

ABSTRACT
Despite the success of CD20 antibody rituximab in immunotherapy, acquired resistance is one of the prime obstacles for the successful treatment of B-cell malignancies. There is an urgent need to intensify efforts against resistance in cancer treatment. Growing evidence indicated that lysosomes may form an “Achilles heel” for cancer cells by sensitizing them to death pathways. Here, we uncover an important role of CD20 in initiation of ceramide/lysosomal membrane permeabilization (LMP)-mediated cell death, showing that colocalization of CD20-TNFR1 after type II CD20 antibody ligation can stimulate de novo ceramide synthesis by ceramide synthase and consequently induce remarkable lysosomal permeabilization (LMP) and lysosome-mediated cell death. Further studies show that the potent lysosome-mediated cell death induced by CD20 antibodies exhibits a profound killing effect against both rituximab-sensitive and -resistant (RR) lymphoma. Furthermore, engineering of rituximab by introducing a point mutation endows it with the ability to induce potent ceramide/LMP-mediated cell death in both RR lymphoma and primary B-cell malignancies from patients with rituximab-refractory, suggesting the potential clinical application to combat rituximab resistance.
KEYWORDS: B-cell lymphoma, CD20 antibody, cell death, rituximab resistance, rituximab variant
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- B-CLL
B cell chronic lymphocytic leukemia
- CDC
complement-dependent cytotoxicity
- CDR
complementarity-determining region
- DEGS1
desaturase, sphingolipid 1
- DLBCL
diffuse large B cell lymphoma
- D-MAPP
D-erythro-2-Tetradecanoylamino-1-phenyl-1-propanol
- FB1
fumonisin B1
- FCM
flow cytometry
- Imip
imipramine
- LMP
lysosomal membrane permeabilization
- PI
Propidium Iodide
- qRT-PCR
quantitative reverse transcriptase PCR
- RR
rituximab resistant
- SIM
structured illumination microscopy
- TEM
transmission EM
- V-ATPases
vacuolar ATPases
- 3-OMe-SM
3-O-Methyl-sphingomyeline
Introduction
Despite rituximab has revolutionized the treatment of lymphoma,1 treatment of refractory or relapsed indolent lymphoma patients with rituximab remains associated with a response rate of only 50%, and close to 60% of prior rituximab responding patients will no longer benefit with retreatment due to acquired resistance.2 Although rituximab resistance mechanisms have been identified in preclinical studies, no effective regimen has been developed to overcome rituximab resistance in patients.3,4 More importantly, rituximab-resistant (RR) cell lines generated in vitro display cross-resistance when are tested against a panel of chemotherapeutic agents,3-5 indicating the alternative survival pathways developed during drug treatment pose additional challenges to the clinical management of patients with RR lymphoma.
Although most CD20 mAbs recognize the larger extracellular loop (only 44 amino acids) of the CD20 molecule,6-8 they are functionally diverse and can be defined as two distinct types: type I mAbs, or rituximab-like, which activate complement and are relatively poor at mediating cell death; and type II mAbs, or 11B8-like, which are relatively inactive in complement activation but tend to promote more cell death.8 Until now, no clear explanation has been offered for why CD20 mAbs are so functionally diverse, especially given their restricted epitope recognition, suggesting that CD20 molecule may still have some significant biological functions that remains uncovered. It is imperative that a better understanding of CD20 biology and the effector mechanisms after antibody ligation will allow more efficient exploitation of CD20 as a therapeutic target against RR NHL.
Numerous studies have revealed that several mechanisms might be involved in providing therapeutic efficacy of CD20 mAbs, including complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and the induction of cell death.9,10 Although Fc-mediated effector functions are important for lymphoma treatment,11-14 the relative contributions of these different mechanisms of action are still a matter of debate.9,10,15 Very recently, type II CD20 mAb-induced lysosome-mediated cell death has been reported to be mediated through a ceramide-dependent pathway.16,17 However, signal transduction pathway of lysosome-mediated cell death induced by type II CD20 mAbs is still elusive. Previously, emerging experimental evidence suggests that such alterations in lysosomes may form an “Achilles heel” for cancer cells by sensitizing them to death pathways involving LMP and the release of cathepsins into the cytosol.18-20 Some lysosome targeting drugs have been shown to possess the ability to resensitize multidrug resistant cells to classical chemotherapy.19,21,22 These data may suggest that lymphoma lysosomes can be specifically utilized for development of antibody to combat resistance.
In the present study, we have characterized the behavior of CD20 molecule after antibody ligation on B-cell lymphoma. Our data reveal that co-localization of CD20 and TNFR1 molecules after type II CD20 antibody ligation could stimulate ceramide synthesis by the enzyme ceramide synthase (DEGS1), which subsequently induced LMP and initiated lysosome-dependent cell death in both rituximab-sensitive and RR B-cell lymphoma. Intriguingly, by introducing a point mutation Tyr102Lys into the complementarity-determining region (CDR), the rituximab variant H102YK could trigger marked LMP-mediated cell death, exhibiting potent therapeutic efficacy against RR lymphoma.
Results
Activation of lysosome-mediated cell death overcomes rituximab resistance in vitro and in vivo
To evaluate whether type II CD20 mAb-mediated cell death can eliminate RR lymphoma, two RR lymphoma Raji-R and Daudi-R generated as previously described 3,4,23 were used. In line with our previous findings,23 both of these RR cells (Raji-R and Daudi-R) exhibited strong resistance to rituximab-mediated CDC (Data not shown). The remarkable cell death initiated by type II CD20 mAb 11B8 can be observed in both of these two RR cells (Fig. 1A). The XTT assays were also used to evaluate the cell death induced by these CD20 mAbs and the similar results can be observed (Data not shown). Further studies showed that caspase and Bcl-2 inhibitors could not block 11B8-induced cell death (Fig. S1A, Fig. 1B). Moreover, cleaved fragments of caspase-3 or caspase-9 were not detected by western blot after treatment with CD20 mAbs for 24 h (Fig. S1B). To further investigate whether autophagy was elicited by type II CD20 mAb, we assessed the expression of several autophagy marker proteins before and after mAb treatment. These data revealed that Beclin-1, Atg 12 and LC-3 were unchanged over the 16-h time course studied (Fig. S1C), indicating that autophagy had not been activated by type II CD20 mAb 11B8. In line with previous findings, 11B8 could elicit homotypic adhesion of lymphoma cells (Fig. S1D), and substantially induce the enlargement of lysosomal compartment and LMP in both Raji and Raji-R cells after treatment for 4 h (Fig. 1C). In line with previous findings reported by other groups,24,25 type II CD20 mAbs could induce LMP and leakage from lysosomes into the more neutral pH of the cytosol, which could subsequently increase pH of lysosome compartment and consequently decrease lysotracker fluorescence (Fig. S1E). A substantial increase in cathepsin B (Fig. 1D) and D (Fig. S1F) staining throughout the cytosol can be observed after 11B8 treatment for 4 h. The similar results can be obtained from other lymphoma cells, including Daudi and Daudi-R (Data not shown). These results have been further validated by FCM analysis (Fig. S1G). To further confirm the involvement of lysosomes in 11B8-induced death process, we utilized the well-characterized inhibitors of vacuolar ATPases (V-ATPases) concanamycin A and bafilomycin A1 (Fig. S1H and S1I). In line with the previous findings,24,25 both concanamycin A and bafilomycin A1 could significantly inhibit the cell death induced by type II CD20 mAb 11B8. The importance of cathepsins in the cell death process was subsequently confirmed by using specific cathepsin inhibitors (cathepsin inhibitor III or E-64d) that virtually ablated 11B8-induced cell death (Fig. 1E) and markedly inhibited the release of cathepsin B from lysosome (Fig. 1F). To further evaluate the specific cell death induced by type II CD20 mAb 11B8, CHO and HT-29 cells expressing human CD20 protein (CHO-CD20 and HT-29-CD20) were used in our experiments (Fig. S1J). After treatment with 11B8, the specific cell death can be observed in both CHO-CD20 and HT-29-CD20 cells. Moreover, the transmission EM (TEM) revealed that no evidence of DNA condensation or apoptotic body formation typical of apoptosis was observed after treatment with 11B8. Instead, we found large cytoplasmic inclusions and vacuoles in 11B8-treated cells that were distinct from control cells (Fig. S1K). The ultrastructural studies confirmed that neither classical apoptotic nor autophagic death was initiated by 11B8 mAbs.
