure 4.

(A-D) Misclassification error rate and heatmap representation of the 15 most significantly altered OTUs in WT mice treated with dietary emulsifier. WT mice were exposed to drinking water containing CMC (A-B) or P80 (C-D) (1.0%) for 12 weeks. (A, C) Misclassification error rate showing that 15 OTUs were sufficient to successfully discriminate microbiota from each experimental group (error rate = 0). (B, D) Heatmap representation of the 15 most significantly altered OTUs in WT mice treated with dietary emulsifier. Colors represent relative expression (white and red for underrepresented and overrepresented, respectively). The 15 OTUs were listed on the right part using their Greengenes Prok_MSA IDs, and assigned taxonomy were labeled starting phylum, then class, order, family, and genus. Dendrogram on the upper part represents sample clustering. (E-H) Misclassification error rate and heatmap representation of the 15 most significantly altered OTUs in IL10−/− mice treated with dietary emulsifier. IL10−/− mice were exposed to drinking water containing CMC (E-F) or P80 (G-H) (1.0%) for 12 weeks. See legend of A-D for more details. (I-L)Misclassification error rate and heatmap representation of the 15 most significantly altered OTUs in TLR5−/− mice treated with dietary emulsifier. TLR5−/− mice were exposed to drinking water containing CMC (I-J) or P80 (K-L) (1.0%) for 12 weeks. See legend of A-D for more details (for A-B, n=5; for C-D, n=5 and 4 for water and P80-treated groups, respectively; for E-F, n=5 and 4 for water and CMC-treated groups, respectively; for G-H, n=5 and 4 for water and P80-treated groups, respectively; for I-J, n=4; for K-L, n=4 and 3 for water and P80-treated groups, respectively).
Extended data figure 5.
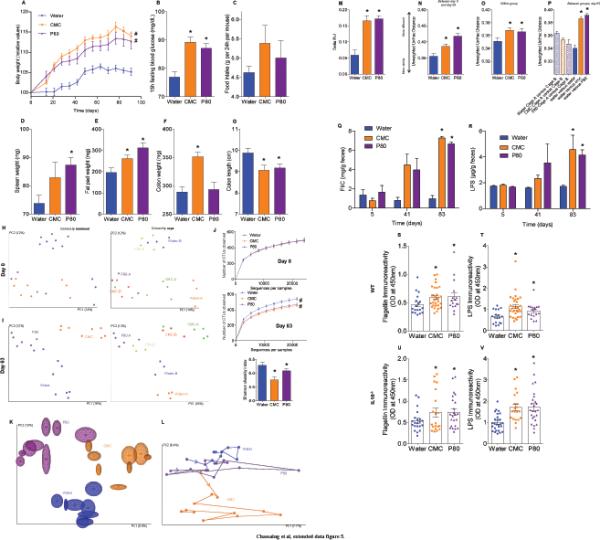
(A-G) Dietary emulsifiers promote metabolic syndrome in adult WT mice. 4 months-old male WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 8 weeks (2 cages per condition). (A) Body weight over time, (B) 15-hours fasting blood glucose concentration, (C) food intake measurement, (D) spleen weights, (E) fat-pad weights, (F) colon weights and (G) colon lengths. Data are the means +/- S.E.M. (n=10). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). (H-P) Dietary emulsifiers alter gut microbiota composition in adult WT mice. 4 months-old male WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 8 weeks (2 cages per condition). (H) Day 0 and (I) day 63 PCoA of the unweighted UniFrac distance matrix of fecal microbiota in WT mice. Treatment of each mouse is indicated by point color (blue = water; orange = CMC; purple = P80). Cage of each mouse is indicated by point color (for H, n=7, 8 and 8 for water, CMC and P80-treated groups, respectively). (J) Day 0 and day 63 microbiota richness and diversity. (K) Day 63 Jackknifed PCoA of the unweighted UniFrac distance matrix of fecal microbiota in WT mice. Treatment of each mouse is indicated by point color (blue = water; orange = CMC; purple = P80) (n=8). (L) After clustering of mouse fecal microbiota using PCoA of the unweighted UniFrac distance matrix, a representative mouse has been used to illustrate the time point evolution of the microbiota (n=1). (M) After clustering of mouse fecal microbiota using PCoA of the unweighted UniFrac distance matrix, evolution of the principal component 1 between day 0 and day 63 has been calculated for each mouse (n=10). (N) Average of the UniFrac unweighted distance for each group (water, CMC and P80) between day 0 and day 63 has been calculated (n=10). (O) Average of the UniFrac unweighted distance within group (water, CMC and P80) has been calculated (n=10). (P) Average of the UniFrac unweighted distance between group (cages, water, CMC and P80) or within group (water) at day 63 has been calculated (n=10). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). (Q-R) Dietary emulsifiers increase pro-inflammatory potential of intestinal microbiota in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Bioactive levels of fecal flagellin (Q) and LPS (R) assayed with TLR5 and TLR4 reporter cells. Data are the means +/- S.E.M. (n=10). Significance was determined using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group. (S-V)Dietary emulsifiers increase serum immune reactivity. WT (S, T) and IL10−/− (U, V) mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (S, U) Serum immune reactivity to flagellin and (T, V) LPS in WT (S, T) and IL10−/− (U, V) mice. Points are from individual mice. Data are the means +/- S.E.M. (for S, n=18, 30 and 16 for water, CMC and P80-treated groups, respectively; for T, n=18, 31 and 17 for water, CMC and P80-treated groups, respectively; for U, n=21, 20 and 23 for water, CMC and P80-treated groups, respectively; for V, n=27, 20 and 25 for water, CMC and P80-treated groups, respectively). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group.
