

ABSTRACT
The factors that determine whether disseminated transformed cells in vivo yield neoplastic lesions have only been partially identified. We established an ad hoc model of peritoneal carcinomatosis by injecting colon carcinoma cells in mice. Tumor cells recruit inflammatory leukocytes, mostly macrophages, and generate neoplastic peritoneal lesions. Phagocyte depletion via clodronate treatment reduces neoplastic growth. Colon carcinoma cells release a prototypic damage-associated molecular pattern (DAMP)/alarmin, High Mobility Group Box1 (HMGB1), which attracts leukocytes. Exogenous HMGB1 accelerates leukocyte recruitment, macrophage infiltration, tumor growth and vascularization. Lentiviral-based HMGB1 knockdown or pharmacological interference with its extracellular impair macrophage recruitment and tumor growth. Our findings provide a preclinical proof of principle that strategies based on preventing HMGB1-driven recruitment of leukocytes could be used for treating peritoneal carcinomatosis.
KEYWORDS: Alarmin, BoxA, DAMP, damage-associated molecular pattern, GFP, green fluorescent protein, HMGB1, High Mobility Box 1, leukocytes, macrophages, MC-38, murine colon adenocarcinoma cell line, peritoneal carcinomatosis, ShRNA, short hairpin RNA, vascularization
Introduction
Peritoneal carcinomatosis is the most common terminal feature of abdominal cancers caused by the propagation of neoplastic cells on peritoneal surfaces. Peritoneal carcinomatosis presents with malignant ascites, abundant neoplastic nodules disseminated in the peritoneal cavity and multiple layers of fibrous tissue involving serosal surfaces and contiguous organs. Carcinomatosis originating from ovarian, colorectal, gastric and pancreatic carcinomas is common, accounting for up to 19% of patients after curative resection of carcinoma and 23–29% of patients with gastric cancer.1,2
Inflammatory leukocytes massively infiltrate human solid tumors3 and actively interact with neoplastic cells. The composition and activation status of tumor-infiltrating leukocytes depend on the tumor type and stage as well as on the anatomic location.4 High numbers of infiltrating cytotoxic T lymphocytes and low numbers of tumor-associated macrophages (TAM) correlate with favorable prognosis in ovarian, breast and colon carcinoma.5,6 Conversely, a high number of TAM correlates with poor prognosis. In colorectal carcinoma, TAMs have been shown to release growth factors, matrix-remodeling enzymes and promote vascularization.7-9 The vascularization is often strictly dependent on macrophages and is fundamental for the tumor growth, invasion and metastatization. New vessels provide oxygen, nutrients and growth factors and allow cancer cells to extravasate and disseminate. The molecular signals by which the neoplastic tissue recruits inflammatory and pro-angiogenic macrophages have been only partially understood so far and could represent novel targets for molecular intervention.
All eukaryotic cells express the nuclear protein HMGB1. HMGB1 is passively released by dying cells and actively generated in inflammatory conditions: it acts in the extracellular environment as a prototypical DAMP/alarmin.10-13 Little is known on the role of HMGB1 in peritoneal carcinogenesis. We have recently observed that HMGB1 released as a consequence of 5-fluorouracil based chemotherapy activates autophagic and apoptotic pathways and might contribute to the resistance of cancer cells to conventional chemotherapy.14
Here we show, using an experimental model of peritoneal carcinomatosis, that colon carcinoma cells use their own HMGB1 to promote the continuous recruitment of leukocytes, in particular macrophages, which are necessary for lesions growth and spreading and foster neovascularization. Blocking HMGB1 activity in vivo with BoxA, a fragment of HMGB1, inhibits leukocyte recruitment and lesion growth, providing proof-of-principle of a new molecularly targeted therapeutic approach to peritoneal carcinomatosis.
Results
Colon carcinoma cells yield neoplastic lesions when injected in the mouse peritoneal cavity and attract inflammatory leukocytes. To mimic human carcinomatosis, which depends on the dissemination in the peritoneal cavity of neoplastic cells derived from tumors of abdominal organs, we injected increasing numbers of MC-38 carcinoma cells, a line which has been derived from a primary poorly immunogenic neoplasm,15 in the peritoneal cavity of syngeneic immunocompetent mice. As little as 205 thousand cells were sufficient to elicit experimental carcinomatosis in all injected mice (Fig. 1A).
Figure 1.
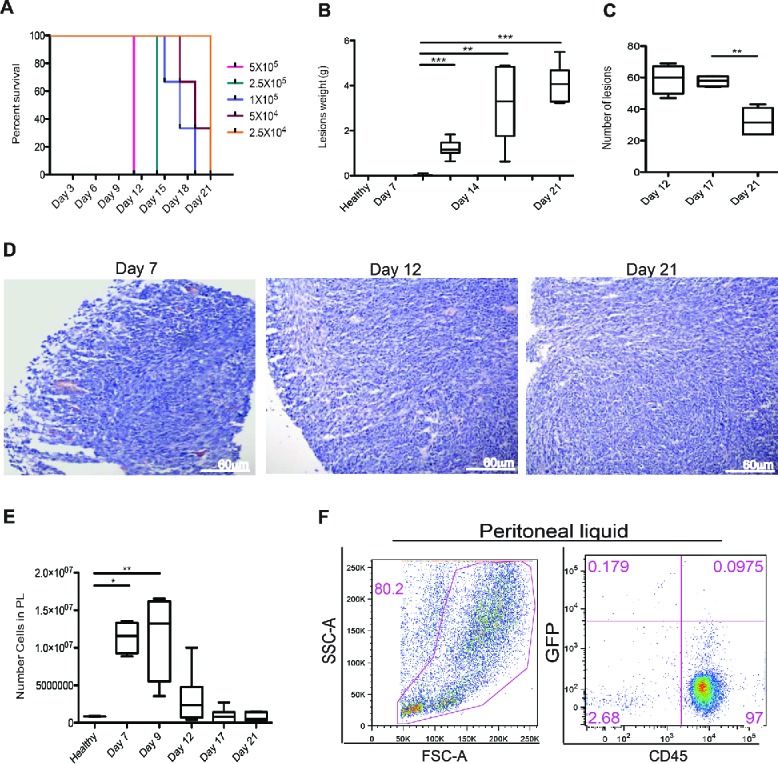
Peritoneal carcinomatosis and leukocyte attraction in the peritoneum. (A) Evaluation of the minimal tumorigenic dose by injection of murine colon cancer cells (MC-38) in the peritoneum of syngeneic mice. Results are expressed as a percentage of survival of n = 5 animals for each time point and dose of MC-38 cells injected in peritoneum. (B) Lesion weight and (C) number of peritoneal lesions at different time points. Results are expressed as mean ± SEM of data pooled from three independent experiments (n = 9 animals per each time point). (D) Representative Hematoxylin and Eosin (H&E) staining of neoplastic lesions obtained after intraperitoneal MC-38 cell injection. 20X magnification. (E) Cell counts in the peritoneal liquid (PL) at different time points after MC-38 cell injection. Results are expressed as mean ± SEM of three independent experiments (n = 9 animals per each time point); **p <0.005, *p <0.05, significantly different from cell counts in the PL of healthy mice (Unpaired T test with Welch correction). All mice received intraperitoneally the minimal tumorigenic dose of MC-38 (2 × 104 cells). (F) Representative dot plot of PL cells retrieved 21 d after intraperitoneal GFP+MC-38 cell injection (5 × 104cells per mouse). Physical parameters (Forward Scatter, FSC-A, x-axis vs. Side Scatter, SSC-A, y-axis, left) and fluorescence analysis (leukocyte CD45 expression, x-axis vs. GFP MC-38 y-axis, right). n = 10 animals for two independent experiments.
Carcinoma cells aggregated, forming lesions that closely resemble human carcinomatosis (Fig. 1B–D). Tumor cells adhered to parietal or visceral peritoneal surfaces, infiltrating and disrupting the Muscularis mucosaevia the submucosa and hence the Muscularis propria. The overall tumor burden progressively grew with time, as assessed by the total lesion weight, until we suppressed mice for ethical reasons at day 21 post injection (Fig. 1B). The number of lesions remained initially stable, while decreased at later time points because of the fusion of expanding serosal lesions (Fig. 1C). Lesions were attached to the peritoneal lining and ranged from small neoplastic nodules to multiple masses of various sizes (consisting of irregular tubular structures characterized by stratification, multiple lumens and reduced stroma) and eventually to multiple layers of confluent tissue that envelop the serosal surfaces and peritoneal organs (Fig. 1D).
Lesions were not or only barely detectable at day 7 post injection. At this time, we already observed massive recruitment of cells in the peritoneal cavity (Fig. 1E). The cell number in the peritoneal fluid progressively decreased at later time points, concomitantly with the growth of neoplastic lesions. More than 95% of the cells floating in the peritoneal liquid of tumor-bearing mice were CD45+ leukocytes, as assessed by flow cytometry (Fig. 1F). These cells were characterized by flow cytometry and in the peritoneal fluid mostly comprised CD11b+, F4/80+ macrophages (31%), CD3+ T lymphocytes (22%), CD19+ B lymphocytes (16%), NK1.1+NK cells (14%) and Ly6G+ neutrophils (17%) (Table 1).