Figure 1.
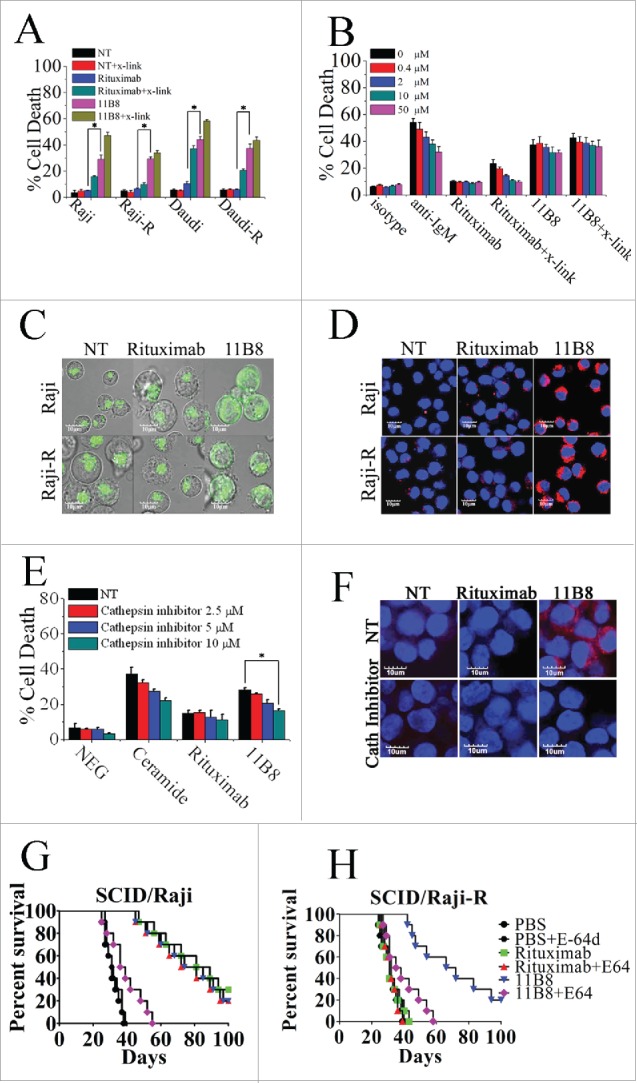
Lysosome-mediated cell death induced by type II CD20 mAbs can overcome rituximab resistance. (A) Evaluation of cell death induced by CD20 mAbs with or without cross-linker on Raji, Raji-R, Daudi and Daudi-R cells. Columns represent mean cell death (Annexin V- and PI-positive cells) (n = 3); bars represent SD. *p < 0.05.(B) Effect of Caspase Inhibitor VI on the cell death induced in 16 h by inhibitor alone, 10 μg/mL rituximab, and 11B8 in the presence or absence of cross-linker (goat anti-human F(ab′)2 fragment, 20 μg/mL) in Raji cells. Inhibitors were added over a range of different concentrations for 2 h before the addition of mAbs. Data are mean ± SD of at least three experiments. (C) Raji cells were incubated with CD20 mAbs as described previously (10 μg/mL). After that, cells were labeled with Lyso-Tracker green and the volume of the lysosomal compartment measured by confocal microscopy after 4 h. (D) Fluorescence microscopy of the lysosomal protease cathepsin B staining (red) of Raji cells 4 h after treatment with mAbs. DNA was counterstained with DAPI (blue; scale bar, 10 μm). (E) The inhibition of CD20 mAb-induced cell death by cathepsin inhibitor III in Raji cells as measured by FCM. Mean ± SD (n = 3). *p < 0.05. (F) Confocal microscopy of cathepsin B staining (red) 4 h after treatment with CD20 mAbs. DNA was counterstained with DAPI (blue). Scale bars: 10 μm. The survival of tumor-bearing SCID mice SCID/Raji (G) and SCID/Raji-R (H) treated with anti-CD20 mAbs. Groups of 10 SCID mice were injected intravenously with 3.5 × 106 Raji or Raji-R cells. Five days after tumor cell inoculation, the mice were treated with rituximab and 11B8 (400 μg/dose). The SCID/Raji and SCID/Raji-R mice were treated with cathepsin inhibitor (E-64d) at a dose of 1 mg/100 g of body weight/day intraperitoneally 3 d a week for 3 weeks.
Although both rituximab and 11B8 were shown to significantly improve the survival of SCID mice bearing disseminated Raji tumor cells (p < .001 for each compared with the PBS control), 11B8 could significantly prolong the survival of SCID/Raji-R mice (Fig. 1H). More importantly, treatment with the cathepsin inhibitor E-64d could markedly decrease the protection of 11B8 in both SCID/Raji and SCID/Raji-R mice, while the significant difference was not observed in the rituximab-treated groups.
De novo synthesis of ceramide is essential for LMP-mediated cell death initiated by type II CD20 mAb
Ceramide, a prototypic sphingolipid, is either synthesized de novo or generated from sphingomyelin breakdown.26 To evaluate the notion that ceramide is involved in the LMP-mediated cell death induced by type II CD20 mAbs, widely used specific inhibitors of A-SMase (Imipramine), N-SMase (3-O-Methyl-sphingomyeline) and ceramide synthase (fumonisin B1) were employed. 11B8-induced cell death can be significantly inhibited by 25 μM fumonisin B1 (FB1), whereas imipramine (Imip) and 3-O-Methyl-sphingomyeline (3-OMe-SM) could not protect the cells (the concentration from 50 μM to 0 μM)(Fig. 2A). The FB1, in the range from 0.2 nM to 25 nM, exhibited a dose-dependent inhibition of cell death induced by 11B8 (Fig. S2A). Exogenous ceramide (either C2 ceramide or ceramide obtained from bovine spinal cord) could induce dose-dependent cell death in both Raji and Ramos cells in the concentrations from 150 μM to 50 μM (Fig. 2B). After treatment with 10 μg/mL CD20 mAbs, the generation of ceramide induced by 11B8 was detectable after 6 h (Fig. 2C). The elevation of intracellular ceramide stimulated by 11B8 could be specifically inhibited by FB1 (Fig. 2D). The generation of Ceramide and the inhibitory effect of FB1 were also confirmed by the confocal fluorescent microscopy analysis (Fig. S2B). Moreover, LMP and the subsequent release of cathepsin B into the cytosol could be markedly inhibited by FB1 (Fig. 2E–G). Treatment with exogenous ceramide (150 μM or 100 μM) could significantly induce LMP and the subsequent release of cathepsin B (Fig. 2E and H).
Figure 2.