Extended data figure 6.

A-Dietary emulsifiers induce histopathologically robust inflammation in IL10−/− mice. IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Colon and small intestine were H&E stained. Bar = 200μm. Pictures are representatives of 15 biological replicates. B-Dietary emulsifiers induce histopathologically robust inflammation in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Colon and small intestine were H&E stained. Bar = 200μm. Pictures are representatives of 5 biological replicates. CHistopathology of emulsifier-treated WT mice. WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Colon and small intestine were H&E stained. Bar = 200μm. Pictures are representatives of 15 biological replicates. (D-H) Dietary emulsifiers elicit low-grade intestinal inflammation in WT and splenomegaly in IL10−/− mice. WT (D-G) and IL10-/- (H) mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (D) Colitis incidence over time, (E, H) spleen weights, (F) epithelial damage and (G) infiltration scores. Points are from individual mice, bar represent the mean. (for E-G, n=14, 27 and 16 for water, CMC and P80-treated groups, respectively; for H, n=11, 18 and 20 for water, CMC and P80-treated groups, respectively). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group. (I-K) Extent of intestinal inflammation correlates with perturbation in microbiota localization in WT and IL10−/− mice. IL10−/− mice were exposed to drinking water containing (I) CMC or (J) P80 (1.0%) for 12 weeks. Fecal levels of the inflammatory marker Lcn2 as well as confocal microscopy analysis of microbiota localization and estimation of the distances of the closest bacteria to intestinal epithelial cells were determined, and plotted in X and Y axis, respectively. Linear regression line was draft and R2 was determined. (n=11). (K) Analysis of bacterial-epithelial distance upon stratification of levels of gut inflammatory marker fecal Lcn2, using both WT and IL10-/- mice exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Mice were grouped according to their fecal Lcn2 levels and bacterial-epithelial distances were then plotted (mean +/- S.E.M.). Significance was determined using oneway ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to X<50 ng/g group).
Extended data figure 7.

(A-G) Dietary emulsifiers promote intestinal inflammation in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A) Fecal levels of the inflammatory marker Lcn2 over time, (B) colitis incidence over time, (C) MPO levels, (D) histological score, (E) colon weights, (F) colon lengths and (G) spleen weights. Data are the means +/- S.E.M. or geometric means with 95% confidence interval (for A) (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group. (H-K) Dietary emulsifiers induce metabolic syndrome in WT and TLR5-/- mice. (H-I) WT and (J-K) TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (H, J) Glucose tolerance and (I, K) insulin sensitivity were analyzed. Data are the means +/- S.E.M. (n=5).Significance was determined using 2-way group ANOVA (# indicates p<0.05) compared to water-treated group. (L-T) Emulsifier-supplemented chow elicits low-grade intestinal inflammation in WT mice. WT mice were given mouse chow containing CMC or P80 (1.0%) for 12 weeks. (L) Fecal levels of the inflammatory marker Lcn2 over time, (M) MPO levels, (N) food intake measurement, (O) 15-hours fasting blood glucose concentration, (P) colon weights, (Q) colon lengths, (R) spleen weights, and PCR-based quantification of (S) total bacterial load and (T) bacterial load adhered to colonic mucosa. Data are the means +/- S.E.M. or geometric means with 95% confidence interval (for A) (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to control group. (U-AH) Dose response characterization of dietary emulsifiers on intestinal inflammation. WT mice were exposed to drinking water containing 0.1-1.0% CMC (U-AA) or P80 (AB-AH) for 12 weeks. (U, AB) Fecal levels of the inflammatory marker Lcn2 over time, (V, AC) MPO levels, (W, AD) food intake measurement, (X, AE) 15-hours fasting blood glucose concentration, (Y, AF) colon weights, (Z, AG) colon lengths and (AA, AH) spleen weights. Data are the means +/- S.E.M. (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to water-treated group.
