

Abstract
Autism spectrum disorder (ASD) is characterized by social and communication impairments as well as by restricted, repetitive patterns of behavior and interests. Genomic studies have not revealed dominant genetic errors common to all forms of ASD. So ASD is assumed to be a complex disorder due to mutations in hundreds of common variants. Other theories argue that spontaneous DNA mutations and/or environmental factors contribute to as much as 50% of ASD. In reviewing potential genetic linkages between autism and alcoholism, it became apparent that all theories of ASD are consistent with aldehyde toxicity, in which endogenous and exogenous aldehydes accumulate as a consequence of mutations in key enzymes. Aldehyde toxicity is characterized by cell-localized, micronutrient deficiencies in sulfur-containing antioxidants, thiamine (B1), pyridoxine (B6), folate, Zn2+, possibly Mg2+, and retinoic acid, causing oxidative stress and a cascade of metabolic disturbances. Aldehydes also react with selective cytosolic and membrane proteins in the cell of origin; then some types migrate to damage neighboring cells. Reactive aldehydes also form adducts with DNA, selectively mutating bases and inducing strand breakage. This article reviews the relevant genomic, biochemical, and nutritional literature, which supports the central hypothesis that most ASD symptoms are consistent with symptoms of aldehyde toxicity. The hypothesis represents a paradigm shift in thinking and has profound implications for clinical detection, treatment, and even prevention of ASD. Insight is offered as to which neurologically afflicted children might successfully be treated with micronutrients and which children are unlikely to be helped. The aldehyde toxicity hypothesis likely applies to other neurological disorders.
Keywords: autism spectrum disorder, aldehyde toxicity, oxidative stress, micronutrient deficiencies, de novo mutations
Introduction
Autism spectrum disorder (ASD) is a complex, devastating, and costly disorder, with the incidence rising to as high as 1 in 68 individuals in developed countries.1 ASD has become a major focus of traditional research and also a target of alternative therapies. On reviewing the medical literature, particularly pursuing a potential link between heritable alcoholism and autism, it has become apparent that all competing theories of autism are consistent with aldehyde toxicity; in other words, symptoms of autism mimic the symptoms of aldehyde toxicity. Regardless of the root genetic cause(s) of ASD, the actual cellular damage is likely caused by an accumulation of very reactive aldehydes. Some aldehydes are endogenous, which are generated enzymatically within cells or nonenzymatically by free radical attacks on polyunsaturated fatty acids (PUFAs) or carbohydrates. Other aldehydes originate from various sources in the environment or in abnormal gut microbiota. It is well established in the medical and scientific literature, particularly in the areas of alcoholism and lipid peroxidation, that aldehydes are very reactive molecules, which form inactivating adducts with selective proteins as well as adducts with DNA that can ultimately result in spontaneous mutations. Often overlooked, yet sufficiently documented, are the observations that aldehydes directly and irreversibly react with a subset of micronutrients, causing severe deficiencies, but only in cells in which the aldehydes are generated. These selective micronutrient deficiencies can severely impair cellular function, yet are not detectable by standard clinical assays, which are capable only of measuring an average of micronutrients in the blood and urine. For lack of suitable assays, deficiencies in a subset of micronutrients are frequently overlooked in ASD children but can cause the myriad of symptoms associated with ASD, including the epigenetic changes often documented. Fortunately, there are some effective treatments for aldehyde toxicity currently available; most are nutritional and are reviewed herein, although potentially better pharmacological options are on the horizon. Some ASD children may be rescued now by the treatment options described herein, but with any type of aldehyde toxicity, time is of the essence to prevent extensive cellular and DNA damage. The review has been written to alert clinicians, researchers, and the autism community to the similarities of ASD and aldehyde toxicity symptoms, so that those with ASD may be treated more effectively and as quickly as possible (see Abbreviations).
Background
Common sources of aldehydes
There is a large diversity of aldehydes in nature, but all cause essentially the same type of cellular damage as a consequence of the shared chemical structure, R–(C=O)–H. The C of the carbonyl group is electrophilic and reacts with nucleophilic groups, such as thiols and amines, found in many biomolecules. R represents a generalized organic group, which is responsible for some diversity in reaction rates and nucleophilic targets. LoPachin and Gavin2 have postulated that, based on hard and soft, acid and base quantum chemical calculations, simple alkanals react better with hard nucleophilic groups, such as lysines, whereas α,β-unsaturated alkenals and α-oxoaldehydes preferentially react with soft nucleophilic groups, such as thiols and cysteines. O’Brien et al3 have comprehensively reviewed various sources of exogenous and endogenous aldehydes. A few categories, particularly related to ASD, will be summarized herein.
Exogenous aldehydes
Aldehydes, such as formaldehyde, acetaldehyde, and acrolein, are ubiquitous in nature,3 as byproducts of photochemical oxidation of plants and fuels, as by-products of industrial manufacturing, and as components in foods. The primary source of aldehydes in the outdoors is motor vehicle exhaust, which emits either aldehydes directly or compounds that can be photochemically oxidized to aldehydes. The indoor concentration of aldehydes, particularly in confined or poorly ventilated areas, is 4- to 10-fold higher and is attributed to smoking, cooking fumes, and industrial products, such as furniture, carpets, fabrics, disinfectants, perfumes, cosmetics, and salon products.3 Particularly noteworthy, are that many foods, including fruits and vegetables, as well as natural food flavorings, such as vanilla, cinnamon, spearmint, peppermint, citrus, and cocoa, contain aldehydes.3 Cooking at temperatures high enough to brown food also increases the concentration of aldehydes. Moreover, numerous pharmaceuticals and xenobiotics either contain aldehydes or are metabolized through aldehyde intermediates. Individuals with normal metabolisms generally have suitable in vivo mechanisms to rapidly detoxify the environmental onslaught of aldehydes. Those with abnormal metabolic antioxidant defense mechanisms or those low in detoxifying sulfur-containing antioxidants are likely to suffer more immediate negative reactions to the environmental toxins.
Nonenzymatic production of endogenous aldehydes
One source of endogenous aldehydes is that generated by the non-enzymatic oxidation of lipids and carbohydrates by the superoxide anion radical (•O2−) and other reactive oxygen species (ROS). Particular examples related to ASD are the lipidic aldehydes generated as a result of ROS attack on PUFAs as shown in Figure 1. The first PUFA metabolites formed are lipid hydroperoxides that are unstable and degrade rapidly to very reactive endogenous α,β-unsaturated aldehydes,4 such as 4-hydroxynonenal (4-HNE) derived from linoleic and arachidonic acids and 4-hydroxyhexenal (4-HHE) derived from docosohexaenoic acid, eicosapentaenoic acid, and linolenic acid. ROS attack on PUFAs ultimately generate over 200 different types of lipidic α,β-unsaturated aldehydes, which are more damaging than ROS because the aldehydes are more stable, amphiphilic, and can diffuse across membranes, modifying proteins in the cytoplasm, nucleus, and cell membrane, far from their site of origin.5 The class of α,β-unsaturated aldehydes is among the most reactive aldehydes known, essentially forming irreversible adducts with other molecules, including proteins6 and DNA.7 Moreover, the biomolecular targets of lipid peroxidation-generated aldehydes are specific to the cell type as well as dependent upon the aldehyde concentration and the pattern of proteins expressed, giving rise to different effects upon specific cell function.5 The lipid peroxidation-derived aldehydes ultimately form adducts with proteins, called advanced lipid peroxidation end-products, which have been implicated in numerous diseases, including atherosclerosis, neurodegenerative (Alzheimer’s, Parkinson’s, and Creutzfeldt–Jakob) diseases, chronic inflammatory and autoimmune diseases, cancer, and aging.8 In a similar manner, •O2− attacks carbohydrates, producing dicarbonyl aldehydes that cross-link and glycate proteins, forming advanced glycation end-products, which are implicated in diabetes,9 various neurodegenerative disorders,10 schizophrenia,11 and possibly autism.12,13
Figure 1.

Aldehydes generated during lipid peroxidation. When ROS react with polyunsaturated fatty acids (PUFAs), unstable lipid peroxides (PUFA-OOH) are generated. The PUFA-OOHs decompose quickly into over 200 very reactive α,β-unsaturated aliphatic aldehydes.4 Shown here is the fate of 4-hydroxynonenal (4-HNE), which forms adducts with accessible lysines, cysteines, histidines, and other reactive amino acids on proteins. 4-HNE can also react with sulfur-containing compounds, such as glutathione. Because the lipid peroxidation-generated aldehydes are aliphatic, the molecules can diffuse throughout the cell, through the membranes, and into the neighboring cells.