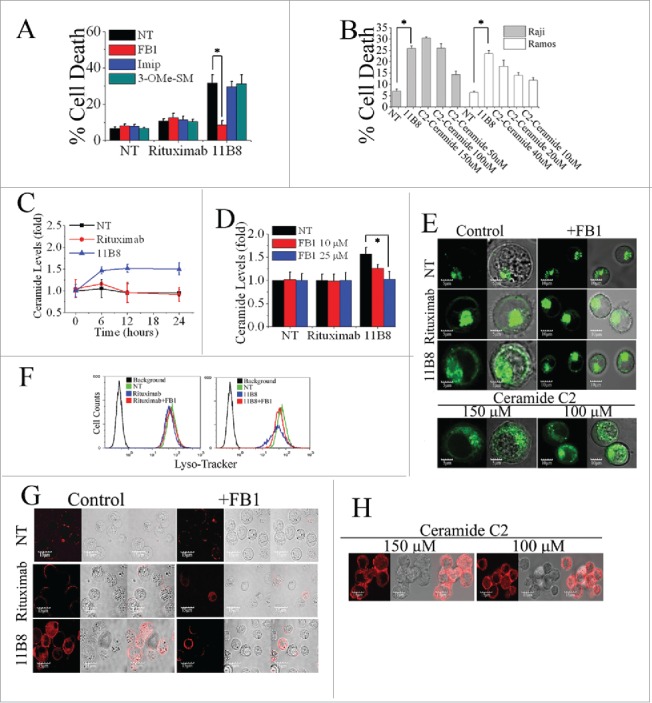
De novo ceramide synthesis involved in LMP-mediated cell death initiated by type II CD20 mAb. (A) The inhibition of cell death in Ramos cells by A-SMase, N-SMase and ceramide synthase inhibitors (Imip, 3-OMe-SM and FB1, respectively) was assessed by FCM. Error bars indicate SD (n = 3). *p < 0.05. (B) The exogenous ceramide (C2-Ceramide) induced cell death in both Raji and Ramos cells in a dose-dependent manner. *p < 0.05. (C) Time course study of ceramide generation in B cells induced by CD20 mAbs. The ceramide levels were quantitated as described in “Supplemental Experimental Procedures.” Results are representative of three independent experiments. (D) The generation of ceramide stimulated by 11B8 was inhibited by FB1. Raji cells were treated with FB1 prior to the addition of CD20 mAbs. *p < 0.05. Detection of total lysosomal volume in cells treated with mAbs and FB1. Cells were incubated with CD20 mAbs (10 μg/mL) and FB1 (25 μM). After that, cells were labeled with LysoTracker green and the volume of the lysosomal compartment measured by confocal microscopy (E) and FCM (F) after 4 h. (G and H) The assessment of LMP by evaluating the release of cathepsin B (red) into cytoplasm. Scale bars: 10 μm.
Dihydroceramide desaturase-1 (DEGS1) is critical to the initiation of LMP-mediated cell death
DEGS1, a key enzyme in the de novo pathway of ceramide generation, is the only dihydroceramide desaturase reported to be present in human cells.27 Here, shRNA against the human desaturase enzyme DEGS1 was used to attenuate its expression and consequently inhibit its activity. Raji and Ramos cells were transfected with DEGS1 shRNA or nonspecific shRNA. Knockdown of DEGS1 mRNA level was confirmed by quantitative reverse transcriptase PCR (qRT-PCR), with about 75% knockdown of DEGS1 mRNA achieved in Raji and Ramos cells (Fig. S3A). The decreased expressions of the DEGS1 protein in DEGS1 shRNA-treated Raji (Raji/DEGS1-) and Ramos (Ramos/DEGS1-) cells were also confirmed by protein gel blotting (Fig. S3B and C). The decrease of DEGS1 expression and consequent reduction of its activity could effectively prevent the cell death induced by 11B8 in both Raji and Ramos cells (Fig. 3A and B). The cathepsin B release induced by 11B8 was not observed in Raji/DEGS1- (Fig. 3C). Moreover, 11B8-stimulated intracellular ceramide elevation could be specifically inhibited by silencing of DEGS1 expression (Fig. 3D), suggesting that attenuation of intracellular ceramide through downregulation of DEGS1 could prevent LMP. Dihydroceramide, an inactive form of ceramide in ceramide de novo synthesis pathway, can be dehydrogenated to form ceramide by DEGS1.28 Addition of C2-dihydroceramide in the range from 5 μM to 100 μM could not induce cell death (Fig. 3E). However, after preincubation of 11B8-treated cells with 50 μM or 100 μM C2-dihydroceramide, a marked increase of cell death was observed (Fig. 3E). The 11B8-treated cells exhibited a much more extensive release of cathepsin B into cytosol after incubation with 100 μM C2-dihydroceramide (Fig. 3F), although C2-dihydroceramide (100 μM) in rituximab-treated cells could not induce cathepsin B release from lysosome, suggesting that DEGS1 can be activated by type II CD20 mAbs.
Figure 3.
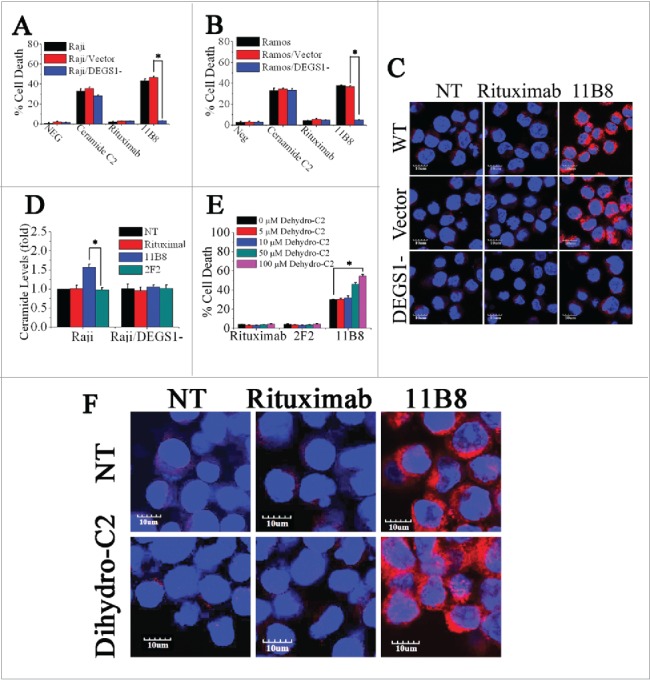
The important role of DEGS1 in the initiation of LMP-mediated cell death. (A and B) After downregulation of DEGS1 in Raji (Raji/DEGS1-) and Ramos (Ramos/DEGS1-) cells, the induction of cell death by CD20 mAbs was evaluated by FCM. Data represent means with SD (n = 3). *p < 0.05. (C) Confocal microscopy of intracellular cathepsin B (red) in Raji and Raji/DEGS1- cells to assess the extent of LMP induced by CD20 mAbs. Raji cells transfected with the control vectors were used as the control. DNA was counterstained with DAPI (blue). Scale bars: 10 μm. (D) The intracellular ceramide level was determined after treatment with CD20 mAbs for 16 h. Columns, mean (n = 3); bars, SD. *p < 0.05. (E) The dihydroceramide-induced death in CD20 mAb-treated cells was determined by staining of SYTOX Red on FCM. Bars represent the mean cell death + SD from three independent experiments. *p < 0.05. (F) Confocal analysis of cathepsin B release after dihydroceramide and CD20 mAbs treatment.