Extended data figure 8.

(A-K) Emulsifiers-induced metabolic syndrome is partially reversible by 6 weeks post-emulsifier treatment. (A) Schematic representation of the experiment. (B, C) Body weight over time, (D) fat-pad weight, (E) fecal levels of the inflammatory marker Lcn2 over time, (F) MPO levels, (G) food intake measurement, (H) 15-hours fasting blood glucose concentration, (I) colon weights, (J) colon lengths and (K) spleen weights. Data are the means +/- S.E.M. (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). (L-S) Sodium sulfite did not induce robust or low-grade intestinal inflammation. WT and IL10−/− mice were exposed to drinking water containing sodium sulfite (1.0%) for 12 weeks. (L, M) Body weight over time, (N) colon weights, (O) colon lengths, (P) spleen weights, (Q) fat-pad weight, (R) MPO levels and (S) serum levels of the inflammatory marker Lcn2. Data are the means +/- S.E.M. (n=5). Points are from individual mice. Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test. (T-W) Dietary emulsifiers promote metabolic syndrome in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (T) Body weight over time, (U) fatpad weight, (V) food intake measurement, and (W) 15-hours fasting blood glucose concentration. Data are the means +/- S.E.M. (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). (X-AF) Emulsifier-supplemented chow promotes intestinal inflammation in TLR5−/− mice. TLR5−/− mice were given mouse chow containing CMC or P80 (1.0%) for 12 weeks. (X) Fecal levels of the inflammatory marker Lcn2 over time, (Y) MPO levels, (Z) food intake measurement, (AA) 15-hours fasting blood glucose concentration, (AB) colon weights, (AC) colon lengths, (AD) spleen weights, and PCR-based quantification of (AE) total bacterial load and (AF) bacterial load adhered to colonic mucosa. Data are the means +/- S.E.M. (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to control group. (AG-AO) Dose response characterization of dietary emulsifiers on intestinal inflammation in TLR5−/− mice. TLR5−/− mice were exposed to drinking water containing 0.1-1.0% P80 for 12 weeks. (AG) Body weight over time, (AH) fat pad weight, (AI) fecal levels of the inflammatory marker Lcn2 over time, (AJ) MPO levels, (AK) food intake measurement, (AL) 15-hours fasting blood glucose concentration, (AM) colon weights, (AN) colon lengths, and (AO) spleen weights. Data are the means +/- S.E.M. (n=3). Points in D are from individual mice. Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group).
Extended data figure 9.