Enzymatic syntheses of endogenous aldehydes
A second source of endogenous aldehydes is that generated enzymatically as intermediates in hundreds of metabolic pathways. These endogenous aldehydes range in chemical type, reactivity, tissue specificity, and concentration, but all share the general chemical properties of aldehydes, with the ability to react, even though at different rates, with the same types of biomolecules. Most endogenous aldehydes are quickly reduced, oxidized, or neutralized by the cellular antioxidant defense system to prevent the random interactions with surrounding biomolecules. Enzymes that reduce the aldehyde moiety to an alcohol (OH) group are members of the alcohol dehydrogenase (ADH), the aldo-keto reductase, or the short-chain dehydrogenase/reductase superfamilies.3 Enzymes that oxidize the aldehyde moiety to an acid group include aldehyde dehydrogenase (ALDH) superfamily members as well as a few other miscellaneous enzymes, such as aldehyde oxidase, xanthine oxidase, and molybdenum hydroxylases.3 Of note, some endogenous aldehydes are first conjugated with glutathione, before being detoxified by specific enzymes in the cellular antioxidant defense system.3 Humans have evolved a variety of mechanisms to neutralize and prevent damage caused by endogenous aldehydes, but an overload of exogenous aldehydes, oxidative stress, and/or faulty genes can sabotage the protective mechanisms. Mutations in the coding or noncoding regions of genes affect either the enzymatic activity or the copy number of the gene product. Some genetic errors, such as those that increase the clearance of intermediate aldehydes, are beneficial. Other genetic errors, which cause an accumulation of intermediate endogenous aldehydes, are detrimental. Most of the current knowledge on the genetic causes of aldehyde accumulation derives from the alcoholism or inborn errors of metabolism literature involving the ADH and ALDH superfamilies.
Endogenous aldehydes involved in alcoholism
As shown in Figure 2, the liver forms of ADH convert ethanol into a toxic acetaldehyde intermediate, and subsequently, ALDH2 converts the toxic acetaldehyde into a water-soluble, nontoxic acid, which is excreted in the urine. A noteworthy feature of the ADH isozymes is that none have a particularly high specificity for ethanol. Members catalyze a wide range of substrates containing an alcohol (OH) group, including but not limited to retinol,14,15 4-hydroxyalkenal,14 and the intermediate metabolites of neurotransmitters,16 all of which are associated with various neurological disorders. The seven human ADH genes, listed in Table 1, are closely clustered on chromosome 4 (4q21-4q25), in the following 5′ to 3′ order: ADH7, ADH1C, ADH1B, ADH1A, ADH6, ADH4, and ADH5.17,18 To date, hundreds of mutations in the coding and noncoding regions of the ADH genes have been identified, which are associated with either alcoholism or an aversion to alcoholism.19 For example, the ADH1B*2 allele, found in high frequency in Asians and a lower frequency in European, African, and Jewish populations, appears to have a protective effect against alcohol dependence because the mutation increases the rapid oxidation of ethanol, thus forming the toxic acetaldehyde intermediate. On the other hand, the ADH1B*3 allele, common among African populations, increases the risk for alcoholism, even though the mutation increases the rate of ethanol oxidation. One explanation appears to be that mutations increasing the ethanol oxidation cannot be interpreted in isolation from information about the corresponding ALDH2 gene, which may or may not rapidly metabolize any excess acetaldehyde generated by mutant ADH genes. An ALDH2 mutation, E487K, is the most infamous of all because the mutation is found in over 500 million people worldwide, mostly in East Asians. The mutation significantly reduces the catalytic activity of the ALDH2 isozyme, increasing the accumulation of the toxic acetaldehyde intermediate when ethanol is consumed. Not only is the acetaldehyde intermediate responsible for the notorious Asian flush reaction and other unpleasant effects, which discourage ethanol ingestion, but the ALDH2 mutation has also been associated with an increased risk of gastrointestinal, esophageal, colon, and liver cancers20 as well as late-onset Alzheimer’s disease,21 even in the absence of long-term ethanol usage.
Figure 2.
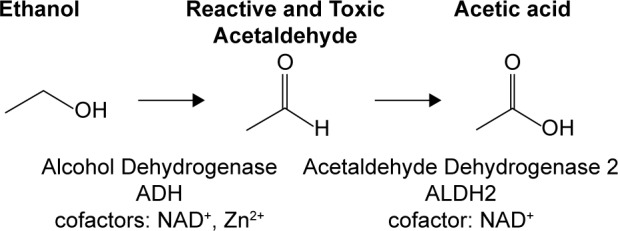
Ethanol metabolism. Ethanol is metabolized to the reactive and toxic acetaldehyde by ADH isozymes. Acetaldehyde is metabolized to acetic acid by acetaldehyde dehydrogenase 2 (ALDH2). An accumulation of acetaldehyde manifests as unpleasant symptoms, including facial flushing, nausea, and rapid heart beat.
Table 1.
Comparison of chromosomal loci of alcohol/aldehyde metabolic genes with selective genes implicated in alcoholism and autism spectrum disorder.*
| ALCOHOL/ALDEHYDE METABOLISM
|
ALCOHOLISM
|
AUTISM SPECTRUM DISORDER
|
||||
|---|---|---|---|---|---|---|
| GENE | LOCI | GENE | LOCI | GENE | LOCI | |
| Risk loci98 | 1q21.1 | |||||
| CHDIL | 1q21.1 | |||||
| ALDH9A1 3rd step of carnitine biosynthesis; also ABA pathway | 1q22-q23 (or 1q24.1) |
|||||
| SERINC2150 | 1p35.2 | |||||
| OPRD1 | 1p35.3 | |||||
| AUTS11 | 1q41 | |||||
|
ALDH4A1§ Hyperprolinemia type II, seizures |
1p36.13 (or 1p36) |
|||||
| NRXN1 | 2p16.3 | |||||
| XDH or XO Acetaldehyde oxidation | 2p23.1 | |||||
| AUTS5 | 2q | |||||
| SCN2A | 2q24.3 | |||||
| ALDH7A1P2 | 2q31.1 | |||||
| ZNF804A113 | 2q32.1 | |||||
| IP 3R151 | 3p26.1 | |||||
| ALDH1L1 Tetrahydrofolate metabolism | 3q21.3 | |||||
| SLC9A9 | 3q24 | |||||
| AUTS8 | 3q25-q27 | |||||
| Risk loci98 | 3q29 | |||||
| GABRA2 | 4p12 | |||||
| GABA4 | 4p12 | |||||
| CD38108 | 4p15.32 | |||||
| SNCA REP1 | 4q22.1 | |||||
| ADH family Ethanol, retinol & neurotransmitter metabolism | 4q21-4q25 | ADH1B | 4q23 | |||
| ADH1C† | 4q23 | ADH1C†,104 | 4q23 | |||
| ADH4 | 4q23 | |||||
| ADH7 | 4q23 | |||||
| DKK2 | 4q25 | |||||
| ALDH7A1P1 | 5q14 (Gene Atlas) |
|||||
|
ALDH7A1§ Antiquitin, seizures Lipid peroxidation metabolism |
5q23.2 | |||||
| GABA-A | 5q34 | |||||
|
ALDH5A1§ SSADH Deficiency |
6p22.3 | |||||
| ALDH8A1 Retinol metabolism | 6q23.3 (6q24.1-q25.1) |
|||||
| NYP | 7p15 | |||||
| Ethanol Consumption†,106 | 7q11.22 | AUTS2†,106 | 7q11.22 | |||
| AUTS1 | 7q22 | |||||
| AUTS9 | 7q31 | |||||
| TAS2R16 | 7q31.32 | |||||
| CHRM2 | 7q35 | |||||
| AUTS10 | 7q36 | |||||
| CNTNAP2 | 7q35-q36 | |||||
| ALDH7A1P3 | 7q36.1 | |||||
| OPRK1 | 8q11.23 | |||||
| ALDH1B1Ethanol detoxification | 9p13.2 | |||||
| ALDH1A1 Ethanol & retinolmetabolism | 9q21.13 | |||||
|
ALDH18A1§ Glutamate metabolism |
10q24.1 | |||||
| CYP2E1 Metabolism of ethanol, drugs, hormones, & xenobiotic toxins | 10q26.3 | CYP2E1†,152,153 | 10q26.3 | CYP2E1†,105 | 10q26.3 | |
| CAT Ethanol metabolism | 11p13 | |||||
| SHANK2 | 11q13 | |||||
| ALDH3B1 Ethanol metabolism Lipid peroxidation | 11q13.2 | |||||
| ALDH3B2 | 11q13.2 | |||||
| AASDHPPT α-aminoadipate dehydrogenase-phosphopante-theinyl transferase Lysine degradation | 11q22 | |||||
| ANKK1 | 11q23 | |||||
| DRD2 | 11q23 | |||||
| PKNOX2150 | 11q24.2 | |||||
| GRIN2B | 12p13.1 | |||||
| AUTS13 | 12q14 | |||||
| ALDH1L2 Tetrafolate metabolism | 12q23.3 | |||||
| ALDH2 Ethanol metabolism | 12q24.2 | ALDH2 | 12q24.2 | |||
| AUTS3 | 13q14.2-q14.1 | |||||
| HTR2A | 13q14.2 | |||||
| RCBTB1 | 13q14.2 | |||||
| CHD8 | 14q11.2 | |||||
|
ALDH6A1§ Methylmalonate semial-dehyde dehydrogenase deficiency |
14q24.3 | |||||
| NRXN3† | 14q24.3-q31.1 | NRXN3† | 14q24.3q31.1 | |||
| AUTS4 | 15q11.2-q13 | |||||
| Risk loci98 | 15q11.2-q13 | |||||
| ALDH1A2 Retinoic acid biosynthesis | 15q21.3 | |||||
|
ALDH1A3§ Microphthalmia; autism; anophthalmia; retinoic acid biosynthesis; and possibly GABA problem in mice |
15q26.3 | ALDH1A3§ | 15q26.3 | |||
| Risk loci98 | 16p11.2 | |||||
| AUTS14A | 16p11.2 | |||||
| AUTS14B | 16p11.2 | |||||
| ALDH3A1 Major corneal protein | 17p11.2 | |||||
|
ALDH3A2§ Oxidation of long chain fatty aldehydes in lipid metabolism; Sjörgren-Larsson syndrome |
17p11.2 | |||||
| SLC6A4† | 17q11.2 | SLC6A4† | 17q11.2 | |||
| AUTS7 | 17q21 | |||||
| ALDH16A1 | 19q13.33 | |||||
| AUTS12 | 21p13-q11 | |||||
| COMT | 22q11 | |||||
| Risk loci98 | 22q11.2 | |||||
| Risk loci98 | 22q13.33 | |||||
| NLGN3 | Xq13.1 | |||||
| NLGN4 | Xp22.32-p22.31 | |||||
| MECP2 | Xq28 Rett syndrome | |||||
| PTCHD1 | Xp22.11 | |||||
| RPL10 | Xq28 | |||||
| TMLHE | Xq28 | |||||
Notes:
The susceptibility chromosomal loci or genes for each disorder were listed in OMIM154 unless otherwise specified.