Induction of LMP is directly mediated by sphingosine
On the basis of its chemical structure and pKa value, sphingosine has been reported to be a lysosomotropic detergent.29 It is only generated from the hydrolysis of ceramides via ceramidases.30 To further explore the role of sphingosine in LMP, we first assessed the cellular sphingosine level induced by CD20 mAbs. As shown in Fig. 4A, the considerable increase of intracellular sphingosine was observed after treatment with 11B8 for 6 h, whereas rituximab was found to have no influence on the level of intracellular sphingosine even after 24 h. Incubation of cells with D-erythro-2-Tetradecanoylamino-1-phenyl-1-propanol (D-MAPP), which specifically blocked the generation of sphingosine by inhibiting ceramidase without suppressing ceramide generation (Fig. S4), could abolish the cellular sphingosine change induced by 11B8 (Fig. 4B) and subsequently inhibit the cell death in 11B8-treated cells (Fig. 4C). Our further analysis showed that D-MAPP in 25 μM could markedly block the release of cathepsin B from lysosomes (Fig. 4D). The substantial release of cathepsin B and the considerable cell death initiated by adding exogenous ceramide (100 μM) could be potently inhibited after D-MAPP treatment (Fig. 4C and D). Increasing the concentration of exogenous sphingosine from 10 μM to 50 μM could induce cathepsin B release from lysosomes and consequently induce cell death in a dose-dependent manner (Fig. 4E and F). The direct and essential role of sphingosine in mediating LMP was further demonstrated by in vitro incubation of lysosomal fraction with sphingosine at different concentrations (Fig. 4G).
Figure 4.

Induction of LMP is directly mediated by sphingosine. (A) A time-course analysis of intracellular sphingosine level after treatment with CD20 mAbs. Points represent mean (n = 3); bars represent SD. (B) The inhibition of sphingosine generation by D-MAPP was evaluated. Data are mean ± SD (n = 3). *p < 0.05. (C) The cell death induced by CD20 mAbs was inhibited by D-MAPP. Cell death was determined by staining of SYTOX Red on FCM. Bars represent the mean cell death + SD from three independent experiments. *p < 0.05. (D) Evaluation of D-MAPP on the release of cathepsin B. Cathepsin B was immunostained (red) and nuclear DNA was stained with DAPI (blue). Scale bars, 10 μm. (F) Dose-dependent induction of cell death by exogenous sphingosine. Cells were stained with SYTOX Red and detected on FCM Data are mean ± SD (n = 3). *p < 0.05. (E) Dose-dependent cathepsin B release determined by confocal analysis. Cathepsin B, red; DAPI, blue. Scale bars, 10 μm. (G) Lysosomal fraction in buffered 300 mM sucrose was exposed for 30 min at 37°C to sphingosine at the concentrations indicated. After centrifugation, supernatant activity was expressed as a percentage of total activity (following treatment with 0.1 M Triton X-100).
Engineering of rituximab by introducing a point mutation could initiate potent ceramide/LMP-mediated cell death against rituximab resistance
Although Rituximab has produced significant tumor regressions in lymphoma patients, it is unable to initiate potent cell death against B-cell malignancies. To our knowledge, the ideal CD20 antibody should have the advantages of both type I and type II CD20 mAbs, exhibiting potent CDC, ADCC and cell death against malignant cells. Interestingly, although introduction of a point mutation Tyr102Lys (H102YK) into the CDR of rituximab could not significantly increase binding avidity (Fig. S5A), the marked enhancement of cell death could be observed (Fig. 5A and B). Although caspase inhibitor (from 0.4 μM to 50 μM) could not block the H102YK-induced cell death (Fig. S5B), cathepsin inhibitor in the range from 1.25 μM to 10 μM could significantly suppress the cell death induced by H102YK (Fig. 5A). Furthermore, the H102YK could induce remarkable cathepsin B release from lysosome, but the cathepsin B release was not observed in rituximab-, H57DE- and L93NR-treated groups (Fig. 5B). In accordance with these observations, H102YK could promote the generation of ceramide (Fig. 5C), and downregulation of DEGS1 (Fig. 5D) could significantly inhibit the cell death triggered by H102YK.
Figure 5.
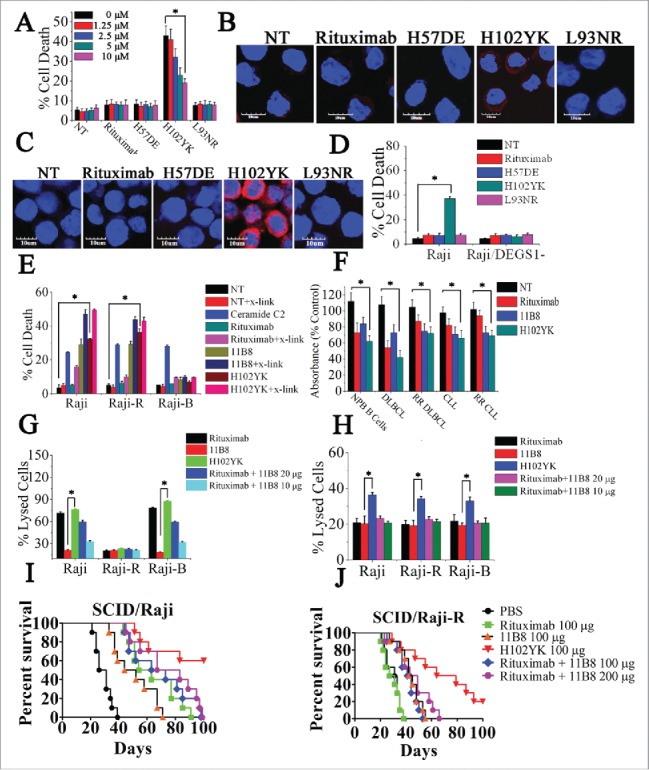
Rituximab variant H102YK exhibiting marked therapeutic efficacy against RR lymphoma. (A) Effect of cathepsin inhibitor on the cell death induced in 48 h by inhibitor alone, 10 μg/mL rituximab, and rituximab variant in Raji cells. Cathepsin inhibitor III was added over a range of different concentrations for 2 h before the addition of mAbs. Data are mean ± SD (n = 3). *p < 0.05. The release of cathepsin B (B) and the intracellular ceramide generation (C) in Raji cells were examined by confocal analysis. Cathepsin B and ceramide were immunostained (red) and nuclear DNA was stained with DAPI (blue). Scale bars, 10 μm. (D) The induction of cell death by CD20 mAbs in Raji and Raji/DEGS1- was evaluated. Data represent means with SD (n = 3). *p < 0.05. (E) Evaluation of cell death induced by CD20 mAbs with or without cross-linker on Raji, rituximab-resistant Raji (Raji-R) and 11B8-resistant Raji (Raji-B) cells. Columns represent mean (n = 3); bars represent SD. *p < 0.05. (F) Induction of cell death on normal peripheral blood (PB) B cells, DLBCL (N = 19), rituximab-refractory DLBCL (n = 17), B-CLL (n = 15), rituximab-refractory B-CLL (n = 14) was evaluated by XTT assay. The absorbance values indicate the cell viability in the experiment conditions. *p < 0.05. Raji and resistant Raji cells were exposed to CD20 mAbs (10 μg/each or 20 μg/each), followed by the addition of NHS (G) or PBMCs (H), respectively. *p < 0.05. The survival of tumor-bearing SCID mice treated with anti-CD20 mAbs . Groups of 10 SCID mice were injected intravenously with 3.5 × 106 Raji (I) or Raji-R cells (J). Five days after tumor cell inoculation, the mice were treated with rituximab, rituximab variant H102YK or simultaneous treatment with rituximab and 11B8 (100 μg/each mice in SCID/Raji).