Table 1.
Characterization of tumor-infiltrating leukocytes in peritoneal lesions and fluid.
| Peritoneal lesions (%) | Peritoneal fluid (%) | |
|---|---|---|
| Mono/Macrophages | 60 | 31 |
| T lymphocytes | 20 | 21 |
| B lymphocytes | 4 | 16 |
| NK cells | 6 | 14 |
| Neutrophils | 10 | 17 |
Leukocytes infiltrating the peritoneal lesions of tumor-bearing mice at day 21 were characterized by immunohistochemistry and the fraction of cells expressing the CD68 marker (mono-macrophages), the CD3 (T lymphocytes), the CD19 (B lymphocytes), the NK1+ (NK cells) and the Ly6G (neutrophils) was determined. Cells floating in the peritoneal cavity of tumor-bearing mice at day 21 were characterized by flow cytometry and the fraction of cells expressing the CD11b and F4/80 markers (mono-macrophages), the CD3 (T lymphocytes), the CD19 (B lymphocytes), the NK1.1+ (NK cells) and the Ly6G (neutrophils) was also determined.
Leukocytes are required for the growth and the vascularization of peritoneal lesions. Peritoneal lesions were highly vascularized (Fig. 2A) and the vascular structure was maintained from establishment throughout the progressive growth of the lesions (Fig. 2B–D). CD45+ leukocytes massively infiltrated tumor lesions at all time points analyzed, as detected by immunohistochemistry (Fig. 2E). At 21 d after the injection of Green Florescence Protein (GFP) colon carcinoma cells, CD45+ leukocytes represent up to 25% of the cell mass lesion (Fig. 2F–G). The majority of tumor-infiltrating leukocytes were mono/macrophages (CD68+, 60%). Other infiltrating cells present in the lesions included T lymphocytes (CD3+, 20%), B lymphocytes (CD19+, 4%), NK cells (NK1.1+, 6%) and neutrophils (Ly6G+, 10%) as assessed by immunohistochemistry (Table 1).
Figure 2.
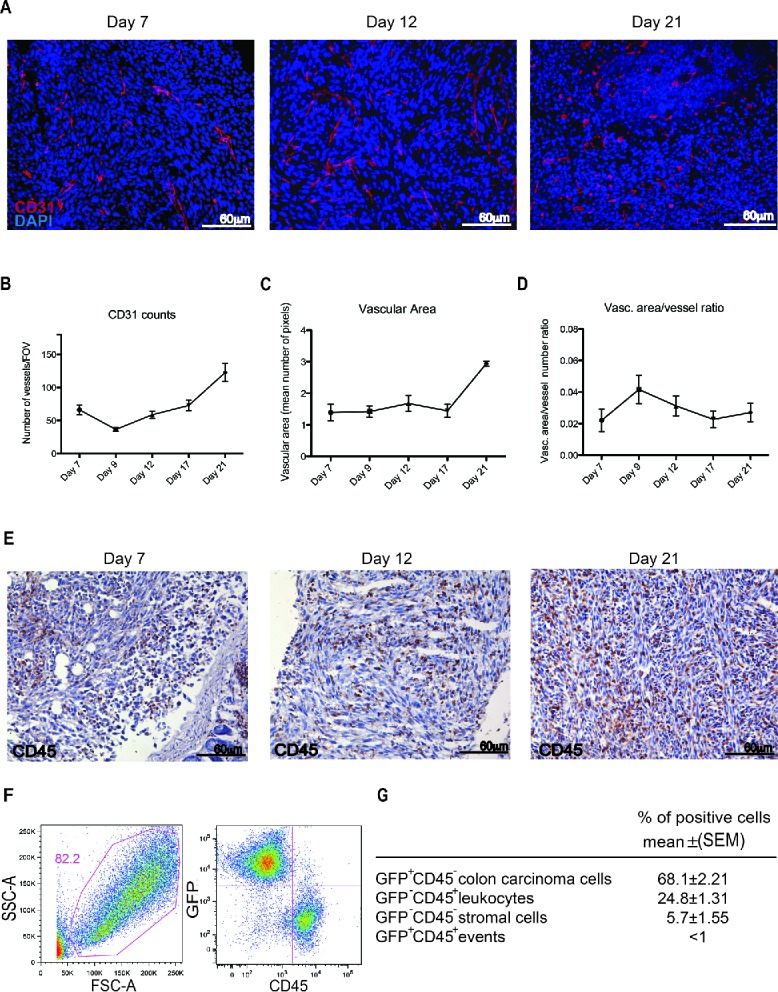
Leukocyte infiltration and vascularization of the peritoneal lesions. (A) CD31-positive endothelial cells (red) in peritoneal lesions. Nuclei were counter-stained with DAPI (blue). 20X magnification. Vessels per Field of Vision (FOV) (B) mean vascular area (C) and ratio between the vascular area and the number of vessels (D) in peritoneal lesions retrieved at various times after intraperitoneal MC-38 cell injection. CD31+ cells were identified by digital image analysis.15 Results are expressed as mean ± SEM of CD31+ cells/FOV, n = 15 animals for each time points for three independent experiment. (E) CD45+ leukocytes were revealed by immunohistochemistry in neoplastic lesions collected at days 7, 12 or 21. n = 9 animals for each time point for three independent experiments. (F) Representative dot plot of cells retrieved after enzymatic digestion of neoplastic peritoneal lesions collected at day 21. Physical parameters (Forward Scatter, FSC-A, x-axis vs. Side Scatter, SSC-A, y-axis left) and fluorescence analysis (leukocyte CD45 expression, x-axis vs. GFP MC-38 y-axis, right). (G) Table shows the percentage of cells population that infiltrate the peritoneal tumor lesions. n = 10 animals for two independent experiments.
Established peritoneal lesions attract phagocytes from the peripheral blood. We then investigated whether tumor-infiltrating leukocytes were mostly derived from local proliferation or were recruited from peripheral blood. Specifically, we used MRI to trace syngeneic phagocytes labeled with super paramagnetic iron oxide (SPIO) particles and injected intravenously in tumor-bearing animals 19 d after tumor cell intraperitoneal injection, i.e. at a time when lesion architecture is well established. Three hours after injection-labeled cells become detectable in neoplastic lesions, as shown by a reduction—albeit limited—in T2* relaxation time. The phenomenon became more evident 24 h later (Fig. 3A–C): The recruitment of SPIO-labeled phagocytes was evident on T2*-weighted images as a progressive reduction of the signal intensity of the tumor lesions. This reflects T2* relaxation time shortening, due to increasing concentration of iron particles (Fig. 3B–C). T2* did not change in other tissues, such as paravertebral skeletal muscle (Fig. 3A–C).
Figure 3.
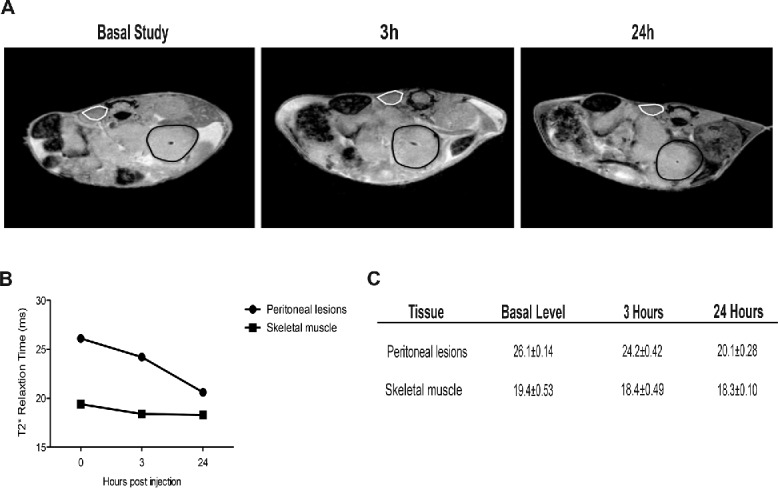
Recruitment of blood leukocytes into established peritoneal carcinomatosis lesions. 7 Tesla, T2*-weighted, MR images of abdomen of a representative mouse from tumor-bearing mice injected with syngeneic leukocytes labeled with SPIO particles (0.22 mg Fe/mL), at different time points (basal, 3 h, 24 h, panel A). At 24 h, the peripheral darkening of peritoneal lesions is strongly evident, without changes in signal intensity of the paravertebral muscle (negative control). A slight reduction of T2* relaxation time is detectable 3 h after SPIO-labeled leukocyte injection, which becomes stronger 24 h after injection (B, C). ROI based analysis of T2-maps: white color, skeletal muscle; black color, peritoneal lesions.