fLoss of function in these genes are associated with single-gene disorders that express symptoms similar to those found in ASD.
These genes have been implicated in both alcoholism and autism.
Endogenous aldehydes generated by inborn errors of metabolism
There are 19 members and several pseudogenes of the ALDH superfamily, which oxidize a diversity of endogenous and exogenous aldehydes, including acetaldehyde and retinal, in a wide range of human tissues.22 Unlike the ADHs, the ALDH family members are coded by genes scattered throughout the chromosomes (Table 1). The only ALDH gene product to participate in ethanol metabolism is ALDH2. The other members of the ALDH superfamily participate in converting an aldehyde intermediate to an acid product in various metabolic pathways, including the biosynthesis of the retinoic acid form of vitamin A, the oxidation of long-chain fatty aldehydes, and the metabolism of several amino acids as well as glutamate, γ-aminobutyric acid (GABA), and inositol. Mutations, which cause a loss of activity in ALDH isozymes, have been associated with cataracts, heart disease, gout, osteoporosis, and seizures. Most relevant to this article are the ALDH polymorphisms associated with inborn errors of metabolism disorders, which often include neurological manifestations, such as mental retardation, spasticity, and seizures.23–27 As shown in Table 2, loss-of-function (LoF) mutations in 7 of the 19 known ALDH genes are either associated with autism or display symptoms that are often observed in individuals with ASD.
Table 2.
Inborn errors of metabolism associated with the loss-of-function mutations in ALDH genes.
| GENE | ALTERNATE NAME(S) | PROTEIN FUNCTION | LoF SYMPTOMS | REFERENCE |
|---|---|---|---|---|
| ALDH1A3 | Retinaldehyde dehydrogenase (RALDH3) | Retinoic acid biosynthesis | Anophthalmia, microphthalmia, autistic traits | 27 |
| ALDH3A2 | Fatty aldehyde dehydrogenase (FALDH) | Oxidation of long chain fatty aldehydes | Sjögren-Larsson mental retardation, spasticity | 23 |
| ALDH4A1 | Pyrroline-5-carboxylate dehydrogenase (P5CDH) | Glutamate biosynthesis from proline | Hyperprolinemia type II seizures | 44, 45 |
| ALDH5A1 | Succinic semialdehyde dehydrogenase (SSADH) | Catabolism of GABA | SSADH deficiency developmental and speech delays, mild autism | 23 |
| ALDH6A1 | Methylmalonate semialdehyde dehydrogenase (MMSDH) | Inositol, valine leucine, isoleucine, propanoate metabolisms | MMSDH deficiency developmental delay, dysmorphic features, dysmeylination | 26 |
| ALDH7A1 | Antiquitin or α-aminoadipic semialdehyde dehydrogenase | Lysine metabolism | Atypical B6-dependent seizures | 24 |
| ALDH18A1 | Glutamate γ-semialdehyde synthetase or 1-pyrroline-5-carboxylate synthetase | Glutamate metabolism, proline biosynthesis | Debarsy syndrome facial dysmorphism, psychomotor retardation | 25 |
Aldehydes generated by gut microbiota
Gastrointestinal abnormalities are common among those suffering from neurodevelopmental disorders, including ASD. In addition to malabsorption problems in unhealthy intestines, abnormal microbiota of the gut appear to be contributing factors in ASD mouse models28 as well as in humans.29 One suggested explanation is that yeast and bacterial gut flora generate toxins, including alcohols and aldehydes, such as methylg-lyoxal,30 during the metabolism of various carbohydrates. Methylglyoxal is a potent aldehyde implicated in numerous disorders.3 Certainly, Candida infections common in ASD31 have long been suspected of converting carbohydrates into ethanol,32 which is subsequently metabolized to the potent neurotoxin, acetaldehyde. Alterations in the normal gut microflora of mice have also been linked to oxidative stress.33 Research into the microbiota–gut–brain axis in neurodevelopmental disorders is in its earliest stages, but aldehydes may play an important role.
Aldehyde toxicity
Aldehydes are very toxic substances, yet there is limited information about the symptoms for all but a few of the environmental aldehydes. For example, the United States Environmental Protection Agency34 reports acute, short-term toxicity symptoms for inhaled, but not oral acetaldehyde as irritation of the eyes, skin, and respiratory tract. The symptoms of chronic long-term exposure of inhaled acetaldehyde resemble those of alcoholism. Moreover, acetaldehyde is a probable carcinogen based on animal and human studies. The toxicity profile of endogenous aldehydes mostly derives from the alcoholism, lipid peroxidation, and glycoxidation literature but is, by no means, comprehensive.
Selective micronutrient deficiencies
Most of the evidence for selective, aldehyde-induced micronutrient deficiencies arises from the alcoholism literature and the study of acetaldehyde, the intermediate of ethanol metabolism. Prolonged ethanol consumption is known to cause oxidative stress35 and induce deficiencies in a number of key nutrients, including but not limited to retinol, glutathione, Zn2+, B1, B6, and folate.36 Although the nutrient-deficient status of an alcoholic is often attributed to a nutrient-poor diet or to ethanol-induced malabsorption, the reality is much more complex.37 The mechanism for some micronutrient deficiencies includes direct reactions with the electrophilic acetaldehyde generated during ethanol metabolism. For example, ethanol is known to induce B138 and B639 deficiencies and to lower hepatic glutathione levels in alcoholics by several mechanisms. In one B1 mechanism demonstrated in vitro, the electrophilic acetaldehyde attacks the C2 adjacent to the sulfur in the thiophene ring of B1, thereby lowering the bioavailability of B1.40 One mechanism for the decrease in hepatic glutathione levels involves the binding of the reactive acetaldehyde, not to glutathione directly, but to the glutathione intermediate cysteinylglycine.41 Similarly, acetaldehyde also reacts directly with selective amino acids and sulfur-containing antioxidants, such as N-acetylcysteine (NAC) and taurine.42 Ethanol ingestion is also known to induce folate deficiencies, with one mechanism demonstrating the acetaldehyde-induced cleavage of folate by xanthine oxidase.43 Although there are no reports of B6-acetaldehyde adducts, the activated form of B6, pyridoxal-5-phosphate (P5P), is a type of aldehyde, which is known to form condensation products with other aldehydes, thereby decreasing the bioavailability of B6. In fact, the evidence for B6-aldehyde condensation products formed in vivo in localized intracellular regions is the strongest and most convincing of all micronutrient studies.44 Two well-established examples include LoF mutations in pyrroline-5-carboxylate dehydrogenase45 and in α-aminoadipic semialdehyde dehydrogenase,24,46,47 also known as antiquitin, which cause the intermediate aldehydes to accumulate. In both examples, the aldehyde intermediates react irreversibly with P5P, forming condensation products that are subsequently detected in the urine. Although a global B6 deficiency is not detected by standard clinical assays, the ensuing, cell-localized B6 deficiency causes atypical B6-dependent seizures in both disorders. The reaction is shown in Figure 3 for antiquitin. Although the investigators show only hydrogen atoms neutralizing the double negative charges on the P5P in their original literature reports, this author suggests that neutralization of the double charge by hydrogen atoms is unlikely at the typical cellular pH. The charge is more likely to be neutralized by divalent metal ions, such as Mg2+ or Zn2+, creating a localized deficiency in the neutralizing atoms. B6 and folate are cofactors in methylation reactions; so chronic deficiencies in one or both disrupt the methylation of DNA,48 which subsequently alters certain transcriptional signaling, DNA repair mechanisms, and chromatin remodeling.49 In addition to these micronutrients, there are suggestions that acetaldehyde or other aldehydes may also react with cobalamin and inositol, but at present, there is a paucity of chemical data to confirm such reactions. Moreover, there are no systematic studies that address the interaction of reactive aldehydes with other micronutrients. Taken together, aldehyde toxicity induces micronutrient deficiencies in sulfur-containing antioxidants, Zn2+, B6, B1, Mg2+, and folate, creating oxidative stress and disruptions in a cascade of metabolic reactions.