To further evaluate the advantages of rituximab variant H102YK, we first established 11B8-resistant Raji cells (Raji-B) according to previously described method 3,4,23 and evaluated CDC, ADCC and induction of cell death among wild type, rituximab- or 11B8-resistant cells. H102YK could induce potent cell death against both Raji and Raji-R cells even without cross-linking (Fig. 5E). To further confirm these findings had relevance to primary tumors, we performed ex vivo experiments on a large subset of rituximab-refractory patient samples from multiple B cell NHL subtypes, including diffuse large B cell lymphoma (DLBCL) and B cell chronic lymphocytic leukemia (B-CLL). The relapsed refractory lymphoma is defined as per criteria proposed by Cheson et al.31 Our data showed that the significant cell death was visibly observed after treatment with 11B8 and H102YK for 16 h (Fig. 5F). Although Raji-B cells exhibited profound resistance to cell death initiated by 11B8 and H102YK (Fig. 5E), H102YK with the benefit of type I CD20 mAbs could promote significant CDC and ADCC against Raji-B cells (Fig. 5G and H). The in vivo therapeutic efficacy of H102YK was studied by comparison with simultaneous administration of rituximab and 11B8 against Raji or Raiji-R cells (Fig. 5I and J). H102YK with the benefits of both type I and type II CD20 mAbs could initiate multiple antitumor mechanisms against RR B-cell malignancies, exhibiting more potent antitumor activities than either type I and type II CD20 mAb treatment alone or combination treatment.
Colocalization of CD20-TNFR1 stimulated by CD20 antibody ligation is responsible for ceramide/LMP-mediated cell death
Here, we used the structured illumination microscopy (SIM) and confocal microscopy to probe the distribution and nanometer scale associations of CD20 on cell membranes and to determine the influence of Tyr102Lys (H102YK) mutation of rituximab on the distribution of CD20 molecules after antibody ligation. The distribution of CD20 in control cells and cells treated with CD20 mAbs for 30 min prior to fixation was assessed by SIM and confocal microscopy. As shown in Fig. 6A, CD20 microcluster formation can be observed after treatment with 11B8 or the rituximab variant H102YK, but rituximab could not have the ability to induce microcluster formation of CD20 molecule. In agreement with these results, the super resolution TIRF-SIM data provided further evidence for CD20 micorcluster formation induced by 11B8 and H102YK (Fig. 6C). To further investigate whether inhibition of de novo ceramide synthesis could have an effect on the microcluster formation of CD20, the ceramide synthase inhibitor FB1 was used. Our data showed that it could not influence the microcluster formation of CD20 (Fig. 6A). Addition of exogenous C2-ceramide (150 μM or 100 μM) could not initiate CD20 microcluster formation (Fig. 6B).
Figure 6.
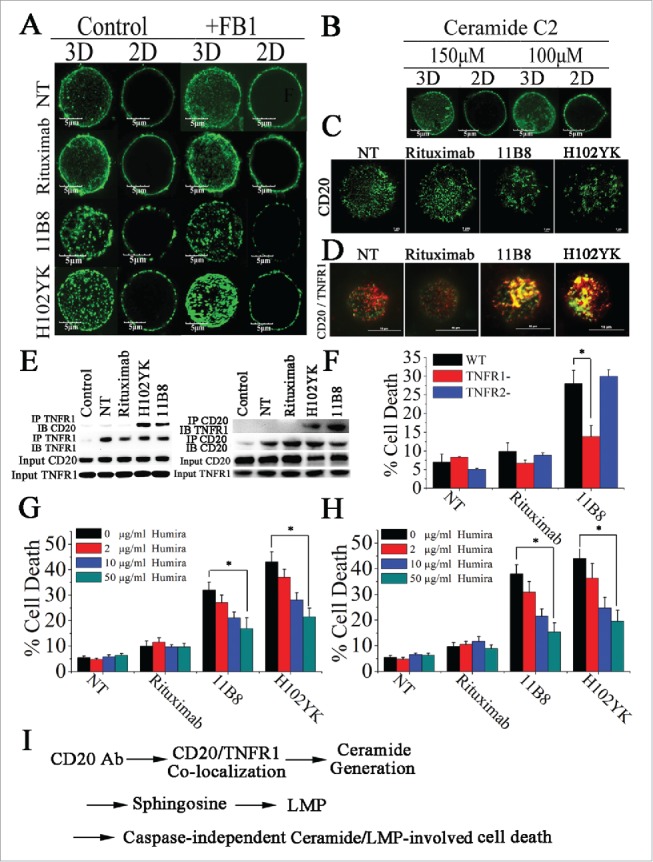
Colocalization of CD20 and TNFR1 initiated by CD20 mAbs is critical for initiation of lysosome-mediated cell death by CD20 mAbs. (A) Confocal images of the distribution of CD20 molecule after treatment with CD20 mAbs for 30 min. 3D reconstruction based on a confocal z-stack. (B) The impact of exogenous C2-ceramide on the spatial arrangement of CD20. Scale bars: 5 μm. (C) TIRF-SIM images of the distribution of CD20 in the bilayer before and after treatment with CD20 mAbs. Scale bars: 1 μm. (D) 3D-SIM images of the colocalization of CD20 (green fluorescence) and TNFR1 (red fluorescence) proteins before and after treatment with CD20 mAbs. The images were acquired by a structured illumination microscopy N-SIM (Nikon). Scale bars: 1 μm. (E) CD20 is recruited to the TNFR-1 signaling complex after treatment with type II CD20 mAbs and H102YK. The recruitment of CD20 protein was determined by western blot. Control: NT group used isotype antibody for IP, CD20 or TNFR1 antibody for IB. (F) The essential role of TNFR1 in induction of cell death triggered by type II CD20 mAbs. *p < 0.05. The inhibition of cell death by Humira after 48 h by Humira alone, 10 μg/mL rituximab, and rituximab variant in Raji (G) and Ramos (H) cells. Humira was added over a range of different concentrations for 2 h before the addition of mAbs. Data are mean ± SD (n = 3). *p < 0.05. (I) A proposed model role for initiation of lysosome-mediated cell death induced by CD20 antibodies.
Furthermore, we used three-dimensional SIM (3D-SIM) to investigate the involvement of TNFR1 in CD20 mAb-induced cell death. As shown in Fig. 6D, after treatment with 11B8 or H102YK, colocalization of CD20 and TNFR1 was observed. In line with these observations, the physical interaction between CD20 and TNFR1 can be observed after treatment with type II CD20 mAbs and H102YK (Fig. 6E). Decrease of TNFR1 expression has been validated by RT-PCR and western-blot (Fig. S6A and B). We found that downregulation of TNFR1 could significantly reduce cathepsin B release (Fig. S7A), inhibit 11B8 or H102YK-induced cell death and decrease the in vivo antitumor protection of type II CD20 mAb (Fig. S7B), whereas downregulation of TNFR2 could not affect the cell death (Fig. 6F). But decrease of TNFR1 expression could not affect the microcluster formation of CD20 by type II CD20 mAbs (Data not shown). In agreement with these findings, Humira, a therapeutic antibody binding to TNF α, was used in our experiments. We found that Humira could markedly inhibit the cell death triggered by H102YK in both Raji and Ramos cells in a dose-dependent manner (Fig. 6G and H).