Peritoneal macrophages are essential for peritoneal carcinomatosis
Since phagocytes are continually recruited into the lesions, we tested the effect of curtailing their recruitment. Tumor-bearing mice were treated with either clodronate to deplete macrophages, or sham-treated with phosphate-buffered saline encapsulated into liposomes (Clodrolip and Shamlip, respectively). Treatments were administered intraperitoneally the day after MC-38 cells injection and every other day until the end of the study at day 21. This treatment was consistently effective in depleting phagocytes, as assessed by staining of cells in the peritoneal fluid for the expression of the F4/80 macrophage marker (Fig. 4A–B), Macrophages, identified by immunohistochemistry for expression of the pan-macrophage marker CD68, were also depleted from neoplastic lesions (Fig. 4B–D).
Figure 4.
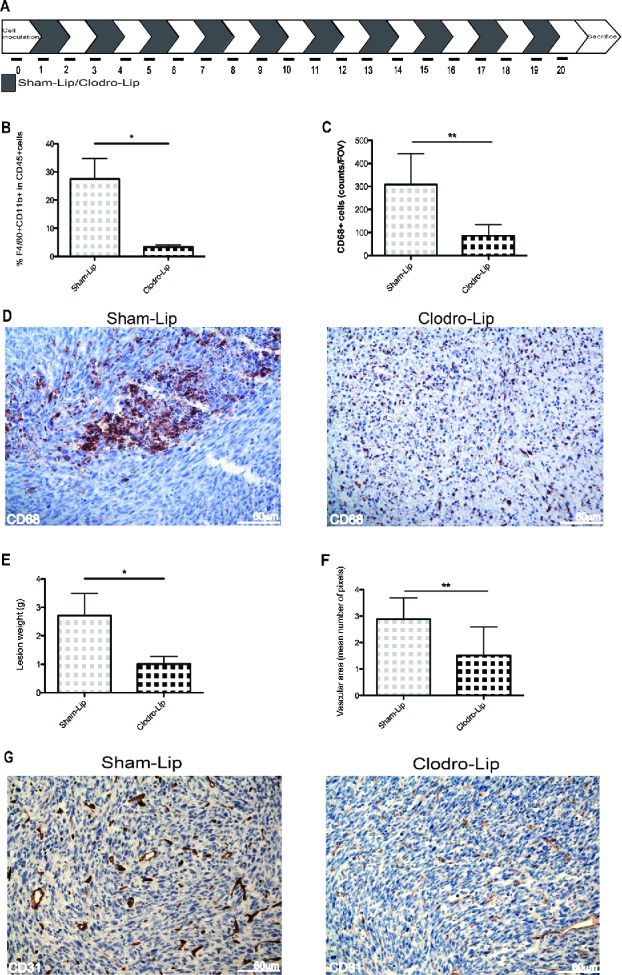
Growth and vascularization of peritoneal lesions upon macrophage depletion. (A) Schematic representation of macrophage depletion by treatment with liposomes containing Clodronate (Clodro-Lip) or saline (Sham-Lip). Treatment started 1 day after MC-38 carcinoma cell injection; mice were treated every other day until animal sacrifice (day 21). (B) Peritoneal lesion weight at sacrifice in the presence or the absence of macrophages was assessed and (C) Efficacy of the depletion was assessed by flow cytometry monitoring the percentage of F4/80+CD11b+ CD45+cells in the peritoneal liquid (D) immunohistochemistry showing infiltration of peritoneal lesions by CD68+ macrophages in Clodro-Lip (macrophage-depleted) and Sham-Lip (control) treated mice. (E) Number of infiltrating CD68+ macrophages per field of view in peritoneal lesions. (F) Mean vascular area in peritoneal lesions, calculated by digital image analysis. In panels (B), (C), (E), (F), data are expressed as mean ± SEM. n = 10 animals per group for two independent experiments. *p < 0.05; **p < 0.005; *p < 0.05; **p < 0.005 (Unpaired T test). (G) CD31+endothelial cells were revealed by immunohistochemistry in peritoneal lesions of Clodro-Lip and Sham-Lip (control) treated mice.
The depletion of macrophages effectively constrained tumor growth (Fig. 4E) indicating that macrophages contribute to the neoplastic progression. The lesion vasculature of mice was disrupted upon macrophage depletion, and vascular area was significantly reduced (Fig. 4F–G), indicating that infiltrating macrophages are involved in tumor angiogenesis.
The DAMP HMGB1 is released by colon carcinoma cells and induces the migration of macrophages, an event that partially depend on its ability to form complexes with the CXCL12 chemokine. Colon carcinoma cells express HMGB1 (Fig. 5). As expected, most intracellular HMGB1 was released upon necrosis induced by freeze-thaw (Fig. 5). The released HMGB1 was biologically active, since the supernatant (Conditioned Medium, CM) of necrotic cells promoted the chemotaxis of murine macrophages, while antagonists of HMGB1 (blocking monoclonal antibodies and the competitive antagonist, BoxA), or of its receptor RAGE abrogated chemotaxis (Fig. 5). HMGB1 action appears to require the integrity of the CXCL12/CXCR4 axis, since it also abates in the presence of the specific CXCR4 antagonist, AMD3100 (Fig. 5).
Figure 5.
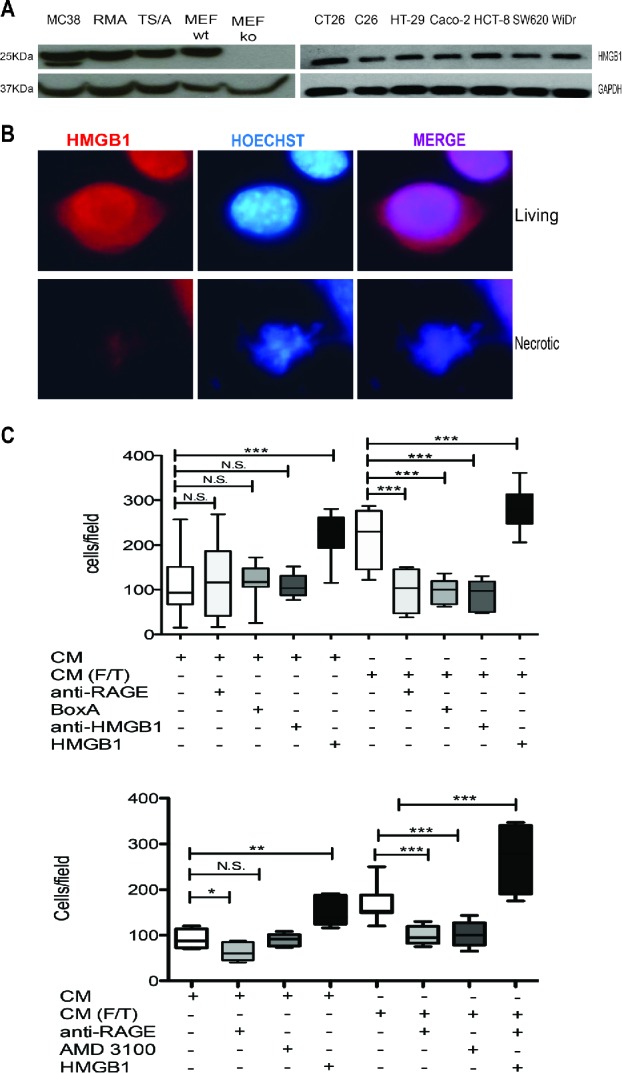
Colon cancer cell HMGB1 expression and biological activity. (A) Western Blot analysis of HMGB1 in murine colon carcinoma cells (MC-38); H-2b mouse lymphoma cells (RMA); mouse mammary adenocarcinoma cell line (TS/A); wild-type mouse Embryonic Fibroblast cells (MEF-wt) and Hmgb1 Knock Out Murine Embryonic Fibroblast cells (MEF-ko); murine colon adenocarcinoma cell lines (CT-26; C-26) and human colon adenocarcinoma cell lines (HT-29; Caco-2; HCT8; SW 620; WiDr). GAPDH was used as loading control. (B) HMGB1 expression (red) was revealed by immunofuorescence in living or freezed-thawed necrotic (F/T) MC-38 cells. Nuclei were counterstained with HOECHST (blue). (C) Murine bone-marrow-derived macrophage migration was assessed in a Boyden Chamber assay in response to conditioned media (CM) of living or necrotic (F/T) MC-38 cells, in the presence or the absence of HMGB1, anti-RAGE antibodies, Box A, anti-HMGB1 antibodies, and (E) the CXCR4 antagonist, AMD3100 as indicated. *p < 0.05; **p < 0.005; ***p < 0.001; significantly different from control (Unpaired T test).