Figure 3.

Semialdehyde reaction with activated B6. LoF mutations in ALDH4A144 or in ALDH7A124 result in an accumulation of a semialdehyde intermediate. Shown here is the α-aminoadipic semialdehyde generated by a LoF mutation in ALDH7A1, which reacts with the P5P aldehyde to form two complexes, A and B. The condensation reaction causes a cell-localized deficiency of P5P, resulting in B6-dependent seizures. It is not known whether the double negative charge on the phosphate group of both complexes is neutralized before excretion to the urine. Because P5P commonly binds to proteins via a complex with Mg2+ or Zn2+, it is possible that Mg2+ or Zn2+ ions neutralize the charges and are lost in the urine as well.
In addition to the direct reactions between aldehydes and micronutrients, the alcoholism literature suggests that alternative mechanisms may induce a deficiency in the retinoic acid form of vitamin A particularly in those individuals with heritable forms of alcoholism. Ethanol is known to compete directly for the retinol-binding site on the ADHs involved in the rate-limiting step of retinol oxidation, thereby decreasing the amount of retinal and retinoic acid that is ultimately produced. Because retinoic acid controls the regulation of fetal development, neuronal growth, differentiation, and limb morphogenesis, ethanol-induced deficiencies of retinoic acid are believed to play a major role in fetal alcohol syndrome disorder (FASD).50,51 Retinoic acid also plays a major role in epigenetic changes in the cell.49 In many ways, the symptoms of FASD mimic those of the complex form of ASD, suggestive of a problem during embryonic development.52 Moreover, many ASD children suffer from hypovitaminosis A,53,54 which is commonly attributed to highly restrictive diets or to intestinal malabsorption. However, some reports indicate that the same children do not have night blindness,53 and some reports suggest that ASD symptoms are reduced by retinol treatment.54 The lack of night blindness in ASD cases of vitamin A deficiencies suggests that retinal is present, but conversion to retinoic acid is blocked. An accumulation of retinal would have the same toxicity consequences as other endogenous aldehydes but with the added problem that a deficiency in retinoic acid would disrupt the retinoic acid response element (RARE)-dependent transcription of many key proteins in embryonic and neuronal development.
Protein and DNA damage
In addition to reacting with small molecules, acetaldehyde is believed to cause long-term cellular damage in chronic alcoholism by reacting with macromolecules.55,56 Acetaldehyde and other types of endogenous aldehydes cause disruptive tissue damage by reacting with accessible lysines, histidines, cysteines, and arginines on proteins57 as well as by forming adducts with mainly deoxyguanine bases of DNA, promoting strand breakage and mutagenesis.7 Curiously, although most proteins contain these amino acids, certain proteins are preferentially modified by acetaldehyde, including hemoglobin, albumin, tubulin, lipoproteins, collagen, cytochrome P450 2E1 (CYP2E1), and ketosteroid reductase.58 High concentrations of ethanol increase levels of microsomal CYP2E1, which mediates the generation of ROS during ethanol oxidation. As shown in Figure 1, ROS then attacks PUFAs and the first metabolites formed are lipid hydroperoxides that are unstable and degrade rapidly to very reactive endogenous α,β-unsaturated aldehydes,4 such as 4-HNE and 4-HHE. The aldehydes generated from lipid peroxidation are more damaging than ROS because the aldehydes are more stable, amphiphilic, and can diffuse across membranes to neighboring cells.5 The α,β-unsaturated aldehydes essentially form irreversible adducts with other molecules, including proteins6 and DNA.7 Moreover, the targets of lipid peroxidation-generated aldehydes are specific to cell types, aldehyde concentration, and the pattern of proteins expressed, giving rise to differential effects upon cell function.5 The lipid peroxidation-derived aldehydes have been implicated in cardiovascular, neurodegenerative, chronic inflammatory, and autoimmune diseases as well as cancer and aging.8 In addition to macromolecular damage, Hao and Maret59 have shown how the lipid peroxidation-generated aldehydes release Zn2+ from proteins by binding to cysteines typically found in Zn2+-binding sites. In this regard, it is noteworthy that zinc supplementation attenuates oxidative stress in mice by suppressing the ethanol-induced CYP2E1 activity but increasing the activity of liver ADH.60 The major enzyme involved in detoxification of lipid peroxidation products is glutathione-S-transferase, which depends upon reduced glutathione (GSH) conjugates generated via ADH5.3 Microsomal ALDH2 also has a role in the detoxification of lipid peroxidation products. In a similar manner, •O2− attacks carbohydrates, producing dicarbonyl aldehydes, such as glyoxal and methylglyoxal, which cross-link and glycate proteins, forming advanced glycation end-products. O’Brien et al3 have comprehensively reviewed the macromolecular damage done by a variety of endogenous and exogenous aldehydes.
Treatment for aldehyde toxicity
With all the cellular damage done by accumulated endogenous aldehydes, it is not surprising that some types of treatment are being devised to counteract the damage. Negre-Salvayre et al8 review some of the pharmacological inhibitors under development to block oxidative stress, lipid peroxidation, and reactive aldehydes. Aldeyra Therapeutics has active clinical trials investigating the effects of a compound called NS2 on aldehyde toxicity in several disorders.61 Until targeted pharmacological therapies become available, the alternative treatment is the use of commercially available antioxidants. The best appear to be the sulfur-containing antioxidants: taurine and the bioavailable form of cysteine, N-acetyl cysteine (NAC). Taurine has been used to mitigate the symptoms of succinic semialdehyde dehydrogenase (SSADH) deficiency induced in Aldh5a1-deficient mice62 as well as in a patient with a genetic defect, resulting in a LoF of ALDH5A1,63 as shown in Figure 4. Some research indicates that taurine interacts directly with aldehydes42 and potentially with some free radicals at physiological concentrations.64 Far more research has been done with NAC, with several reviews citing the therapeutic value of NAC in treating psychiatric and neurological disorders, including but not limited to ASD, addictions, obsessive–compulsive disorder, and Alzheimer’s disease.65,66 Several mechanisms have been proposed for the therapeutic value of NAC, including its role as a modulator of glutamate transmission, precursor and protector of GSH levels in oxidative stress, and interactions with inflammatory mediators.67 During oxidative stress, microsomal cytochrome P450s reduce O2 in the presence or absence of substrates to generate •O2−, hydrogen peroxide (H2O2), and in the presence of a chelated iron, a hydroxyl radical (•OH).68 Most recent reviews assume that NAC functions as an excellent scavenger of all ROS generated during oxidative stress and, thus, protects the GSH stores. However, the chemical data69 demonstrate that NAC only reacts quickly with •OH, very little with hydrogen peroxide, and not at all with •O2−. Whether NAC reacts directly with any type of ROS in vivo or not, NAC has been shown to provide the best protection against acetaldehyde toxicity in rats, even better than taurine.70 Other studies have shown that NAC reacts strongly with the endogenous aldehydes, such as 4-HNE generated during human lipid peroxidation.5,71 Lipoic acid, another sulfur-containing antioxidant, also seems to be effective in protecting against 4-HNE-mediated oxidative stress.72
Figure 4.