(A-F) Dietary emulsifiers promotes metabolic syndrome in IL10−/− mice. IL10−/− mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (A-C) Body weight over time, (D) fat-pad weight, (E) food intake measurement, and (F) 15-hours fasting blood glucose concentration. Data are the means +/- S.E.M. (for A, n=24, 18 and 21 for water, CMC and P80-treated groups, respectively; for B, n=14, 11 and 8 for water, CMC and P80-treated groups, respectively; for C, n=14, 9 and 9 for water, CMC and P80-treated groups, respectively; for D, n=14, 18 and 20 for water, CMC and P80-treated groups, respectively; for E, n=15, 11 and 12 for water, CMC and P80-treated groups, respectively; for F, n=21, 17 and 20 for water, CMC and P80-treated groups, respectively). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test (* indicates p<0.05 compared to water-treated group) or 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). Points are from individual mice and red points in F represent mice with overt colitis. (G-P) Emulsifiers-induced low-grade intestinal inflammation was abolished under germ-free conditions. (G-K) Conventionally-housed and germ-free (L-P) Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (G, L) Fecal levels of the inflammatory marker Lcn2 over time, (H, M) MPO levels, (I, N) colon weights, (J, O) colon lengths and (K, P) spleen weights. Data are the means +/- S.E.M. (n=8 for conventionally-housed mice and n=4 for germ-free mice). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to control group. (Q-W) Dietary emulsifiers induce perturbations in fecal short-chain fatty acid composition. Fecal short-chain fatty acids composition was analyzed at the Metabolomics Core of the University of Michigan. (Q) Acetate, (R) propionate, (S) butyrate, (T) isovalerate, (U) valerate (V) hexanoate and (W) heptanoate were analyzed. Data are the means +/- S.E.M. (n=5, 8 and 7 for water, CMC and P80 conventional mice-treated groups and n=4, 4 and 5 for water, CMC and P80 germfree mice-treated groups, respectively). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group. (X-AA) Dietary emulsifiers promote metabolic syndrome in mice from different vendors. WT mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (X, Y) Body weight over time of Bl/6 mice used upon receipt from Jackson Laboratories (X) or bred at Georgia State University (Y). (Z, AA) Body weight over time of Swiss Webster mice used upon receipt from Charles River company (Z) or bred at Georgia State University (AA). Data in X are not used elsewhere in report, while data in Y, Z, and AA are from Figs 3A, extended data figure 8A-K, and 4A). Data are the means +/- S.E.M. (n=8 for Bl/6 mice used upon receipt from Jackson Laboratories, n=16 for Bl/6 mice bred at Georgia State University, n=10 for Swiss Webster mice used upon receipt from Charles River company, n=8 for Swiss Webster mice bred at Georgia State University). Significance was determined using 2-way group ANOVA (# indicates p<0.05 compared to water-treated group). (AB-AT) Dietary emulsifiers induce perturbations in fecal bile acids composition. Fecal bile acids composition was analyzed at the Metabolomics Core of the University of Michigan. (AB) Lithocholic acid (LCA), (AC) chenodeoxycholic acid (CDCA), (AD) deoxycholic acid (DCA), (AE) hyodeoxycholic acid / ursodeoxycholic acid (HDCA/UDCA), (AF) α-muricholic acid (α-MCA), (AG) β-muricholic acid (β -MCA), (AH) cholic acid (CA), (AI) hyocholic acid (HCA), (AJ) ω-muricholic acid (ω-MCA), (AK) glycolithocholic acid (GLCA), (AL) glycochenodeoxycholic acid (GCDCA), (AM) glycodeoxycholic acid (GDCA), (AN) hyodeoxycholic acid / glycoursodeoxycholic acid (HDCA/GUDCA), (AO) glycocholic acid (GCA), (AP) taurolithocholic acid (TLCA), (AQ) taurine-conjugated chenodeoxycholic acid (TCDCA), (AR) taurodeoxycholic acid / Tauroursodeoxycholic acid (TDCA/TUDCA), (AS) taurohyodeoxycholic acid, and (AT) taurocholic acid (TCA) were analyzed. Data are the means +/- S.E.M. (n=5, 8 and 7 for water, CMC and P80 conventional mice-treated groups and n=4, 4 and 5 for water, CMC and P80 germfree mice-treated groups, respectively). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group.
Extended data figure 10.