Exogenous HMGB1 supports peritoneal carcinomatosis
We thus verified whether exogenous HMGB1 could influence the establishment of peritoneal carcinomatosis. Mice were injected intraperitoneally with minute amounts of HMGB1 (1 µg/animal) or with saline, starting 2 d after intraperitoneal injection of colon carcinoma cells and then every other day until the time of sacrifice (day 15, Fig. 6A). We monitored disease progression by [18F]-FDG PET at 6, 8, 12 and 14 d (Fig. 6B). At day 12, neoplastic lesions of HMGB1-treated mice showed evident 18F-FDG uptake, which was substantially higher than in mock-treated mice. Scattered positive spots of [18F]-FDG uptake rapidly increased in intensity and extent thereafter, from day 12 to day 14 (Fig. 6B). Sites of [18F]-FDG uptake corresponded to serosal neoplastic lesions, as confirmed by necroscopy after animal suppression that was carried out for ethical reasons at day 15 (Fig. 6C). The [18F]-FDG volume and the tumor weight of mice treated with HMGB1 were dramatically higher than those of sham-treated mice (Fig. 6D–ssE).
Figure 6.
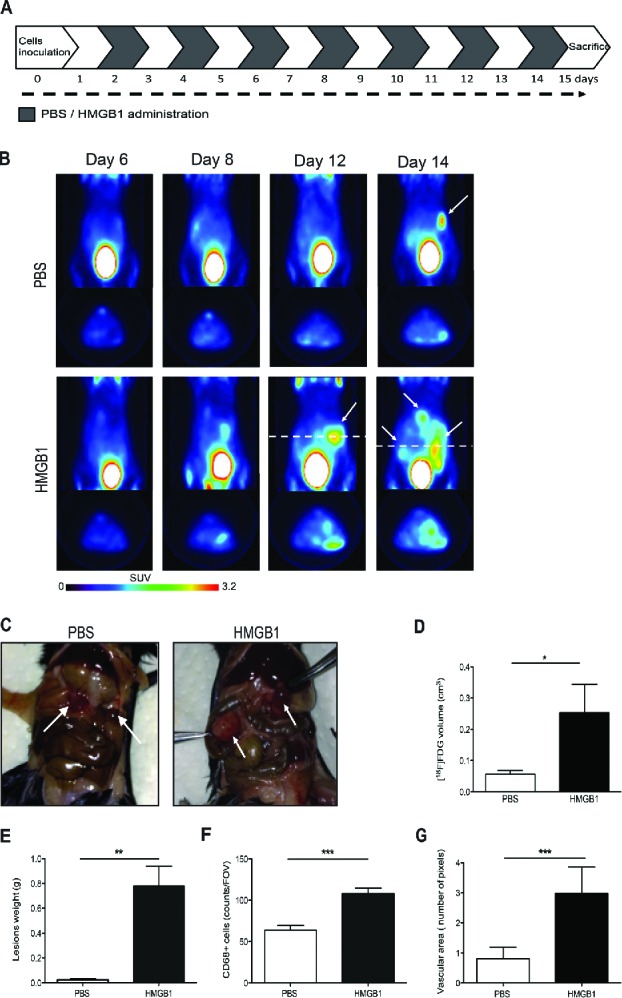
Exogenous HMGB1 promotes leukocyte recruitment into the peritoneal fluid and supports tumor growth. (A) Schematic representation of mice treatment with HMGB1. Treatment (1 µg/mouse) started 2 d after MC-38 carcinoma cell injection. Mice were treated every other day until animal sacrifice (day 15). (B) Tumor growth was monitored by [18F]FDG scans of HMGB1- or PBS-treated mice. Coronal (a) and transaxial (b) view of reconstructed images at the level of tumor location (dashed white line) are reported. Sites of [18F]FDG accumulation are indicated with arrows. Images were presented with the same scale and corrected for injected dose and animal weight. (C) Representative pictures of abdomens of HMGB1- or PBS-treated mice. Arrows indicate neoplastic lesions. (D) Volume of neoplastic lesions of HMGB1- or PBS-treated mice measured at PET (cm3, y-axis). Results are expressed as mean ± SEM. n = 5 per PBS- and n = 4 per HMGB1-treated groups, two independent experiments. (E) Weight of neoplastic lesions of HMGB1- or PBS-treated mice retrieved at sacrifice. Results are expressed as mean ± SEM of four independent experiments, n = 20. (F) CD68+ macrophages per Field of Vision (FOV) were identified by immunohistochemistry (y-axis) in peritoneal lesions of HMGB1- or PBS- treated mice retrieved at sacrifice (x-axis). Results are expressed as mean ± SEM of CD68+ cells/FOV. n = 10 animals per group, two independent experiments. (F) The mean vascular area (y-axis) was calculated in peritoneal lesions of HMGB1- or PBS-treated mice retrieved at sacrifice (x-axis) by digital image analysis. Data are expressed as mean ± SEM of results obtained. n = 10 animals per group, two independent experiments. *p < 0.05, **p < 0.005, ***p < 0.001, significantly different from control (Unpaired T test).
Exogenous HMGB1 also dramatically increased macrophage recruitment within the peritoneal lesions, as assessed by immunohistochemistry (Fig. 6F). Lesions of HMGB1-treated mice were also significantly more vascularized at day 15, with a vascular area >3-fold larger than in untreated tumor-bearing mice (Fig. 6G).
Blockade of HMGB1 reverts peritoneal carcinomatosis
Thus HMGB1 is expressed and released in a bioactive form by colon cancer cells (see Fig. 5), and appears to promote leukocytes recruitment and lesions vascularization and growth (see Fig. 6). Moreover, both HMGB1 and the CXCR4 ligand, CXCL12 which is known to license HMGB1 ability to attract inflammatory leukocytes, were present in the peritoneal fluid of mice with carcinomatosis at the end of the follow up (day 21, 55.25 ± 6.19 ng/mL and 0.84 ± 0.010 ng/mL, respectively), suggesting that the signal might play a role in the natural history of peritoneal carcinomatosis. To verify this hypothesis we intraperitoneally injected the HMGB1 antagonist BoxA every other day, starting from day 7 until animal suppression (day 21, Fig. 7A). BoxA treatment significantly reduced leukocyte attraction in the peritoneal fluid as assessed by flow cytometry verifying the fraction of CD45+ cells expressing the F4/80 (p < 0.05, Fig. 7B), tumor growth (p < 0.05, Fig. 7C) and lesion infiltration by macrophages as assessed by immunohistochemistry (p < 0.001, Fig. 7D–E). The vascular area of lesions of BoxA-treated mice was significantly reduced (p < 0.005, Fig. 7F–G).
Figure 7.
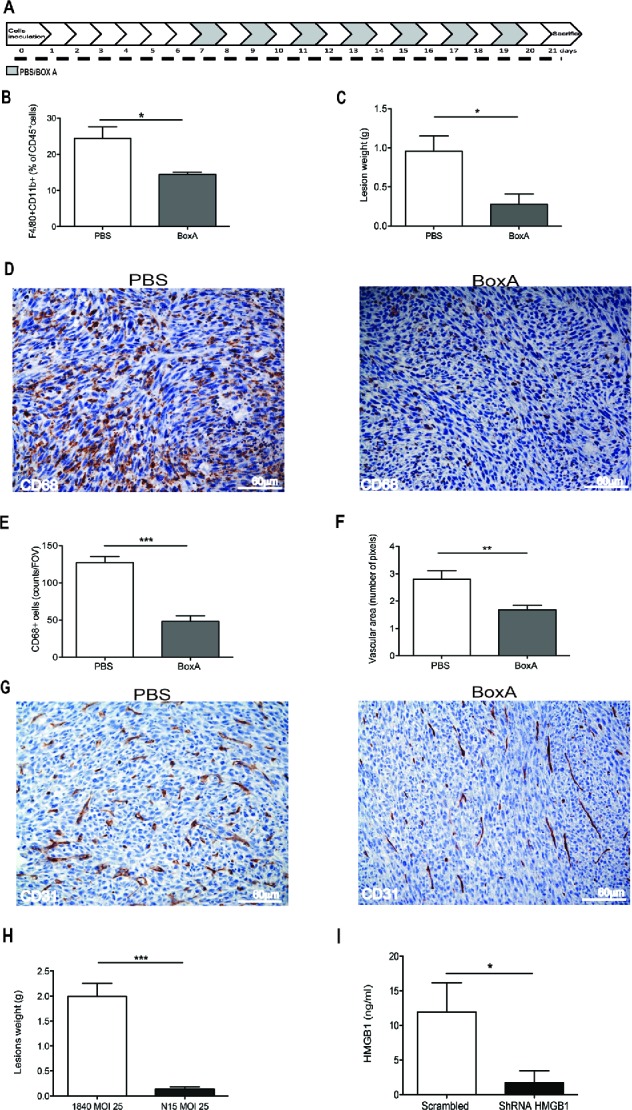
HMGB1 blockade reduces peritoneal carcinomatosis. (A) Schematic representation of mice treatment with BoxA. Treatment (300 µg/mouse) started 7 d after MC-38 carcinoma cell injection. Mice were treated every other day until suppression (day 21). Two independent experiments, n = 10 animals per group. (B) The percentage of F4/80+CD11b+ CD45+ cells in the peritoneal liquid of BoxA- or PBS-treated mice was assessed by flow cytometry and (C) peritoneal lesion weight at sacrifice of BoxA- or PBS-treated mice. (D) CD68+ macrophages in peritoneal lesions from BoxA- and PBS-treated mice. (E) Number of CD68+ macrophages/FOV in peritoneal lesions of BoxA- and PBS-treated mice. (F) Mean vascular area in peritoneal lesions, evaluated by digital image analysis. G: CD31+ endothelial cells in peritoneal lesions of BoxA- and PBS-treated mice. (H, I) Lesion weight (H) and the amount of HMGB1 in the peritoneal fluid (I) of mice transplanted with colon carcinoma cells expressing HMGB1 shRNA or LacZ a-miR LV-transduced were determined at the time of sacrifice (21 d). Three independent experiments, n = 15 animals per group. In panels (B), (C), (E), (F), (H), (I), data are expressed as mean ± SEM. *p < 0.05; **p < 0.005; ***p < 0.001, significantly different from control mice (PBS-treated mice in (B)–(G), mice injected with LacZ a-miR LV-transduced MC-38 colon carcinoma cells in (H) and (I).