SSADH deficiency. A LoF mutation in ALDH5A1 results in an accumulation of succinic semialdehyde, which is subsequently metabolized to γ-hydroxybutyric acid (GHB), the date rape drug. Taurine probably interacts with succinic semialdehyde42 and prevents the formation of GHB. Taurine therapy was effective in treating a child with SSADH deficiency.63
Autism spectrum disorder
Description
ASD is a heterogeneous group of neurodevelopmental disorders characterized by impairments in social interactions and communications as well as the presence of restricted, repetitive patterns of behavior and interests.73,74 The types and severity of phenotypic symptoms represent a very heterogeneous spectrum of behavior that may be a result of abnormalities in numerous underlying biochemical mechanisms.75 Hand flapping, head banging, and body rocking are examples of repetitive behaviors seen in some, but not all, individuals diagnosed with ASD. Delays in verbal language, staring, and a high degree of irritability or agitation (rage tantrums) are examples of communication or social impairment skills. Mental retardation afflicts as many as 70% of ASD individuals, though the severity varies greatly. Miles52 summarizes a comprehensive list of behaviors. In one common type of autism, a child develops normally for up to two years and then regresses suddenly over the course of a week or two, sometimes without any identifiable trigger.76 Since 2013, the definition of ASD has broadened to include Asperger’s syndrome as well as pervasive developmental disorder not otherwise specified.74 With the broader definition as well as better early diagnostics, it is estimated that as many as 1 in 68 individuals are afflicted with ASD.1 An additional challenge to the diagnosis of ASD is that many ASD children have comorbid medical conditions, including gastrointestinal symptoms, seizures and epilepsy, attention deficit hyperactivity disorder, anxiety, allergies, and mitochondrial disease.77 Although much emphasis has been placed on identifying the genetic basis of ASD, it is well appreciated that environmental factors, such as air pollution and heavy metals,78 also play a role. To date, the broad heterogeneity of symptoms, all grouped into ASD, has confounded attempts to identify abnormal chromosomal loci common to all, specific environmental risk factors, unique biochemical markers, or rational treatment plans applicable to all. The largest subgroupings include those individuals with immune dysregulation and/or inflammation, oxidative stress, environmental toxicant exposures, mitochondrial dysfunction, an inhibitory–excitatory systems’ imbalance, and folic acid deficiencies.79–82 Related to the central hypothesis of this article are reports of oxidative stress in autistic children by several researchers who have identified elevated urinary aldehydes, 4-HNE, and malondialdehyde (MDA), among other changes in proteins and small molecular markers associated with oxidative stress.83–86 In one study, the severity of autism was directly correlated with the urinary level of the urinary aldehyde, 4-HNE.84 Although many researchers in the autism field note that ROS, perhaps meaning all its downstream manifestations, is responsible for the cellular damage, Damodaran and Arumugam84 specifically state that the lipid hydroperoxides, which are generated when ROS react with PUFAs as shown in Figure 1, are too labile to measure and decompose rapidly to aldehyde products. In their review, Signorini et al87 emphasize that aldehyde products of PUFAs are likely responsible for intracellular and membrane protein damage, depletion of antioxidants, and DNA damage associated with oxidative stress in autism. Other urinary aldehyde markers are found in some autistic children with comorbid manifestations and are a consequence of abnormalities associated with ALDH genes.23,24,46,47,88
Genetics
Early twin studies suggest the heritability of ASD to be approximately 90%, but with additional larger populations studies, the heritable factor is now commonly estimated to be 50%.89 Some believe that environmental factors also contribute to the development of the disorder in genetically predisposed individuals.78 In the last decade, there has been an explosion of genetic studies of ASD, using numerous methodologies.90 Early genetic studies of twins and small populations indicated a significant heritability of ASD. Indeed, a number of single-gene disorders, with symptoms similar to autism, have identified several syndromes, including but not limited to Fragile X, Rett, Timothy, and Joubert syndromes.52 As the cohort samples became larger, consistent results between candidate gene studies and genome-wide association studies (GWAS) have not emerged. Not surprisingly, due to the heterogeneous nature of ASD, no single mutation in a gene or small cluster of genes has been identified as the causative factor in all cases. Instead, prevailing belief is that common variants of hundreds of genes each contribute a small percentage to ASD.91 Although a number of intriguing candidate genes have been identified by GWAS, a comprehensive meta-analysis of common genetic variants in ASD has failed to identify any that are statistically significant.92 The theory that combinations of common variant genes lead to autism has been challenged by a different theory in which de novo mutations occur in the ASD individual that were not present in either parent.93–96 Using a statistical analysis of the de novo, likely gene-disruptive mutations, upward of 50% of ASD is postulated to be caused by spontaneous mutations.97 In addition to single-nucleotide polymorphisms, de novo copy number variations have been implicated in autism at six chromosomal locations, as shown in Table 1, implicating 65 risk genes in several pathways including, but not limited to, chromatin remodeling involving lysine methylation/demethylation, zinc fingers, transcription factors, and synaptic proteins.98 With so many possible gene candidates, efforts are turning toward biological network and pathway analyses to identify functional pathways implicated by genomic studies as well as associated with specific phenotypes,99 but it may require another generation or more before ASD is better understood. It is not the objective herein to summarize all of the genetic studies relevant to ASD but simply to highlight a few interesting genetic candidates as well as to describe recent genetic findings that support the central hypothesis of this article.
The percentage of ASD children estimated to have a history of familial alcoholism varies greatly, from 4% to as high as 55%.100–103 The broad range depends on the homogeneity or heterogeneity of the genetic population under study, as well as on the methodology for ascertaining the presence of alcoholism in the family genes of autistic individuals. The lower numbers consider only whether a parent is an alcoholic. The higher estimates consider the number of alcoholics among the first- and second-degree relatives to insure that the ASD children have a pedigree consistent with the heritable form of alcoholism.100–102 Because the studies all use vastly different techniques, cohort groups, and alcoholism definitions, a link between autism and alcoholism is controversial. To clarify, a comprehensive analysis examined the linkage between the ADH variants associated with alcoholism and the risk of other psychiatric disorders, including autism.104 The study found single-nucleotide polymorphisms in the ADH genes to be significantly correlated with schizophrenia in African-Americans and autism in European Americans. The most common mutations are found in ADH1C, which codes for a protein also involved in the turnover of serotonin, one of the neurotransmitters implicated in some forms of autism. No one has yet conducted an analogous study looking at the associations between autism and ALDH2/CYP2E1 polymorphisms involved in the metabolism of ethanol or a combination of ADH and ALDH2/CYP2E1 polymorphisms.