Discussion
Recent studies have highlighted a scenario in which, upon prolonged treatment by a highly specific targeted therapy, tumor cells reprogram themselves to develop alternative compensatory pathways to sustain cell proliferation, ultimately leading to drug resistance. Previous studies showed that RR cells exhibited constitutive hyperactivation of the nuclear factor-κB and extracellular signal-regulated kinase 1/2 pathways, leading to overexpression of Bcl-2 and Bcl-2-related gene.4 Thus, attention has been driven toward other signaling pathways that circumvent the signaling events of classical apoptosis to eliminate cancer cells. However, due to the heterogeneous nature of tumors, different resistance mechanisms may coexist in the same patient. Targeting one molecule or one resistance mechanism is usually ineffective. Simultaneous activation of multiple killing mechanisms could be a promising strategy against drug-resistant tumor. In this study, we have optimized rituximab by introducing a point mutation Tyr102Lys, which endows rituximab variant with the advantages of both type I and type II CD20 mAbs, exhibiting significant CDC, ADCC and ceramide/LMP-mediated cell death against RR B-cell lymphoma. The two RR lymphoma cell lines were previously characterized in both others and our own group, which have shared some common characteristics with the primary RR samples from patients.15,23,32,33 To our knowledge, the resistance mechanisms of these RR lymphoma cell lines could be at least partially relate to real RR tumor cells generated in vivo. Furthermore, these observations have been confirmed by our ex vivo experiments with the primary patient samples from RR DLBCL and RR B-CLL, showing that H102YK could induce significant lysosome-mediated cell death against RR primary B-NHL cells. No significant difference in TNFR1 expression of these primary samples was observed (Data not shown). Recently, Nakayama and her colleagues interestingly found that TNFR1 expression variation could be correlated with the poor prognosis of DLBCL.34 In my opinion, although TNFR1-positive cases of DLBCL were reported to be significantly correlated with a poorer overall survival,34 these NHL patients with TNFR1 expression might potentially benefit from the administration of type II CD20 mAb and H102YK.
By using the F(ab′)2 fragments of CD20 antibodies, we have previously demonstrated that the direct caspase-independent cell death triggered by rituximab variant could not be related to the Fc part of antibody.23 Here, our results indicate that the caspase-independent cell death induced by rituximab variant belongs to ceramide/LMP-mediated cell death, suggested that combating rituximab resistance can be achieved through induction of potent ceramide/LMP-mediated cell death and simultaneous activation of multiple killing mechanisms could be an effective way to eradicate resistant B-cell lymphoma.
Although Golay and her colleagues have raised several important points regarding the possible pitfalls in assessing CD20 mAb-induced cell death by flow cytometry 35, Cragg and his colleagues have used four different techniques to evaluate the cell death.36 More importantly, the clinical trials have further confirmed the antitumor effects of GA101,37 which recently have approved by FDA for first-line CLL treatment.38 As we known, GA101 is a type II CD20 mAb with enhancement of hFcγRIII-dependent effector functions through glycomodification.39-41 In agreement with our present study, their results showed that there was no difference in in vivo B-cell depletion when comparing GA101, non-glycoengineered GA101 and non-glycoengineered mIgG2a versions of GA101,42 suggesting that the direct cell death induced by type II CD20 mAb GA101 may play an important role in its in vivo antitumor protection.
Growing evidences suggest that lysosomes can be considered as an “Achilles heel” for cancer cells and some lysosome targeting drugs have the ability to resensitize multidrug resistant cells to classical chemotherapy,18,19,21,22 suggesting that lysosome could be considered as an attractive target for combating rituximab resistance. Previous studies have revealed that type II CD20 mAbs can elicit lysosome-mediated cell death against lymphoma.24,25 Very recently, ceramide has been reported to be involved in type II CD20 mAb-induced cell death, although the mechanisms and the signal transduction pathways remain unclear.16,17 As we all known, microcluster formation of receptors has attracted increasing attention in recent years due to it is one of the earliest steps in receptor activation, which can initiate the early signaling events.43,44 To further investigate how to initiate ceramide/LMP-mediated cell death after CD20 mAb ligation, super-resolution SIM and confocal microscopy have been used to visualize the nanoscale organization of CD20 at the plasma membrane of human B lymphoma cells before and after CD20 mAb binding. Notably, type II CD20 mAb 11B8 could induce CD20 microcluster formation and colocalization of CD20-TNFR1 within 30 min, which exhibits the ability to initiate potent lysosome-mediated cell death. The relationship between CD20-TNFR1 colocalization and lysosome-mediated cell death has been further demonstrated by the comparative study of rituximab and rituximab variant H102YK. Our data show that CD20 microcluster formation and colocalization of CD20-TNFR1 can be accomplished by introducing only a point mutation Tyr102Lys in the CDR of rituximab, which endows the rituximab variant H102YK with the ability to trigger substantial lysosome-mediated cell death. These data indicate that CD20 microcluster formation and the involvement of TNFR1 are responsible for ceramide/LMP-mediated cell death initiated by CD20 mAbs.
In conclusion, the co-localization of CD20-TNFR1 initiated by CD20 mAbs stimulates de novo ceramide synthesis. Sphingosine subsequently produced from ceramide by ceramidase is directly responsible for LMP and consequent cell death, which has been demonstrated to be an effective way to combate rituximab resistance of B-cell lymphoma. In addition, the rituximab variant H102YK with the benefits of both type I and type II CD20 mAbs exhibits potent antitumor activities, even in RR lymphoma mouse models, suggesting that it may serve as a promising therapeutic agent for the treatment of human B-cell lymphoproliferative disorders.
Materials and methods
Patient samples
Human NHL samples were collected with the informed consent of the patients and the experiments were approved by Institute Research Ethics Committee at Cancer Center, PLA General Hospital; ethics committee of Affiliated Hospital of Academy of Military Medical Science; ethics committee of Second Military Medical University. The primary cells were maintained in RPMI 1640 medium (Hyclone Laboratories, USA), supplemented with 10% fetal bovine serum (Hyclone Laboratories, USA), and incubated at 37°C in 5% CO2humidified air.
Cell death assay
Cells were incubated with CD20 mAbs (10 μg/mL) or different concentrations of C2-ceramide (Sigma-Aldrich) at 37°C, with 5% CO2, for 16 h. Various inhibitors were added before CD20 mAb adding to the culture media. After washing, cells were treated with Annexin-V Alexa Fluor 488 & Propidium Iodide (PI) (Invitrogen) on ice for 15 min protected from light. Flow cytometry (FCM) were performed to analyze cell death (Annexin V- and PtdIns-positive cells). Alternative assays used to detect mAb-induced cell death. Raji cells were treated with mAb (10 μg/mL) for 48 h and stained with SYTOX red (Invitrogen). Cell death was then analyzed by FCM.
In XTT assays, antibody-treated cells (100-µL samples) were harvested and incubated with 50 µL of XTT reagent for 4 h at 37°C in 96-well plates, then absorbance was detected at 485 nm (reference wavelength: 680 nm) using the multi-detection plate reader and normalized relative to untreated controls (determine the average value from the triplicate readings and subtract the average value for the blank wells as well as the average value of the non-specific readings).
Lysosomal permeability assessment
The method we used are followed as previously described.24,25 In brief, cells were labeled with 200 nM Lyso-Tracker probe (Invitrogen) at 37°C after treatment with CD20 mAbs with or without fumonisin B1 (FB1) (25μM, Sigma-Aldrich) adding to the culture media before for 16 h. Fluorescence of Lyso-Tracker labeled cells was assessed by FCM and confocal microscopy. Unlabeled cells were used as a background control.