HMGB1 expressed by tumor cells represents a relevant target: the growth of peritoneal lesions abated upon effective knockdown of HMGB1 expression in tumor cells using an artificial microRNA-expressing lentiviral vector platform (Fig. 7H–I and Fig. S1), while a similar approach using a control Scrambled short hairpin RNA (scrambled ShRNA) was not effective. Downregulation of HMGB1 expression via this system per se did not detectably influence carcinoma cell characteristics in vitro, including their viability and proliferation rate (Fig. S1).
Discussion
The interaction between cancer and immune cells in the peritoneal environment is a critical area and the identification of new molecular targets is mandatory for treating peritoneal carcinomatosis, a largely unmet medical need. In this study we make three significant observations. First, HMGB1 released by colon carcinoma cells actively stimulate the chemotaxis of macrophages, possibly exploiting a CXL12/CXCR4 dependent pathway. Secondly, addition of minute amounts of HMGB1 in the peritoneal cavity potently amplifies attraction of leukocytes in the peritoneal cavity and in the neoplastic lesions, which develop a structured neo-vasculature and grow with strikingly accelerated kinetics. Thirdly, interference with HMGB1 expression or release by genetic or pharmacological tools restricts tumor growth. These results suggest that HMGB1 represents a major player in the crosstalk between cancer cells and the peritoneal environment, a non-redundant signal for the growth and spreading of peritoneal carcinomatosis and a potentially interesting target for novel treatments.
Several preclinical studies have been conducted using xenograft models that rely on human cancer cells transferred into immunodeficient mice.16,17 These models do not fully reproduce the environment of peritoneal carcinomatosis, and to overcome this limitation we have established a murine model based on the injection of murine MC-38 colon adenocarcinoma cells in the peritoneum of syngeneic mice. MC-38 is a cell line derived from a primary colon adenocarcinoma in C57Bl/6 mice18 and well reproduces the main histopathological features of human peritoneal lesions derived from adenocarcinomas of various origins, in terms of architecture, angiogenesis, abundance and characteristics of infiltrating inflammatory cells. Macrophages are an early, abundant and conserved constituent of the peritoneal neoplastic lesions that arouse in experimental mice after MC-38 colon carcinoma cell injection.
Macrophages recruited within the lesions adopt a throphic, supportive phenotype that facilitates the cancer growth and vascularization. This is in agreement with what has been previously shown in human and in experimental model of peritoneal disease, including ovarian cancer19,20,21 and endometriosis.15,22 In the latter condition, macrophages deliver signals that contribute to the attraction of neo-vessels, possibly facilitating the survival of ectopic endometrial cells in the peritoneal cavity, a relatively hypoxic environment.23 A license from macrophages could as well be required when cancer cells derived from abdominal organs adhere to the serosal lining. If this were the case, one would expect that only a minority of exfoliated tumor cells yield lesions. Indeed, peritoneal carcinomatosis is synchronously detected during primary resection in about 5% of patients with colorectal cancer. It is supposed to develop metachronously in 4–19% of patients,24 but the reported incidence at autopsy ranges between 40 and 80%.25,26 The molecular signals generated in the peritoneal cavity, and specifically the ability to attract supportive macrophages at serosal sites, might play a role in the ability of neoplastic cells to yield peritoneal lesions and in their kinetics.
Indeed, relatively few colon carcinoma cells injected into the peritoneal cavity of healthy syngeneic mice are sufficient to cause a swift local attraction of inflammatory phagocytes, which are an integral constituent of the developing neoplastic lesions and are required for their growth and vascularization. Moreover, lesions apparently keep attracting macrophages, as demonstrated by the preferential migration of labeled peripheral blood leukocytes revealed by MRI.
The ability of HMGB1 to regulate cell migration has been extensively investigated in the last decade27-29 and appears to involve actin-based cytoskeleton rearrangement as well as polarization of various cell surface molecules involved in cell adhesion/transmigration, including various integrins and CD44. Several pathways, more importantly those related to the RAGE and CXCR4 receptors, are particularly important for the regulation of leukocyte migration by HMGB127,28and both receptors appear in our system to be involved in the response of murine macrophages to MC-38 colon carcinoma-derived HMGB1. Further studies will be necessary to identify the actual contribution of these molecules, as well as of carcinomatosis-associated environmental conditions, such as redox and hypoxia to the attraction of cancer-promoting macrophages within the peritoneum. Recently, the ability of HMGB1 to bind to mitochondrial DNA in the cytoplasm of hypoxic tumor cells, activate signaling events downstream the TLR9 and to promote the growth of subcutaneously implanted hepatocellular carcinoma in vivo has been described.30 Little is known so far on the possible relevance of this event for peritoneal carcinomatosis.
The selective interference with HMGB1 resulted in a substantial anti-neoplastic effect. This was achieved by genetic interference with HMGB1 expression in carcinoma cells or by interference with the extracellular action using BoxA, which has been extensively used to antagonize HMGB1 biological effects in various disease models, including established sepsis, myocardial infarction, rheumatoid arthritis and skeletal muscle damage. HMGB1 contributes to the development of malignant mesotheliomain response to the exposure of mesothelial cells to asbestos.31,32 Conversely viable mesothelioma cells actively secrete HMGB1: interference with HMGB1 or with the RAGE receptors limits the growth of human xenografts in immunodeficient mice,33 supporting the contention that the molecule can play cancer-supporting actions in the extracellular environment.34 Cell death occurring after chemotherapy with cytotoxic agents substantially increases the extent of HMGB1 released from MC-38 cells.14 Some HMGB1 is released even in the absence of elicited cell necrosis, possibly as a consequence of normal MC-38 cell turnover (not shown).14 Indeed, we observed HMGB1 accumulation in the peritoneal liquid of mice with MC-38 carcinomatosis, at concentrations at which the signal is certainly bioactive and could justify the recruitment of inflammatory leukocytes in the lesions. Leukocytes could in turn contribute to neoplastic growth and vascular remodeling using their own HMGB1,35 establishing a self-sustaining positive feed-forward loop. Taken together, our findings indicate that peritoneal carcinomatosis relies on HMGB1 and they offer a preclinical proof-of-principle that pharmacological ablation of HMBG1 is sufficient to constrain it, suggesting a novel therapeutic target.
Patients/material and methods
Animal care and use
C57Bl/6 female mice (6–7 weeks of age) were purchased from Harlan. Animals were kept under specific pathogen-free conditions, handled and maintained according to San Raffaele Institutional Animal Care and Use Committee (IACUC) ethical regulations. Animals were sacrificed by CO2 asphyxiation. The IACUC also approved the experiments described in this study (approval number 2011_510).