Other interesting ASD gene candidates are found in ALDH family members, related to ALDH2 but not involved in ethanol metabolism. Of the 19 known ALDH isozymes, two have been linked to autism: the first via retinoic acid metabolism (ALDH1A3)27 and the second via GABA metabolism (ALDH5A1).23 An additional five ALDH genes have been linked to inborn errors of metabolism, which display symptoms that are often observed in individuals with ASD. As shown in Table 2, LoF of ALDH3A2, ALDH4A1, ALDH6A1, ALDH7A1, or ALDH18A1 results in intellectual disabilities, Sjögren–Larsson syndrome, hyperprolinemia type II, γ-hydroxybutyric aciduria (also known as SSADH deficiency), developmental delay, or atypical B6-dependent seizures, respectively.23–26 Moreover, one of the pseudogenes, ALDH7A1P3 of unknown function, falls within the chromosomal region 7q36, which is strongly associated with autism. Although there are no known inborn errors of metabolism linked to the remaining 12 ALDH genes, some are involved in ethanol-unrelated metabolic pathways that are errant in ASD individuals. For example, ALDH9A1 catalyzes the oxidation of a broad range of aldehydes and has a role in l-carnitine biosynthesis and GABA metabolism. In addition to those already noted for inborn errors of metabolism, several more are involved in folate metabolism (ALDH1L1 and ALDH1L2), lipid peroxidation (ALDH3B1), and retinol metabolism (ALDHA1). In addition to the ADH and ALDH genes, another ethanol metabolizing gene, CYP2E1, has recently been linked to ASD.105 Also, the autism susceptibility gene (AUTS2) has been linked to the regulation of alcohol consumption.106
Also noteworthy is that retinoic acid, via RARE, plays a role in the transcription of ALDH1A3, which is linked to autism. RARE is known to be associated with other genes linked to ASD, such as the inositol 1,4,5-triphosphate receptor (IP3R1) disabled in Timothy syndrome107 and the CD38 antigen.108 Although the presence of RARE has not been studied in most genes associated with ASD, some genes, such as GRIN2B, belong to a super-family coding for ion channels,109,110 which generally include RARE as a part of transcriptional regulation. Other ASD-associated genes, such as the synaptic neurexins, neuroligins, contactins, Down syndrome cell adhesion molecule (DSCAM), and SH3 and multiple ankyrin repeat domains 3 (SHANK3) are members of the cell adhesion protein family, some of which involve RARE in mouse embryonic development.111,112 RARE is extremely important in fetal developmental and neuronal differentiation. A number of candidate genes associated with ASD involve methylation, such as methyl-CpG-binding protein 2 (MeCP2),52 which binds to methylated CpGs as well as to zinc-finger transcription factors, such as zinc finger protein 804A (ZNF804A).113
Treatment
Currently, there are no known effective medications to treat the core symptoms of autism. Medications are used to treat only severe symptoms of comorbid disorders because autistic children generally respond less favorably and experience more side effects than peers without ASD.114 For example, the most common medications include the antipsychotics, such as risperidone and aripiprazole, to treat the extreme irritability manifested as aggression, self-injury, and severe tantrums. NAC added to risperidone has been shown to be more effective than risperidone alone in reducing irritability symptoms of ASD.115,116 Other medications are reviewed by Ji and Find-ling.114 Given the adverse side effects of medications in children with ASD, it is not surprising that caregivers have focused on alternative treatments. The most effective appears to be early intensive behavioral therapy, based on the principles of applied behavior analysis and given for multiple years at an intensity of 20–40 hours per week.117 There is also a large body of research investigating the nutritional status of individuals with ASD as well as the potential benefit of micronutrient supplementation. Autistic children are notorious picky eaters, leading to some documented deficiencies in individual case studies, such as vitamin A deficiencies.53 Nutritional status based on dietary record keeping, not surprisingly, gives conflicting results,118 in part because autistic children often suffer from gastrointestinal mal-absorption problems77 or because genetic factors may play a role in nutritional deficiency. There are a myriad of studies in which single micronutrient supplementation has been assessed, with several reporting mitigation, but not complete elimination, of some ASD symptoms. Many of the micronutrients studies have been based on plasma and urinary clinical results, which suggest the elevation of lipid peroxide-generated aldehydes (HNE, MDA)84–86 as well as deficiencies in antioxidants, such as vitamin E, sulfur-containing compounds, such as cysteine, methionine, and GSH,119 carnitine,120 biotin,121 and (n-3) PUFAs.122 Given the depressed levels of sulfur-containing compounds in ASD individuals, it is not surprising that these children are more sensitive than neurotypicals to the exposure of mercury and other heavy metals.78 The antioxidant supplements, which show symptom improvements include, but are not limited to ubiquinol,123 NAC,124 carnosine, and vitamin C.125 Other micronutrients, including B1,126 B6-Mg2+,127 folate, vitamin E, retinol, and Zn2+, have been selected for a variety of reasons relating to the involvement in abnormal metabolic pathways implicated in ASD,1 even though the clinical assays do not always show a deficiency in ASD individuals compared to neurotypical controls.119 With all of the single micronutrient studies, there are conflicting results regarding efficacy.119 There are a few studies, which utilize broad-spectrum micronutrient supplementation and report improvements in clinical biomarkers and/or various autism-rating scales.76,121,128 The broad-spectrum micronutrient studies are very promising because all demonstrate a significant correlation between nutritional deficiencies and severity of autistic symptoms.
Discussion
Autism symptoms mimic the symptoms of aldehyde toxicity
Acetaldehyde toxicity
As early as 1975,129 the disease of alcoholism was believed to be a disease of acetaldehyde toxicity. Acetaldehyde reacts directly with a select group of micronutrients, eliminating their bioavailability. These micronutrients include many sulfur-containing antioxidants, resulting in oxidative stress, as well as B1, B6, and folate, causing a cascade of metabolic disturbances in hundreds of reactions. Other micro-nutrients, such as cobalamin, inositol, and other carbohydrate aldoses, may possibly react with acetaldehyde, but no in vitro studies are available. Acetaldehyde also forms covalent adducts with accessible reactive amino acids on proteins, not only inactivating the proteins, but releasing Zn2+. Acetaldehyde reacts with DNA, forming covalent adducts and eventually mutagenizing selective bases. In addition to acetaldehyde toxicity, ethanol also effectively competes with retinol for binding to certain ADHs, inducing a retinoic acid deficiency that can disrupt the RARE-dependent transcription of many proteins. This feature is particularly relevant to FASD,52 in which the ethanol-induced retinoic acid deficiency affects fetal development, neuronal growth, differentiation, and limb morphogenesis.
ASD vs aldehydism
In reviewing all facets of autism, the author notes that many manifestations of ASD mimic those of acetaldehyde toxicity, but occurring at an earlier age and in the absence of ethanol. Because all aldehydes share a similar chemical structure and reactivity profile with acetaldehyde, the author suggests that all the different types of aldehydes, no matter the source, cause the cellular damage and symptoms most commonly associated with ASD. There is already a plethora of evidence suggesting that aldehydes generated by lipid peroxidation are involved in ASD.84–87 This author expands the concept to a much broader range of aldehydes, including exogenous aldehydes in food and the environment3 and endogenous aldehydes, which accumulate as a result of genetic abnormalities that alter the enzymes that produce or clear aldehydes. In addition to specific inborn errors of metabolism that cause LoF in seven ALDH family members,23–27 other sources of aldehydes include those generated by ROS-induced peroxidation of lipids,3 ROS-induced glycoxidation of carbohydrates,3 genetic mutations that sabotage the normal cellular protection mechanisms that neutralize aldehydes,3 genetic mutations related to the heritable forms of alcoholism,19,130,131 and possibly aldehydes generated by abnormal gut microflora.30
In traditional medical literature, the strongest evidence for the role of aldehydes in ASD comes from inborn errors of metabolism associated with LoF in seven ALDH isozymes.23–27 Each disorder accumulates an intermediate aldehyde, which then expresses a symptom found in ASD (Table 2). In the case of SSADH deficiency, taurine supplementation ameliorates developmental and speech delays.63 In the case of hyperprolinemia type II44 and α-aminoadipic semialdehyde dehydrogenase deficiency,24 B6 supplementation reduces the seizures caused by the intracellular aldehyde-induced deficiencies in B6. A clinical trial,61 using a pharmacological aldehyde scavenger, is ongoing for Sjögren–Larsson syndrome to reduce the symptoms of mental retardation and spasticity.
In the orthomolecular medical literature, the strongest evidence for the role of aldehydes in ASD arises from the studies of urinary aldehydes, generated from lipid per-oxidation of PUFAs, resulting in an accumulation of potent aliphatic aldehydes, such as 4-HNE and 4-HHE.84–86 The aliphatic aldehydes can diffuse throughout the cell, through the cell membrane, and into neighboring cells.5 During diffusion, neuronal receptors and ion channels with accessible reactive amino acids form adducts with the aliphatic aldehydes, destroying their function. The aliphatic aldehydes also form adducts with DNA and cause mutations at select sites.132 The damage caused by the aliphatic aldehydes generated during lipid peroxidation occurs slowly in degenerative diseases, such as inflammatory, autoimmune, Parkinson’s, and Alzheimer’s diseases.8 In ASD, the damage occurs very quickly, suggesting that the concentration and/or reactivity of the accumulated endogenous aldehydes are much greater. A similar situation may occur in schizophrenia, but with an intermediate concentration/reactivity of accumulated aldehydes and an onset of 17–20 years.
In the alcoholism literature, acetaldehyde accumulation results, not only from an excess of ethanol ingestion, but also from the genetic errors, which predispose an individual to alcoholism. Some of the known genetic errors involve the primary ethanol-metabolizing genes: the ADH isozymes and ALDH2.19,128,129 In the absence of ethanol, the ADHs and ALDH2 catalyze other substrates, primarily involved in retinol metabolism14,15 and neurotransmitter catabolism.16 If an individual has inherited mutations, which lead to an accumulation of acetaldehyde during ethanol ingestion, these same mutations will lead to an accumulation of other types of aldehydes in the absence of ethanol. The best evidence arises from the studies of the ALDH2 mutation (E487K), which causes an accumulation of acetaldehyde in the presence of ethanol. The resulting acetaldehyde toxicity is so unpleasant that the mutation is believed to be protective and reduce alcoholism. Nevertheless, the ALDH2 mutation, even in the absence of ethanol ingestion, causes long-term damage to the same individuals, increasing the risk of Alzheimer’s disease20 as well as gastrointestinal, esophageal, colon, and liver cancers,21 all associated with DNA damage. Although the estimates vary greatly, some autistic children come from families who display genetic traits of heritable alcoholism. If some subsets of autistic children have inherited genes, which predispose them to alcoholism, then in the absence of ethanol, they are likely to have metabolic reactions that result in an accumulation of endogenous aldehydes with similar consequences to that of acetaldehyde toxicity.