(A-H) Dietary emulsifiers do not alter mucus thickness under germ-free conditions. (A-C, G-H) Conventionally-housed and germ-free (D-F, H) Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. (AF) Confocal microscopy analysis of microbiota localization; Muc2 (green), actin (purple), bacteria (red), and DNA (Blue). Bar = 20μm. (G) Distances of closest bacteria to intestinal epithelial cells per condition over 5 high-powered fields per mouse. Pictures are representatives of 5 biological replicates. (H) Mucus thickness over 5 high-powered fields per mouse. Data are the means +/- S.E.M. (n=5). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test, * indicates p<0.05 compared to water-treated group. (I-L)Dietary emulsifiers do not induce drastic perturbations of mucus layer integrity under germ-free conditions. Germ-free Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 8 weeks. Mice were removed from isolator, inoculated with 0.5 μm green fluorescent beads (Polysciences, Warrington, PA), and euthanized 7h post inoculation. (I-K) Confocal microscopy analysis of fluorescent beads localization; Muc2 (green), actin (purple), fluorescent beads (red), and DNA (Blue). Bar = 20μm. (L) Distances of closest fluorescent beads to intestinal epithelial cells per condition over 5 high-powered fields per mouse. Pictures are representatives of 5 biological replicates. Data are the means +/- S.E.M. (n=4). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test. (M-R) Dietary emulsifiers increase pro-inflammatory potential of intestinal microbiota in SW mice, transferable to germfree mice recipient. (M, N) Germ-free Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Bioactive levels of fecal flagellin (M) and LPS (N) assayed with TLR5 and TLR4 reporter cells. (O, P) Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. Bioactive levels of fecal flagellin (O) and LPS (P) assayed with TLR5 and TLR4 reporter cells. (Q-R) Germ-free Swiss-Webster mice were conventionalized via microbiota transplant from the Swiss-Webster mice treated with emulsifiers described above. Bioactive levels of fecal flagellin (Q) and LPS (R) assayed with TLR5 and TLR4 reporter cells. Data are the means +/- S.E.M. (n=4 for germ-free mice, n=8 for conventionally-housed mice and n=5 for conventionalized mice). Significance was determined using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to control group. (S-Y)Microbiota transplant transfers emulsifier-induced low-grade intestinal inflammation. Germ-free Swiss-Webster mice were conventionalized via microbiota transplant from mice that received standard drinking water or drinking water containing CMC or P80 (1.0%). (S) Schematic representation of the experiment. (T) Fecal levels of the inflammatory marker Lcn2 over time, (U) MPO levels, (V) colon weights, (W) colon lengths, (X) spleen weights, and (Y) food intake measurement. Data are the means +/- S.E.M. (n=4). Significance was determined using one-way ANOVA corrected for multiple comparisons with Sidak test or using two-way ANOVA corrected for multiple comparisons with Bonferroni test, * indicates p<0.05 compared to control group. (Z-AE) Dietary emulsifiers induce profound alterations in gut microbiota composition in SW mice, transferable to germfree mice recipient. (Z-AB) Swiss-Webster mice were exposed to drinking water containing CMC or P80 (1.0%) for 12 weeks. PCoA of the unweighted UniFrac distance matrix of fecal microbiota at (Z) day 0, (AA) day 42 and (AB) day 98. (AC-AE) Germ-free Swiss-Webster mice were conventionalized via microbiota transplant from the Swiss-Webster mice treated with emulsifiers described above. PCoA of the unweighted UniFrac distances of fecal microbiota at (AC) day 14, (AD) day 42 and (AE) day 98 post transplant (for Z-AB, n=5, 8 and 7 for water, CMC and P80-treated groups, respectively; for ACAE, n=5, 4 and 3 for water, CMC and P80-treated groups, respectively). Treatment of each mouse is indicated by point color and matching colored circles indicate mice receiving same treatment (blue = water; orange = CMC; purple = P80). Black dashed circles represent mice sharing a cage.
Supplementary Material
Acknowledgements
This work was supported by NIH grant DK099071 and DK083890. BC is a recipient of the Research Fellowship award from the Crohn's and Colitis Foundation of America (CCFA). We thank Benyue Zhang, Lucie Etienne-Mesmin, Hao Q. Tran and Emilie Viennois for technical assistance.
Footnotes
Primary accessions Sequencing data are deposited in the European Nucleotide Archive accession PRJEB8035.
Author contributions: B.C. and A.T.G conceived the project designed the experiments interpreted the results and wrote the manuscript. B.C. performed all experiments and analysis with advice and guidance from O.K. J.G. and A.P. S.S and R.E.L guided experimental design and data interpretation.
References
- 1.Johansson ME, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. doi:10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts CL, et al. Translocation of Crohn's disease Escherichia coli across M-cells: contrasting effects of soluble plant fibres and emulsifiers. Gut. 2010;59:1331–1339. doi: 10.1136/gut.2009.195370. doi:10.1136/gut.2009.195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swidsinski A, Loening-Baucke V, Herber A. Mucosal flora in Crohn's disease and ulcerative colitis - an overview. J Physiol Pharmacol. 2009;60(Suppl 6):61–71. [PubMed] [Google Scholar]
- 4. https://http://www.fsc.go.jp/english/evaluationreports/foodadditive/polysorbate_report.pdf.