Adenocarcinoma cell lines and genetic manipulation
MC-38 is a murine colon adenocarcinoma line that was induced by the injection of dimethylhydrazine in syngeneic C57BL/6 mice and was passaged subcutaneously. It was generously provided by Dr Mario P. Colombo (Milano). TS/A mammary adenocarcinoma cells, CT-26 and C-26 murine colon adenocarcinoma cell lines, HT-29, Caco-2, HCT8, SW 620 and WiDr human colon adenocarcinoma cell lines were kindly provided by Dr Maria Pia Protti, Milano. Murine RMA lymphoma cells, Hmgb1−/−murine embryonic fibroblasts and their wild-type counterparts were also used.36 Cells were grown in DMEM (Gibco BRL, Grand Island, NY) containing 10% FBS (South American Origin, BioWhittaker-Lonza, B-4800 Verviers, Belgium), 2 mM glutamine, 100 IU/mL penicillin, 100 g/mL streptomycin (Gibco BRL, Grand Island, NY). GFP-MC-38 cells were generated by transduction with a lentiviral vector carrying GFP under the PGK promoter (PGK-GFP-LV). Vector infectivity was 1 × 104 transducing units per ng of HIV capsid p24 by vector DNA titration of 293T cells. For transduction, cells were plated at 2.5 × 106 cells/mL and infected for 24 h. Cells were then passaged and expanded: transduction was checked by flow cytometry analyzing GFP expression. The ShRNA sequence targeting the Hmgb1 gene was cloned into integrating lentiviral backbone pCCLsin.PPT.SFFV.EF Intron.mO2.Wpre.37 This vector co-expresses the HMGB1 a-miR and the reporter transgene mOrange from the Spleen Focus Forming Virus promoter. The sequence of the siRNA used to generate the HMGB1 a-miR is: 5′-CAATATCCTTCTCATACTTCT-3′. As a specificity control for the Hmgb1 knockdown experiment, we used a lentiviral vector expressing an a-miR targeting LacZ.37 VSV-G pseudotyped third generation lentiviral vectors expressing the a-miRs were produced by transient four plasmid cotransfection into HEK293T cells and concentrated by ultracentrifugation.38 Tumor cells were transduced with increasing multiplicity of infection (MOI) of the Hmgb1 or the LacZ a-miR LV. Infected cells (MOI 25) were sorted for mOrange. The efficiency of knockdown and the expression of HMGB1 were assessed by western blotting. Cell lines were tested for Mycoplasma by MycoAlert→ Mycoplasma detection kit (BioWhittaker-Lonza, B-4800Verviers, Belgium).
Peritoneal carcinomatosis
C57BL/6 mice were intraperitoneally implanted with MC-38 cells (2.5 × 104 cells/mouse or 4 × 104 cells/mouse), MC-38-GFP cells (5 × 104cells/mouse), HMGB1 shRNA or scrambled shRNA MC-38 (4 × 104cells/mouse), i.e., with the minimum dose that elicited the growth of neoplastic lesions in 100% of animals. The growth of tumor was stopped at 21 d for ethical reason. Peritoneal liquid was retrieved and cells were counted using the vital dye Trypan Blue. Tumor lesions were micro-dissected and, when indicated, dehydrated or fixed in 4% PFA, incubated overnight in 30% sucrose, cryopreserved in liquid N2 cooled isopentane after embedding in optimal cutting temperature (OCT) (Killik, Bio-Optica) for immunohistochemistry (see below). For single cell preparation, lesions were disrupted and digested in Collagenase A+B+D (Roche, final concentration 1.5 mg/mL) supplemented with DNase I (from bovine pancreas, Roche, final concentration 30 μL/mL) at 37°C for 1.5 h. PerCP-conjugated anti-mouse CD45 mAb (clone 30-F11, BD PharMingen), and GFP positivity. Analysis was carried out using FACS Canto and Diva Software (BD Biosciences) and elaboration with FlowJo Software (Tree Star, v. 9.4.4).
Magnetic resonance imaging
Magnetic Resonance Imaging (MRI) was performed on a 7T preclinical magnetic resonance scanner (Bruker, BioSpec 70/30 USR, Paravision 5.1), equipped with 450/675 mT/m gradients (slew-rate: 3400–4500T/m/s; rise-time 140 µs) and a mouse body volume coil. Mice were under general anesthesia by 1.5–2% isoflurane vaporized in 100% oxygen (flow: 1 L/min). Breathing and body temperature were monitored (SA Instruments, Inc., Stony Brook, NY, USA) and maintained around 30 breaths-per-minute and 37°C, respectively. MRI studies included a multigradient echo T2-star sequence (MGE-T2*) (relaxation time = 1500 ms; 16 echo times between 3.5 ms and 66.7 ms; flip angle 30°; 18 slices with a thickness of 0.6 mm; field of view 3 × 1.6 cm; matrix 192 × 136). Tumor-bearing mice (day 19) were studied before and 3 or 24 h after i.v. injection of syngeneic leukocytes labeled with SPIO particles (0.22 mg).39
Pharmacological treatments
To deplete peritoneal macrophages, animals were treated with dichloromethylenebisphosphonic acid (clodronate; 1 mg/20 mg body weight in 200 μL of suspension), to reach macrophages depletion, or PBS encapsulated into liposomes (“Clodrolip” and “Shamlip,” respectively) as control and purchased from http://clodronateliposomes.org. Treatments were administered the day after MC-38 cell injection and every other day until the end of study. We treated mice with HMGB1 (1 μg/mouse/100 uL, HMG Biotech, Milano), its antagonist Box-A (0.3 mg/mouse/100 μL, HMGBiotech) or with PBS. HMGB1 was administered intraperitoneally from the second day post tumor injection every other day until day 15. Box A was given intraperitoneally every other day starting from 7 d post tumor injection until the end of experiment at day 21.
Chemotaxis assays
Chemotaxis assays were performed using Boyden chambers.40 Briefly, polycarbonate filters (8 μm pores, Neuro Probe, Inc., Gaithersburg, MD, USA) were coated with porcine skin gelatin (2 mg/mL, Sigma-Aldrich). DMEM with 10% FCS (positive control), DMEM alone (negative control), or media conditioned by MC-38 cells were placed in the lower chamber (50 μL). HMGB1 (1 μg/mL, HMGBiotech); BoxA (100 ng/mL, HMGBiotech) or monoclonal antibodies against HMGB1 or RAGE (anti-HMGB1, 1 ng/mL, DPH1.1; anti-RAGE 1 μg/mL; H300; Dia.Pro Diagnostics) or AMD3100 (1 μM, Sigma-Aldrich) were added to carcinoma cells conditioned media before the experiment. 50 × 103 murine bone-marrow-derived macrophages, isolated from femurs and tibiae of C57Bl/6 mice in 200 μL of DMEM were placed in the upper chamber. Chambers were then incubated at 37°C in 5% CO2 for 6 h. Cells remaining on the upper section of the filters were removed mechanically. Macrophages migrated to the lower section of the filters were fixed with absolute ethanol, stained with Giemsa (Sigma-Aldrich), and counted at 40X in 10 random fields per filter. Each assay was performed in triplicate and repeated for two independent experiments.
PET studies
In vivo imaging studies were performed using a small animal tomograph, the YAP-(S)-PET II (I.SE s.r.l., Pisa, Italy).41 [18F]FDG is routinely prepared in our facility for clinical use (European Pharmacopeia VIII ed.) and was injected with a radiochemical purity greater than 99%. After slight anesthesia with ether animals were injected in the tail vein with 4.25 ± 0.3 MBq of [18F]FDG in a volume of 50 μL of saline. Immediately before PET acquisition, mice were anesthetized with 2% isoflurane vaporized in air and positioned prone on the tomograph bed with the abdomen centered in the tomograph field of view. PET scans started 60 min after tracer injection and lasted 30 min (six frames of 5 min each). PET data were acquired in list mode using the full axial acceptance angle of the scanner (3D mode) and then reconstructed with the expectation maximization (EM) algorithm.41
For quantification analysis, data were corrected for the physical decay of fluorine 18 (t1/2: 109.8 min) and transformed in absolute radioactivity concentration values (MBq/gr) after calibration of the tomograph using a standard phantom and considering tissue density equal to that of water. For each animal, maximal standardized uptake values (SUVmax) and volume of radioactivity uptake (Volmetab) were measured. To this aim, PET images were thresholded to create masks for the automatic extraction of the volume of tracer distribution. A visual inspection analysis was initially applied to PET images and [18F]FDG positive slices, depending on the presence of abnormal areas of radioactivity accumulation, were selected. After that, using Region of Interest (ROI), we calculated maximum radioligands concentration in tumor (MBq/g) (upper threshold value) and mean radioligands distribution in background (thorax muscle, MBq/g) (background value). Then lower threshold was calculated as the halfway value between upper threshold value and background value. This method allowed to automatically extract the metabolic tumor volume (cm3) and the maximum and mean uptake of the tumor. Standardized Uptake Value (SUV) was calculated according to the formula: SUV = tumor concentration activity [MBq/g]/(injected activity [MBq]/animal weight [g]).