Expression of micronutrient deficiencies in ASD
Frequently overlooked are the data demonstrating that endogenous aldehydes induce localized, intracellular deficiencies in a subset of micronutrients, which either initiate oxidative stress (multiple sulfur-containing antioxidants, Zn2+) or serve as cofactors (B1, B6, folate, and Zn2+) in the metabolic pathways implicated in autism: neurotransmitter functions, DNA methylation, chromatin remodeling, transcriptional regulation, and neuronal development. Thiamine pyrophosphate (TPP), the activated form of B1, and Mg2+ are cofactors of the E1 subunit of pyruvate dehydrogenase. The latter enzyme converts pyruvate to acetate and is the rate-limiting step in the ultimate synthesis of the neurotransmitter acetylcholine. When the conversion of pyruvate to acetate is blocked, the accumulating pyruvate is converted to lactate. Curiously, a subset of ASD individuals exhibit classical mitochondrial dysfunction as well as an atypical form without the classic features associated with mitochondrial disease.133 No one has addressed the possibility that a B1 deficiency elevates plasma lactate levels and is commonly mistaken for mitochondrial dysfunction.134 The activated form of B6, usually in combination with Zn2+ or Mg2+, is a cofactor in over 300 enzymes, including those involved in the production of neurotransmitters, such as GABA, glutamate, dopamine, and serotonin. B6 and folate are involved in methylation reactions; so chronic deficiencies in one or both disrupt the methylation of DNA,48 which subsequently alters certain transcriptional signaling, DNA repair mechanisms, and chromatin remodeling.49 In addition to the methylation of DNA, folate is essential in DNA and RNA syntheses, repair of DNA, cell division, and proper neural tube formation. Zn2+ also serves a role in the maintenance of DNA integrity, but a more important function may be its role in oxidative stress. Zn2+ has long been known to have a protective effect against oxidative stress, not directly as an electron transfer agent, but indirectly by acting as a Lewis acid that accelerates the transfer of electrons during the catalytic activity of Zn2+-binding enzymes.135 Zn2+ binds to cysteines, protecting thiol groups from oxidation. Accumulated endogenous aldehydes react with cysteines, releasing Zn2+ from enzymes, including those involved in ROS and aldehyde detoxification, creating increasing oxidative stress. The frequent deficiencies in sulfur-containing nutrients also contribute to oxidative stress, for which the broadest definition is used: an imbalance between oxidants and antioxidants, in favor of oxidants.136
Simple, regressive form of autism
The author postulates that endogenous and exogenous aldehyde toxicity explains many features associated with the simple form of autism,52 including oxidative stress, regression, de novo mutations, environmental risk factors, and genetic complexity, involving hundreds of common and rare variants. Aldehydes react with and deplete sulfur-containing antioxidants, initiating a downward spiral of increasing oxidative stress. Aldehydes induce deficiencies in selective micronutrients, which are cofactors in the metabolic pathways implicated in autism. Each metabolic disruption caused by a micronutrient deficiency can be mimicked by mutations in one or more genes. Thus, induced deficiencies in the set of selective micronutrients can be mimicked by hundreds of common and rare genetic variants, which have already been identified by various types of genomic studies. As modeled in Figure 5, the intracellular damage occurs first in a localized area near the origination site of the endogenous aldehydes, creating the selective nutrient deficiencies and inactivating both large and small molecules involved in antioxidant defense mechanisms. As the concentration of accumulated endogenous aldehyde increases, the downward spiral accelerates, disabling the cellular protection mechanisms against oxidative stress further and further from the origination site of aldehyde formation. The endogenous aldehydes also form adducts with DNA, which eventually lead to strand breakage and select cases of spontaneous mutations as the repair mechanisms are disabled by Zn2+, folate, and other micronutrient deficiencies. The aldehyde-induced DNA damage explains how the de novo mutations observed in approximately 50% of autistic individuals can occur. Because lipid peroxidation is likely to be involved in ASD, the aliphatic aldehydes, which are insufficiently neutralized by the cellular defense mechanisms, diffuse and begin to impair the function of membrane proteins and spread the damage to other neighboring cells. Moreover, environmental risk factors can contribute to the downward spiral. Many of the environmental risk factors, such as air pollution, new industrial products, and food flavorings, contain exogenous aldehydes, which can accelerate a decline in the antioxidant defense mechanisms. Other risk factors, including heavy metals, such as mercury and lead, are not cleared quickly if the individual has become low in sulfur-containing compounds.
Figure 5.

Effect of aldehyde accumulation in a cell. A normal cell is shown in (A), with the ★ representing both micro- and macromolecules of the antioxidant defense system as well as micronutrients B1, B6, and folate. A cell, which is under oxidative stress by an accumulation of aldehydes (Image), is shown in (B). The aldehydes are more concentrated at the site of origin. The aldehydes react with selective cellular molecules ★. The unreacted aldehydes continue to diffuse throughout the cell, reacting with whatever molecules they encounter and eventually diffuse through the membrane to other cells.
Complex form of autism
The complex form of autism involves some type of problem during embryonic development.52 Some complex forms of autism express symptoms that can be attributed to single-gene disorders, such as Timothy syndrome, tuberous sclerosis complex, and Rett syndrome.52 However, many cases cannot be explained by mutations in a single gene. The observation that some autistic children have a history of heritable alcoholism raises the possibility that a combination of ADH and ALDH genetic errors might be responsible for cell-localized folate or retinoic acid deficiency in some complex cases of ASD with unknown etiology. Folate deficiencies are known to disrupt various aspects of fetal developmental. A retinoic acid deficiency could also occur, even in the absence of heritable alcoholism, if errors in a gene pair involved in retinol metabolism produce an accumulation of retinal and a lower amount of retinoic acid. A retinoic acid deficiency would exhibit many of the same symptoms of FASD. However, unlike FASD in which ethanol induces a retinol deficiency through substrate competition, a retinoic acid deficiency in ASD is more likely to involve mutations associated with ALDHs, resulting in an accumulation of retinal and a much lower production of retinoic acid. Hypovitaminosis A in autistic children has frequently been observed,53,54 but is commonly attributed to highly restrictive diets or to intestinal malabsorption. However, some reports indicate that the same children do not have night blindness53 and some suggest that ASD symptoms are reduced by retinol treatment.54 The lack of night blindness in ASD cases of vitamin A deficiencies suggests that retinal is present, but conversion to retinoic acid is blocked. An accumulation of retinal would have the same toxicity consequences as other endogenous aldehydes described for simple autism but with the added problem that a deficiency in retinoic acid would disrupt the RARE-dependent transcription of many key proteins in embryonic and neuronal development.
Potential early symptoms of aldehyde toxicity
At present, most cases of ASD are detected by behavioral abnormalities manifested as early as 18 months. However, if aldehyde toxicity has a pivotal role in ASD, it is essential to initiate the treatment earlier to avoid irreversible cellular damage. Unfortunately, with the exception of the lipid peroxidation-generated aldehydes,83,137 there are no widely available clinical tests for other types of endogenous aldehydes. As described, aldehyde toxicity will initially express as selective micronutrient deficiencies and oxidative stress. Because the micronutrient deficiencies will occur in those cells, which accumulate aldehydes, localized intracellular deficiencies are not likely to be amenable to detection by erythrocyte and urine clinical assays that only measure the average nutritional status. Thus, other early observable symptoms of potential deficiencies must be considered. Moreover, the notion must be dispelled that there will ever be a unique diagnostic test that distinguishes ASD from other neurological disorders. ASD is a spectrum disorder, with overlapping symptoms shared by other disorders diagnosed at later ages. Aldehyde toxicity may also play a role in other neurological disorders, but the symptoms of ASD appear earlier, possibly due to higher concentration, greater reactivity, specific type, and cellular locations of accumulated aldehydes.
Potential early clinical symptoms of micronutrient deficiencies
Expressed clinical symptoms of micronutrient deficiencies would be dependent on the cellular location of the accumulated aldehydes and would not be the same as those observed for global deficiencies in malnourished individuals. Excessive nausea during pregnancy may, in some cases, be one of the first signs of a B6 deficiency, possibly due to an accumulation of endogenous aldehydes for genetic reasons. The treatment for excessive nausea is the medication Diclegis, a combination of the antihistamine doxylamine and vitamin B6. The second symptom to consider is a weak autonomic reflex, which suggests oxidative stress and a B1 deficiency. A deficiency in activated B1 (TPP) decreases the production of the neurotransmitter controlling autonomic reflexes, acetylcholine, by affecting the rate-limiting step via the TPP, Mg2+-dependent pyruvate dehydrogenase complex. Third, in cases of mild-to-moderate micronutrient deficiencies, initial symptoms usually express bilaterally in the epithelial layer where blood flow is limited. Thus, any type of symmetrical, not asymmetrical, dermatological abnormality, such as rashes, petechiae, eczema, and follicular hyperkeratosis, will provide clues as to the deficiency. Should any epithelial symptoms appear, infections and intestinal malabsorption maladies must first be considered and treated. The fourth clue to the presence of micronutrient deficiencies is the persistent nature of the symptoms. Waxing and waning symptoms, often observed in some neurological disorders, reflect a fluctuation in dietary or environmental factors. In addition to identifying and removing environmental triggers, supplementation is most likely to alleviate symptoms in these individuals by providing a constant source of micronutrients to overcome the effects of fluctuating triggers. The fifth clue is the response of the symptoms to micronutrient supplementation. For waxing and waning symptoms, trial and error approaches are less expensive, as long as nutrients are administered in safe combinations, such as B1 and Mg2+ and B6 and Zn2+/Mg2+, to prevent inadvertent induction of deficiencies in other nutrients. Consultation with professionals trained in nutritional supplementation is strongly recommended.