- 5.NTP Toxicology and Carcinogenesis Studies of Polysorbate 80 (CAS No. 9005-65-6) in F344/N Rats and B6C3F1 Mice (Feed Studies). Natl Toxicol Program Tech Rep Ser. 1992;415:1–225. [PubMed] [Google Scholar]
- 6.Swidsinski A, et al. Bacterial overgrowth and inflammation of small intestine after carboxymethylcellulose ingestion in genetically susceptible mice. Inflamm Bowel Dis. 2009;15:359–364. doi: 10.1002/ibd.20763. doi:10.1002/ibd.20763. [DOI] [PubMed] [Google Scholar]
- 7.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 8.Vijay-Kumar M, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. doi:10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ubeda C, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–1456. doi: 10.1084/jem.20120504. doi:10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chassaing B, Ley RE, Gewirtz AT. Intestinal Epithelial Cell Toll-like Receptor 5 Regulates the Intestinal Microbiota to Prevent Low-Grade Inflammation and Metabolic Syndrome in Mice. Gastroenterology. 2014 doi: 10.1053/j.gastro.2014.08.033. doi:10.1053/j.gastro.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson ME, Hansson GC. Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods Mol Biol. 2012;842:229–235. doi: 10.1007/978-1-61779-513-8_13. doi:10.1007/978-1-61779-513-8_13. [DOI] [PubMed] [Google Scholar]
- 12.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. doi:10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. doi:10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 14.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. doi:10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 15.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. doi:10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Png CW, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. doi:10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 17.Arthur JC, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. doi:10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carvalho FA, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–152. doi: 10.1016/j.chom.2012.07.004. doi:10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012;9:219–230. doi: 10.1038/nrgastro.2012.14. doi:10.1038/nrgastro.2012.14. [DOI] [PubMed] [Google Scholar]
- 20.Cox LM, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. doi:10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut. 2014;63:1069–1080. doi: 10.1136/gutjnl-2013-304909. doi:10.1136/gutjnl-2013-304909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziegler TR, et al. Detectable serum flagellin and lipopolysaccharide and upregulated anti-flagellin and lipopolysaccharide immunoglobulins in human short bowel syndrome. Am J Physiol Regul Integr Comp Physiol. 2008;294:R402–410. doi: 10.1152/ajpregu.00650.2007. doi:10.1152/ajpregu.00650.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chassaing B, et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. doi:10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. doi:10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kullberg MC, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. doi:10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 27.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. doi:10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong T, et al. Divergent metabolic adaptations to intestinal parasitic nematode infection in mice susceptible or resistant to obesity. Gastroenterology. 2007;133:1979–1988. doi: 10.1053/j.gastro.2007.09.006. doi:10.1053/j.gastro.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. doi:10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 30.Suez J, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514:181–186. doi: 10.1038/nature13793. doi:10.1038/nature13793. [DOI] [PubMed] [Google Scholar]
- 31.Uematsu S, et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat Immunol. 2006;7:868–874. doi: 10.1038/ni1362. doi:10.1038/ni1362. [DOI] [PubMed] [Google Scholar]
- 32.Denizot J, et al. Adherent-invasive Escherichia coli induce claudin-2 expression and barrier defect in CEABAC10 mice and crohn's disease patients. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21787. [DOI] [PubMed] [Google Scholar]
- 33.Castaneda FE, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. doi:10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 34.Katakura K, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. doi:10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sitaraman SV, et al. Elevated flagellin-specific immunoglobulins in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2005;288:G403–406. doi: 10.1152/ajpgi.00357.2004. doi:10.1152/ajpgi.00357.2004. [DOI] [PubMed] [Google Scholar]
- 36.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. doi:10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilbert JA, et al. The Earth Microbiome Project: Meeting report of the “1 EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6 2010. Stand Genomic Sci. 2010;3:249–253. doi: 10.4056/aigs.1443528. doi:10.4056/aigs.1443528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. doi:10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Med. 2010;7:335–336. doi: 10.1038/nmeth.f.303. doi:10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aronesty E. Comparison of Sequencing Utility Programs. The Open Bioinformatics Journal. 2013;7:1–8. [Google Scholar]
- 41.Aronesty E. Command-line tools for processing biological sequencing data. 2011 http://code.google.com/p/ea-utils.
- 42.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. doi:10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 43.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. doi:10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. doi:10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. doi:10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. doi:10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.