Structured illumination microscopy
Cells were imaged by using Nikon's N-SIM microscopy system, which can achieve a resolution of 100 nm along the x–y axis and 300 nm along the z-axis. TIRF-SIM and 3D-SIM Images were acquired with Andor Technology iXon3 897 EMCCD camera coupled to a Nikon Motorized inverted microscope ECLIPSE Ti-E. Images were recorded and processed with NIS-Elements software (Nikon).
Immunotherapy
Groups of 10 8-week-old female SCID mice were injected via the tail vein with 3.5 × 106 Raji or Raji-R cells on day 0, followed 7 d later by the intravenous injection of CD20 mAb IgG (100 or 400 μg/mouse). For the Rituximab and 11B8 simultaneous treatment groups, groups of 10 SCID mice were injected with 3.5 × 106 Raji-R cells intravenously on day 0 and then treated with 100 μg or 200 μg each of mAbs intravenously on day 7. The mice were observed daily and killed at the onset of hind-leg paralysis.
Statistics
Data represent mean ± SD of representative experiments, unless otherwise stated. Statistical significance was calculated by Student unpaired t test to identify significant differences unless otherwise indicated. Survival of mice was analyzed by log-rank tests. Differences were considered significant at a p value of less than 0.05.
Please refer to Supplementary Data for complete details of Methods.
Supplementary Material
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (81301956, 81402552, 81471578), National Major Scientific and Technological Special Project for “Significant New Drugs Development,” Beijing Science & Technology Nova Program, Innovation Program of Shanghai Municipal Education Commission (15ZZ040), the Natural Science Foundation of Beijing, China (7162177, 7154238, 7162179), Roche's Scientific Corporation Program and Young Scholar Program of Second Military Medical University (2014QN01).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are grateful to Nikon instruments (Beijing) and the Nikon-Tsinghua bio-imaging Core Facility for providing technical support. We are deeply thankful to Prof. Yajun Guo for his valuable comments and suggestions.
Author contributions
Conception and design: L. Zhao, X. Wang. Development of methodology: F. Zhang, J. Yang, H. Li, M. Liu, J. Zhang, L. Zhao, L. Wang, R. Linghu, F. Feng, X. Gao, B. Dong, X. Liu, J. Zi, W. Zhang, Y. Hu, J. Pan, L. Tian, Y. Hu, Z. Han, H. Zhang. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): F. Zhang, J. Yang, H. Li, M. Liu, J. Zhang, L. Zhao, L. Wang, R. Linghu, F. Feng, X. Gao, B. Dong, X. Liu, J. Zi, W. Zhang, Y. Hu, J. Pan, L. Tian, Y. Hu, Z. Han, H. Zhang. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): F. Zhang, J. Yang, H. Li, M. Liu, J. Zhang, L. Zhao, L. Wang, R. Linghu, F. Feng, X. Gao, B. Dong, X. Liu, J. Zi, W. Zhang, Y. Hu, J. Pan, L. Tian, Y. Hu, Z. Han, H. Zhang. Writing, review, and/or revision of the manuscript: L. Zhao, X. Wang. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): L. Zhao.
References
- 1.Boye J, Elter T, Engert A. An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann Oncol 2003; 14:520-35; PMID:12649096; http://dx.doi.org/ 10.1093/annonc/mdg175 [DOI] [PubMed] [Google Scholar]
- 2.Bello C, Sotomayor EM. Monoclonal antibodies for B-cell lymphomas: rituximab and beyond. Hematology Am Soc Hematol Educ Program 2007; 2007:233-42; PMID:18024635; http://dx.doi.org/ 10.1182/asheducation-2007.1.233 [DOI] [PubMed] [Google Scholar]
- 3.Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 2008; 14:1561-70; PMID:18316581; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1254 [DOI] [PubMed] [Google Scholar]
- 4.Jazirehi AR, Vega MI, Bonavida B. Development of rituximab-resistant lymphoma clones with altered cell signaling and cross-resistance to chemotherapy. Cancer Res 2007; 67:1270-81; PMID:17283164; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2184 [DOI] [PubMed] [Google Scholar]
- 5.Olejniczak SH, Hernandez-Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired resistance to rituximab is associated with hemotherapy resistance resulting from decreased bax and bak expression. Clin Cancer Res 2008; 14:1550-60; PMID:18316580; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1255 [DOI] [PubMed] [Google Scholar]
- 6.Du J, Wang H, Zhong C, Peng B, Zhang M, Li B, Huo S, Guo Y, Ding J. Structural basis for recognition of CD20 by therapeutic antibody Rituximab. 2007; 282:15073-80; PMID:17395584 [DOI] [PubMed] [Google Scholar]
- 7.Perosa F, Favoino E, Vicenti C, Guarnera A, Racanelli V, De Pinto V, Dammacco F. Two structurally different rituximab-specific CD20 mimotope peptides reveal that rituximab recognizes two different CD20-associated epitopes. J Immunol 2009; 182:416-23; PMID:19109173; http://dx.doi.org/ 10.4049/jimmunol.182.1.416 [DOI] [PubMed] [Google Scholar]
- 8.Teeling JL, Mackus WJM, Wiegman LJJM, van den Brakel JHN, Beers SA, French RR, van Meerten T, Ebeling S, Vink T, Slootstra JW et al.. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 2006; 177:362-71; PMID:16785532; http://dx.doi.org/ 10.4049/jimmunol.177.1.362 [DOI] [PubMed] [Google Scholar]
- 9.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood 2004; 104:2635-42; PMID:15226177; http://dx.doi.org/ 10.1182/blood-2004-03-1110 [DOI] [PubMed] [Google Scholar]
- 10.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol 2007; 44:3823-37; PMID:17768100; http://dx.doi.org/ 10.1016/j.molimm.2007.06.151 [DOI] [PubMed] [Google Scholar]
- 11.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC et al.. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005-10; PMID:16537476; http://dx.doi.org/ 10.1073/pnas.0508123103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan HTC, Beers SA, French RR, Cox KL, Davies AJ et al.. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood 2011; 118:2530-40; PMID:21768293; http://dx.doi.org/ 10.1182/blood-2011-01-330357 [DOI] [PubMed] [Google Scholar]
- 13.Bowles JA, Wang S-Y, Link BK, Allan B, Beuerlein G, Campbell M-A, Marquis D, Ondek B, Wooldridge JE, Smith BJ et al.. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood 2006; 108:2648-54; PMID:16825493; http://dx.doi.org/ 10.1182/blood-2006-04-020057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002; 99:754-8; PMID:11806974; http://dx.doi.org/ 10.1182/blood.V99.3.754 [DOI] [PubMed] [Google Scholar]
- 15.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene 2003; 22:7359-68; PMID:14576843; http://dx.doi.org/ 10.1038/sj.onc.1206939 [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Shu L, Wu J. Ceramide participates in lysosome-mediated cell death induced by type II anti-CD20 monoclonal antibodies. Leuk Lymphoma 2015; 56:1863-8; PMID:25393677; http://dx.doi.org/ 10.3109/10428194.2014.981179 [DOI] [PubMed] [Google Scholar]
- 17.Ren H, Zhang C, Su L, Bi X, Wang C, Wang L, Wu B. Type II anti-CD20 mAb-induced lysosome mediated cell death is mediated through a ceramide-dependent pathway. Biochem Biophys Res Commun 2015; 457:572-7; PMID:25603047; http://dx.doi.org/ 10.1016/j.bbrc.2015.01.026 [DOI] [PubMed] [Google Scholar]