Histological analysis
For the immunohistochemical (IHC) analysis serial, 6 μm thick sections of PFA-fixed, sucrose-protected, OCT-embedded tissues were treated with 0.3% H2O2 to quench endogenous peroxidase activity. To evaluate leukocyte infiltration and vascular structures, tissue sections were incubated in PBS + 4% BSA (SIGMA) + 0.1% Triton (BDH) and then with biotinilated anti-mouse CD68 mAb (FA-11, AbDSerotec) or anti-mouse NK1.1 (PK 136 BD Bioscience) or anti-mouse Ly6G (1A8, Biolegend), purified anti-mouse CD19 (1D3, BD Bioscience) or anti-mouse CD3 (145-2C11 BD Bioscience) or purified anti-mouse CD31 (BD Biosciences, clone MEC 13.3). Signals were revealed with R.T.U. horseradish peroxidase streptavidin (Vector Laboratories, Burlingame, CA), which was detected using Vector NovaRED substrate kit (Vector Laboratories, Burlingame, CA). Slides were counterstained with Mayer's hematoxylin and examined under a Nikon Eclipse 55i microscope (Nikon, Tokyo, Japan). Images were captured with Digital Sight DS-5 M digital camera (Nikon) using Lucia G software (Laboratory Imaging, Prague, CZ). Parallel slides in which the primary mAb had been omitted were identically processed and used as negative controls. Stained sections were examined by three independent operators blinded to experimental design. Immunofluorescence was performed on serial 7 μm thick sections of PFA-fixed, sucrose-protected, OCT-embedded tissues. Slides were hydrated in PBS and auto-fluorescence was quenched incubating in 0.1 M Glycine (pH 7.7). For HMGB1, antigen retrieval was performed using the citrate-based VECTOR® Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Slides were permeabilized/blocked for 1 h with PBS containing 2.5% BSA, 5% FBS, and 0.1% Triton-X100 (Fisher Scientific). Primary antibodies diluted in PBS containing 2.5% BSA, 5% FBS incubated on sections overnight at 4°C, included: anti-mouse purified anti-mouse CD31 (BD Biosciences, clone MEC 13.3). After washing, secondary antibodies from Invitrogen were used at 1:500, including anti-rat Alexa 594 Slides were counterstained with DAPI (Invitrogen), mounted in H20 and glycerol, and images were captured as above.
For the quantification of CD31 vessels, at least 10 fields at 20X magnification were counted using ImageJ software. For the quantification of the vascular area ImageJ software and the algorithm described in15,41 were used.
Immunofluorescence
Colon carcinoma cells were plated on cover glasses (5 × 105/mL/well in 12-well Multiwell Plates, Corning Inc.). After overnight culture, cell were fixed in 4% PFA, quenched with 0.1 M Glycine (pH 7.7) and permeabilized/blocked for one hour with PBS containing 4% BSA, 0.2% Triton-X100 (Fisher Scientific). Primary antibody purified anti-HMGB1 (polyclonal, Abcam) diluted in PBS containing 0.2% BSA was incubated overnight at 4°C and revealed using an anti-rabbit Alexa 594 (Invitrogen). Slides were counterstained with Hoechst 33348 (Invitrogen). Cells were incubated with Triton 0.5% in PBS for 10 min at RT to induce necrosis. Immunofluorescence was also performed on serial 7 μm thick sections of PFA-fixed, sucrose-protected, OCT-embedded tissues. Slides were hydrated in PBS and auto-fluorescence was quenched incubating in 0.1 M Glycine (pH 7.7). For HMGB1, antigen retrieval was performed using the citrate-based VECTOR® Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Slides were permeabilized/blocked for 1 h with PBS containing 2.5% BSA, 5% FBS, and 0.1% Triton-X100 (Fisher Scientific). Primary antibodies diluted in PBS containing 2.5% BSA, 5% FBS incubated on sections overnight at 4°C, included anti-mouse purified anti-mouse CD31 (BD Biosciences, clone MEC 13.3). After washing, secondary antibodies from Invitrogen were used at 1:500, including anti-rat Alexa 594. Slides were counterstained with DAPI (Invitrogen), mounted in H20 and glycerol, and images were captured as above. For the quantification of CD31 vessels and assessment of the vascular area, at least 10 fields at 20X magnification were counted using ImageJ software.
Flow cytometry
Samples were incubated with PerCP-conjugated anti-mouse CD45 mAb (clone 30-F11, BD PharMingen) for 20 min after saturation with PBS containing 10% FBS. Cells were then washed once, fixed in 4% PFA for 20 min on ice, washed again, and stored at 4°C until analysis with an LSR Fortessa flow cytometer with Diva Software (BD Bioscience). For multi-color analysis 1–5 × 105 cells were incubated 30 min on ice with Rat Anti-Mouse Fcγ III/II Receptor (2.4G2 BD PharMingen, 2 μg/mL), to block nonspecific binding, and with LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (1:500, Invitrogen) to exclude dead cells and debris. After washing, samples were incubated in PBS containing 5% FCS and 0.1 mM EDTA on ice using appropriate combinations of the following monoclonal antibodies: Alexa 700 anti-CD45 for leukocytes gating (BD Biosciences, clone 30-F11, dilution 1:65), PE-CY7 anti-CD3 (BD Biosciences, clone 145-2C11, dilution 1:65), FITC anti-Ly6G (Biolegend, clone1A8, dilution 1:200), FITC anti-CD19 (BD Biosciences, clone 1D3, dilution 1:200), PERCP anti-NK1.1 (BD Biosciences, clone PK136, dilution 1:100), APC anti-CD11b (Biolegend, clone M1/70, dilution 1:125) and APC-CY7 anti-F4/80 (Biolegend, clone BM8, dilution 1:50). Samples were analyzed at FACS Canto with Diva Software (BD Biosciences). Analysis was performed with FlowJo Software (Tree Star, v. 9.4.4).
ELISA assay
The concentration of HMGB1 and CXCL-12 was determined testing peritoneal fluid of tumor bearing mice at the end of tumor growth (day 21) by ELISA according to manufacturer's instructions (Shino Test and R&D System).
Western blot analysis
Cell or tissue lysates were centrifuged at 16,000 g for 15 min at 4°C. Equal amounts of protein (10–20 μg for cell lysates) were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto Immobilon-P (Millipore). After Ponceau S (Sigma-Aldrich) staining, membranes were saturated in 20 mM Tris-HCl, pH 7.6, 150 mM NaCl (Tris-buffered saline, TBST), containing nonfat milk 5%. Antigens were detected using mouse monoclonal anti- HMGB1 (final concentration 0.1 μg/mL, kindly provided by Dia. Pro. Diagnostic Bioprobes) and mouse monoclonal anti-GAPDH (final concentration 0.1 μg/mL; Sigma-Aldrich, clone GAPDH-71.1) antibodies. All antibodies were diluted in TBST 5% nonfat milk. Incubation was performed overnight at 4°C for primary antibodies that were revealed with HRP-conjugated secondary antibodies (final concentration 0.2 μg/mL; GE Healthcare Europe GmbH) and a chemiluminescence kit (ECL, Western blotting detection reagents; GE Healthcare Europe GmbH).
Statistical analysis
Results are expressed as mean ± SD, Standard Deviation, unless differently stated (mean ± SEM, Standard Error Mean). Experimental differences were tested for statistical significance using Student's t test (unpaired, two sided, p considered statistically significant when <0.05), by Graph Pad PRISM Software (version 4.0c for Macintosh by Software MacKiev), when n/replicates where equal or more than 3. Differences significance and the p value are represented in figures by asterisks as follows: *<0.05; **<0.005; ***<0.0001. Survival curve was calculated with the Kaplan–Meier survival plot using Graph Pad PRISM Software (version 4.0c for Macintosh by Software MacKiev).
Supplementary Material
Disclosure of potential conflicts of interest
The authors have no conflicting financial interests. However, M.E. Bianchi is founder and part owner of HMG Biotech, a company that provides goods and services related to HMGB proteins.
Funding
This work was supported by the Italian Association for Cancer Research (AIRC), -IG11761 project (to A. Manfredi), the Ministry of Higher Education and Research (MIUR), by the Ministry of Health and by the Ricerca Finalizzata RF-2011-02352291 (to A. Manfredi).
References
- 1.D'Angelica M, Gonen M, Brennan MF, Turnbull AD, Bains M, Karpeh MS. Patterns of initial recurrence in completely resected gastric adenocarcinoma. Ann Surg 2004; 240:808-16; PMID:15492562; http://dx.doi.org/ 10.1097/01.sla.0000143245.28656.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunisaki C, Akiyama H, Nomura M, Matsuda G, Otsuka Y, Ono H, Nagahori Y, Hosoi H, Takahashi M, Kito F et al.. Comparison of surgical results of D2 versus D3 gastrectomy (para-aortic lymph node dissection) for advanced gastric carcinoma: a multi-institutional study. Ann Surg Oncol 2006; 13:659-67; PMID:16538414; http://dx.doi.org/ 10.1245/ASO.2006.07.015 [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 2010; 22:231-7; PMID:20144856; http://dx.doi.org/ 10.1016/j.coi.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 4.Erez N, Coussens LM. Leukocytes as paracrine regulators of metastasis and determinants of organ-specific colonization. Int J Cancer J Int du Cancer 2011; 128:2536-44; PMID:21387299; http://dx.doi.org/ 10.1002/ijc.26032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al.. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; http://dx.doi.org/ 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 6.Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, Patsouris ES, Peros G, Horcic M, Tornillo L et al.. Tumor infiltration by FcgammaRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer 2011; 128:2663-72; PMID:20715106; http://dx.doi.org/ 10.1002/ijc.25609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiol 2013; 218:1402-10; PMID:23891329; http://dx.doi.org/ 10.1016/j.imbio.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 8.Bosurgi L, Bernink JH, Delgado Cuevas V, Gagliani N, Joannas L, Schmid ET, Booth CJ, Ghosh S, Rothlin CV. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A 2013; 110:13091-6; PMID:23878224; http://dx.doi.org/ 10.1073/pnas.1302507110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014; 41:49-61; PMID:25035953; http://dx.doi.org/ 10.1016/j.immuni.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19:4046-57; PMID:23723299; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Wu K, Zhao E, Shi L, Li R, Zhang P, Yin Y, Shuai X, Wang G, Tao K. HMGB1 recruits myeloid derived suppressor cells to promote peritoneal dissemination of colon cancer after resection. Biochem Biophys Res Commun 2013; 436:156-61; PMID:23707808; http://dx.doi.org/ 10.1016/j.bbrc.2013.04.109 [DOI] [PubMed] [Google Scholar]
- 12.Bianchi ME, Manfredi AA. How macrophages ring the inflammation alarm. Proc Natl Acad Sci U S A 2014; 111:2866-7; PMID:24532661; http://dx.doi.org/ 10.1073/pnas.1324285111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J et al.. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A 2014; 111:3068-73; PMID:24469805; http://dx.doi.org/ 10.1073/pnas.1316925111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cottone L, Capobianco A, Gualteroni C, Perrotta C, Bianchi ME, Rovere-Querini P, Manfredi AA. 5-Fluorouracil causes leukocytes attraction in the peritoneal cavity by activating autophagy and HMGB1 release in colon carcinoma cells. Int J Cancer 2015; 136:1381-9; PMID:25098891; http://dx.doi.org/ 10.1002/ijc.29125 [DOI] [PubMed] [Google Scholar]
- 15.Capobianco A, Monno A, Cottone L, Venneri MA, Biziato D, Di Puppo F, Ferrari S, De Palma M, Manfredi AA, Rovere-Querini P. Proangiogenic Tie2(+) macrophages infiltrate human and murine endometriotic lesions and dictate their growth in a mouse model of the disease. Am J Pathol 2011; 179:2651-9; PMID:21924227; http://dx.doi.org/ 10.1016/j.ajpath.2011.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong MY KS. Ovarian cancer mouse models: A summary of current models and their limitations. J Ovarian Res 2009; 2(1):12; PMID:19781107; http://dx.doi.org/ 10.1186/1757-2215-2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colvin EK, Scarlett CJ. A historical perspective of pancreatic cancer mouse models. Semin Cell Dev Biol 2014; 27:96-105; PMID:24685616; http://dx.doi.org/ 10.1016/j.semcdb.2014.03.025 [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986; 233:1318-21; PMID:3489291; http://dx.doi.org/ 10.1126/science.3489291 [DOI] [PubMed] [Google Scholar]
- 19.Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, McFarland-Mancini MM, Drew AF. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res 2007; 67:5708-16; PMID:17575137; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4375 [DOI] [PubMed] [Google Scholar]
- 20.Rei M, Goncalves-Sousa N, Lanca T, Thompson RG, Mensurado S, Balkwill FR, Kulbe H, Pennington DJ, Silva-Santos B. Murine CD27(-) Vgamma6(+) gammadelta T cells producing IL-17A promote ovarian cancer growth via mobilization of protumor small peritoneal macrophages. Proc Natl Acad Sci U S A 2014; 111:E3562-70; PMID:25114209; http://dx.doi.org/ 10.1073/pnas.1403424111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colvin EK. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front Oncol 2014; 4:137; PMID:24936477; http://dx.doi.org/19574425 10.3389/fonc.2014.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bacci M, Capobianco A, Monno A, Cottone L, Di Puppo F, Camisa B, Mariani M, Brignole C, Ponzoni M, Ferrari S et al.. Macrophages are alternatively activated in patients with endometriosis and required for growth and vascularization of lesions in a mouse model of disease. Am J Pathol 2009; 175:547-56; PMID:19574425; http://dx.doi.org/ 10.2353/ajpath.2009.081011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capobianco A, Rovere-Querini P. Endometriosis, a disease of the macrophage. Front Immunol 2013; 4:9; PMID:23372570; http://dx.doi.org/ 10.3389/fimmu.2013.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segelman J, Granath F, Holm T, Machado M, Mahteme H, Martling A. Incidence, prevalence and risk factors for peritoneal carcinomatosis from colorectal cancer. Br J Surg 2012; 99:699-705; PMID:22287157; http://dx.doi.org/ 10.1002/bjs.8679 [DOI] [PubMed] [Google Scholar]
- 25.Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP. Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Ann Surg 2006; 243:212-22; PMID:16432354; http://dx.doi.org/ 10.1097/01.sla.0000197702.46394.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sloothaak DA, Mirck B, Punt CJ, Bemelman WA, van der Bilt JD, D'Hoore A, Tanis PJ. Intraperitoneal chemotherapy as adjuvant treatment to prevent peritoneal carcinomatosis of colorectal cancer origin: a systematic review. Br J Cancer 2014; 111:1112-21; PMID:25025964; http://dx.doi.org/ 10.1038/bjc.2014.369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A et al.. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 2012; 209:551-63; PMID:22370717; http://dx.doi.org/ 10.1084/jem.20111739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol 2013; 55:76-82; PMID:23207101; http://dx.doi.org/ 10.1016/j.molimm.2012.10.037 [DOI] [PubMed] [Google Scholar]
- 29.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol 2013; 93:865-73; PMID:23446148; http://dx.doi.org/ 10.1189/jlb.1212662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll Like Receptor 9. J Hepatol 2015; 63(1):114-21; PMID:25681553; http://dx.doi.org/20616036 10.1016/j.jhep.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, Rivera Z, Jube S, Nasu M, Bertino P, Goparaju C, Franzoso G, Lotze MT, Krausz T et al.. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc Natl Acad Sci U S A 2010; 107:12611-6; PMID:20616036; http://dx.doi.org/ 10.1073/pnas.1006542107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi F, Okimoto G, Jube S, Napolitano A, Pass HI, Laczko R, Demay RM, Khan G, Tiirikainen M, Rinaudo C et al.. Continuous exposure to chrysotile asbestos can cause transformation of human mesothelial cells via HMGB1 and TNF-α signaling. Am J Pathol 2013; 183:1654-66; PMID:24160326; http://dx.doi.org/ 10.1016/j.ajpath.2013.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res 2012; 72:3290-301; PMID:22552293; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res 2007; 13:2836-48; PMID:17504981; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1953 [DOI] [PubMed] [Google Scholar]
- 35.Campana L, Santarella F, Esposito A, Maugeri N, Rigamonti E, Monno A, Canu T, Del Maschio A, Bianchi ME, Manfredi AA et al.. Leukocyte HMGB1 Is Required for Vessel Remodeling in Regenerating Muscles. J Immunol 2014; 192:5257-64; PMID:24752445; http://dx.doi.org/ 10.4049/jimmunol.1300938 [DOI] [PubMed] [Google Scholar]
- 36.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Müller S, Iannacone M, Traversari C et al.. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004; 5:825-30; PMID:15272298; http://dx.doi.org/ 10.1038/sj.embor.7400205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amendola M, Passerini L, Pucci F, Gentner B, Bacchetta R, Naldini L. Regulated and multiple miRNA and siRNA delivery into primary cells by a lentiviral platform. Mol Ther 2009; 17:1039-52; PMID:19293777; http://dx.doi.org/ 10.1038/mt.2009.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantore A, Ranzani M, Bartholomae CC, Volpin M, Valle PD, Sanvito F, Sergi LS, Gallina P, Benedicenti F, Bellinger D et al.. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med 2015; 7:277ra28; PMID:25739762; http://dx.doi.org/18250435 10.1126/scitranslmed.aaa1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manfredi AA, Capobianco A, Esposito A, De Cobelli F, Canu T, Monno A, Raucci A, Sanvito F, Doglioni C, Nawroth PP et al.. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol 2008; 180:2270-5; PMID:18250435; http://dx.doi.org/ 10.4049/jimmunol.180.4.2270 [DOI] [PubMed] [Google Scholar]
- 40.Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukocyte Biol 2009; 86:609-15; PMID:19414537; http://dx.doi.org/ 10.1189/jlb.0908576 [DOI] [PubMed] [Google Scholar]
- 41.Cottone L, Valtorta S, Capobianco A, Belloli S, Rovere-Querini P, Fazio F, Manfredi AA, Moresco RM. Evaluation of the role of tumor-associated macrophages in an experimental model of peritoneal carcinomatosis using (18)F-FDG PET. J Nuclear Med 2011; 52:1770-7; PMID:22045707; http://dx.doi.org/ 10.2967/jnumed.111.089177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.