- 18.Petersen NHT, Olsen OD, Groth-Pedersen L, Ellegaard A-M, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lønborg A et al.. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of Acid sphingomyelinase. Cancer Cell 2013; 24:379-93; PMID:24029234; http://dx.doi.org/ 10.1016/j.ccr.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 19.Fehrenbacher N, Jäättelä M. Lysosomes as targets for cancer therapy. Cancer Res 2005; 65:2993-5; PMID:15833821 [DOI] [PubMed] [Google Scholar]
- 20.Mathiasen IS, Jäättelä M. Triggering caspase-independent cell death to combat cancer. Trends Mol Med 2002; 8:212-20; PMID:12067630; http://dx.doi.org/ 10.1016/S1471-4914(02)02328-6 [DOI] [PubMed] [Google Scholar]
- 21.Groth-Pedersen L, Jäättelä M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett 2013; 332:265-74; PMID:20598437; http://dx.doi.org/ 10.1016/j.canlet.2010.05.021 [DOI] [PubMed] [Google Scholar]
- 22.Česen MH, Pegan K, Špes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res 2012; 318:1245-51; PMID:22465226; http://dx.doi.org/ 10.1016/j.yexcr.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 23.Li B, Zhao L, Guo H, Wang C, Zhang X, Wu L, Chen L, Tong Q, Qian W, Wang H et al.. Characterization of a rituximab variant with potent antitumor activity against rituximab-resistant B-cell lymphoma. Blood 2009; 114:5007-15; PMID:19828699; http://dx.doi.org/ 10.1182/blood-2009-06-225474 [DOI] [PubMed] [Google Scholar]
- 24.Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, Shimada K, Chan CHT, Tutt A, Beers SA et al.. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011; 117:4519-29; PMID:21378274; http://dx.doi.org/ 10.1182/blood-2010-07-296913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL, Potter KN, Murray S, Chan CHT, Klymenko T et al.. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest 2009; 119:2143-59; PMID:19620786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 2004; 4:604-16; PMID:15286740; http://dx.doi.org/ 10.1038/nrc1411 [DOI] [PubMed] [Google Scholar]
- 27.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9:139-50; PMID:18216770; http://dx.doi.org/ 10.1038/nrm2329 [DOI] [PubMed] [Google Scholar]
- 28.Bielawska A, Crane HM, Liotta D, Obeid LM, Hannun YA. Selectivity of ceramide-mediated biology. Lack of activity of erythro-dihydroceramide. 1993; 268:26226-32; PMID:8253743 [PubMed] [Google Scholar]
- 29.Kågedal K, Zhao M, Svensson I, Brunk UT. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J 2001; 359:335-43; PMID:11583579; http://dx.doi.org/ 10.1042/bj3590335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu R, Sun W, Jin J, Obeid LM, Mao C. Role of alkaline ceramidases in the generation of sphingosine and its phosphate in erythrocytes. The FASEB Journal 2010; 24:2507-15; PMID:20207939; http://dx.doi.org/ 10.1096/fj.09-153635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheson BD, Cheson BD, Horning SJ, Horning SJ, Coiffier B, Shipp MA, Shipp MA, Fisher RI, Connors JM, Connors JM et al.. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. 1999. page 1244 [DOI] [PubMed] [Google Scholar]
- 32.Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, Kiyoi H, Kinoshita T, Naoe T. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood [Internet] 2009; 113:4885-93. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19246561&retmode=ref&cmd=prlinks; PMID:19246561; http://dx.doi.org/ 10.1182/blood-2008-08-175208 [DOI] [PubMed] [Google Scholar]
- 33.Jilani I, O'Brien S, Manshuri T, Thomas DA, Thomazy VA, Imam M, Naeem S, Verstovsek S, Kantarjian H, Giles F et al.. Transient down-modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood 2003; 102:3514-20; PMID:12893761; http://dx.doi.org/ 10.1182/blood-2003-01-0055 [DOI] [PubMed] [Google Scholar]
- 34.Nakayama S, Yokote T, Tsuji M, Akioka T, Miyoshi T, Hirata Y, Hiraoka N, Iwaki K, Takayama A, Nishiwaki U et al.. TNF-α receptor 1 expression predicts poor prognosis of diffuse large B-cell lymphoma, not otherwise specified. Am J Surg Pathol 2014; 38:1138-46; PMID:24805855; http://dx.doi.org/ 10.1097/PAS.0000000000000094 [DOI] [PubMed] [Google Scholar]
- 35.Golay J, Bologna L, André P-A, Buchegger F, Mach JP, Boumsell L, Introna M. Possible misinterpretation of the mode of action of therapeutic antibodies in vitro: homotypic adhesion and flow cytometry result in artefactual direct cell death. Blood 2010; 116:3372–3–authorreply3373-4; PMID:21030571; http://dx.doi.org/ 10.1182/blood-2010-06-289736 [DOI] [PubMed] [Google Scholar]
- 36.Cragg MS, Alduaij W, Klein C, Umana P, Glennie MJ, Illidge TM. Response: novel lysosomal-dependent cell death following homotypic adhesion occurs within cell aggregates. Blood 2010; 116:3373-4; http://dx.doi.org/ 10.1182/blood-2010-07-291054 [DOI] [Google Scholar]
- 37.Morschhauser FA, Cartron G, Thieblemont C, Solal-Celigny P, Haioun C, Bouabdallah R, Feugier P, Bouabdallah K, Asikanius E, Lei G et al.. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol [Internet] 2013; 31:2912-9. Available from: http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2012.46.9585; PMID:23835718; http://dx.doi.org/ 10.1200/JCO.2012.46.9585 [DOI] [PubMed] [Google Scholar]
- 38.Lee H-Z, Miller BW, Kwitkowski VE, Ricci S, DelValle P, Saber H, Grillo J, Bullock J, Florian J, Mehrotra N et al.. U.S. Food and drug administration approval: obinutuzumab in combination with chlorambucil for the treatment of previously untreated chronic lymphocytic leukemia. Clin Cancer Res 2014; 20:3902-7; PMID:24824310; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-0516 [DOI] [PubMed] [Google Scholar]
- 39.Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E et al.. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393-402; PMID:20194898; http://dx.doi.org/ 10.1182/blood-2009-06-225979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, Klein C, Introna M. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 2013; 122:3482-91; PMID:24106207; http://dx.doi.org/ 10.1182/blood-2013-05-504043 [DOI] [PubMed] [Google Scholar]
- 41.Herter S, Birk MC, Klein C, Gerdes C, Umaña P, Bacac M. Glycoengineering of therapeutic antibodies enhances monocyte/macrophage-mediated phagocytosis and cytotoxicity. J Immunol 2014; 192:2252-60; PMID:24489098; http://dx.doi.org/ 10.4049/jimmunol.1301249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tipton TRW, Roghanian A, Oldham RJ, Carter MJ, Cox KL, Mockridge CI, French RR, Dahal LN, Duriez PJ, Hargreaves PG et al.. Antigenic modulation limits the effector cell mechanisms employed by type I anti-CD20 monoclonal antibodies. Blood 2015; 125:1901-9; PMID:25631769; http://dx.doi.org/ 10.1182/blood-2014-07-588376 [DOI] [PubMed] [Google Scholar]
- 43.Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VLJ, Batista FD. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol 2008; 9:63-72; PMID:18059271; http://dx.doi.org/ 10.1038/ni1547 [DOI] [PubMed] [Google Scholar]
- 44.Gadella TW, Jovin TM. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J Cell Biol 1995; 129:1543-58; PMID:7790353; http://dx.doi.org/ 10.1083/jcb.129.6.1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.