Potential early clinical symptoms of oxidative stress
There are a few plasma and urinary biomarkers for oxidative stress, including plasma levels of glutathione, glutathione peroxidase, methionine, and cysteine.119 However, the best early clue to oxidative stress is mild-to-moderate neonatal jaundice. As many as 60% of infants develop clinical jaundice, but a much smaller percentage (~6%) exhibit extreme levels of neurotoxic bilirubin (>25 mg/dL) causally linked to kernicterus causing devastating neurologic sequelae, including mental retardation.138,139 In cases of mild-to-moderate levels of hyperbilirubinemia (≤25 mg/dL), the temporary episode is not believed to be harmful. However, several studies have reported clinical results suggesting a relationship between neonatal jaundice and oxidative stress caused by ROS damage as determined by, among other factors, elevated levels of plasma MDA and reduced levels of GSH.140–142 Given the association of neonatal jaundice with oxidative stress and the association of oxidative stress with ASD, it is not surprising that two large population studies, one including 56,019 infants from Nova Scotia138 and the other including 733,826 Danish infants,142 found an association between moderate hyperbilirubinemia and an increased risk for ASD. Moderate neonatal jaundice may be a marker for oxidative stress, which is present in a subset of individuals suffering from many types of neurological problems, not just ASD. If the central hypothesis is correct, the source of the oxidative stress is likely due to an accumulation of endogenous aldehydes, which reduce the GSH levels to the point at which the clearance of unconjugated bilirubin is impaired. Although it would take further studies to prove such a hypothesis, the significance of such a finding would suggest that the administration of antioxidants, such as GSH, taurine, and NAC, might be useful as a cotreatment with phototherapy for hyperbilirubinemia.
Micronutrient therapies for ASD
Individuals most likely to be helped by micronutrient supplementation are those 50%–70% with the simple, regressive form of autism.52 For those 30%–50% of individuals with the complex form of autism, considerable damage is believed to have occurred during embryonic development.52 Such damage may be caused by rare single-gene inborn errors of metabolism, by folate or retinoic acid deficiency, possibly related to ALDH mutations or ethanol ingestion during pregnancy, or by other, as yet, unidentified problems. As suggested by the success of taurine supplementation in a patient with SSADH deficiency,63 an inborn genetic error might be treatable if diagnosed early.
Aldehyde neutralization
Regardless the source(s), reactive aldehydes appear to carry out the primary cellular damage associated with the simple, regressive form of autism. The best treatment approach is to neutralize the aldehydes and, simultaneously, to replace the micronutrients that have become deficient by aldehyde toxicity. For treatments targeted to a particular aldehyde type, the only guidelines currently available are those postulated by LoPachin and Gavin2: simple alkanals react better with hard nucleophilic groups, such as lysines, whereas α,β-unsaturated alkenals and α-oxoaldehydes preferentially react with soft nucleophilic groups, such as thiols and cysteines. In the absence of targeted pharmacological interventions for specific aldehyde types, the core of any treatment plan necessitates the continuous use of one or more strong sulfur-containing antioxidants, such as NAC, taurine, GSH, and lipoic acid, to react with and neutralize the accumulated aldehydes before additional cellular damage occurs. Although the alcoholism literature suggests that NAC is the best antioxidant to halt acetaldehyde damage,70 there are insufficient data in the literature to recommend one sulfur-containing antioxidant over another for other types of aldehydes found in different cellular locations. Nonsulfur-containing antioxidants, such as vitamin C and vitamin E, are not likely to be as effective in neutralizing aldehyde toxicity and would require much higher doses. The suggested core treatment is similar to a regimen that Walsh76 advocates, although he does not differentiate between sulfur- and nonsulfur-containing antioxidants in his publications.
Replenishment of micronutrients
To be most effective, any supplementation plan should include a broad-spectrum micronutrient combination to replenish depleted micronutrients as well as to prevent micronutrient imbalances, such as an excess in one micronutrient causing a deficiency in another. Depending on the symptoms and clinical tests, extra doses of B6, Zn2+, Mg2+, B1, and folate may be necessary to replenish the deficiencies created before the aldehyde toxicity was treated. It is particularly noteworthy that the chemical structure of B6 is an aldehyde and most susceptible to forming condensation reactions with other aldehydes, as reported for hyperprolinemia type II44 and other B6-dependent seizure disorders.24,46,47 Moreover, in the activated form of P5P, the negatively charged phosphate groups are likely to be neutralized by Mg2+ or Zn2+ and not by H+ at the normal pH of a cell to transport any P5P–aldehyde complex through a cell membrane and ultimately into the urine. Other aldehyde micronutrients include inositol and the retinal form of vitamin A. In some cases, supplementation with retinol and essential fatty acids, all contained in cod liver oil, may be useful.
Necessity of early treatment
Unfortunately, with very reactive aldehydes, such as those belonging to the groupings of α,β−unsaturated alkenals or the α-oxoaldehydes,2,3 adduct formation with proteins and DNA is not reversible, and the alterations, including spontaneous DNA mutations, become permanent. Unless these aldehydes are neutralized early, cellular damage ultimately reaches a threshold beyond which repair is not possible with a supplement plan, though the treatment of specific ASD symptoms, such as aggression, can still be alleviated by micronutrient supplementation.115,116,124 Thus, time is a critical factor in treating aldehyde toxicity—the earlier, the better. Ideally, treatment in those individuals prone to ASD should start during pregnancy. The typical prenatal vitamins contain C and E, but not sulfur-containing antioxidants. Although safety issues with regard to elevated levels of taurine143 and methionine144 as prenatal supplements have been raised, NAC appears to be safe, at least in cases of chorioamnionitis during pregnancy.145 When a mother begins breastfeeding, the child receives the first direct dose of sulfur-containing antioxidants in breast milk assuming, of course, that the mother does not have errant genes that would lower her antioxidant levels. Taurine, which is present in human but not cow’s milk, may be a contributing factor to the observations that children who have been breastfed for six months or more have a lower risk of developing ADHD and ASD.146 For those with the complex form of autism, retinoic acid supplementation during pregnancy might be better than retinol supplementation, but additional studies would need to identify definitive biomarkers to justify the use of retinoic acid as a supplement for a pregnant woman. If doses are too high, retinoic acid is teratogenic.
Administration of micronutrients
It would be inappropriate to suggest specific doses of micronutrients for ASD because the doses depend on body weight and individual metabolism. However, for a given age and body weight, the maximum recommended dose of a broad-spectrum commercial supplement, which includes a mix of vitamins and activating minerals, is likely safe. Additionally, sulfur-containing antioxidants, such as taurine and NAC, are superior to vitamin C and form the essential core of any supplementation plan to counteract aldehyde toxicity or oxidative stress. l-carnitine, but not acetyl-carnitine, is likely beneficial for many and can be included. These are water-soluble micronutrients, and there are, as yet, no upper limits with regard to safety. Nevertheless, as with any supplementation plan, prudence and careful monitoring of the individual for adverse symptoms is advised. Finally, the dietary reference intake for retinol, in the form of mercury-free cod liver oil that also includes essential fatty acids and vitamin D, should be included for most. Unless one is using supplements containing an extended-release compound, it is best to administer water-soluble micronutrients thrice a day to a child as 50% of water-soluble vitamins pass into the urine within four hours. Once a supplementation plan has been optimized, micronutrients can be compounded with extended-release formulations to reduce the treatment to twice daily. Caution about expectations with lower dietary reference intake doses is advised. It may take several months to observe a positive response with a low-dose supplementation regimen due to the up- and downregulation of genes as enzymes adjust to new micronutrient levels. With higher pharmacological doses of micronutrients, definitive improvement may be observed within days. However, it must be noted that very high micronutrient doses are absorbed by passive diffusion mechanisms, while the active diffusion mechanisms are inactivated by the downregulation of nutrient transporters.147 Thus, an individual must be weaned slowly from high micronutrient doses to allow enough time to upregulate the active transporter mechanisms again. Obviously, before any supplement plan is initiated, triggers of symptoms including infections, allergens, and environmental risk factors, should be identified, if possible, and eliminated.
Future directions
Bioaldehyde interactions with micronutrients/carbohydrates
The central hypothesis that some forms of ASD are a manifestation of aldehyde toxicity, of course, needs to be tested rigorously, even though there are ample data, already published and reviewed herein, to support the hypothesis. First and foremost, evidence for direct interactions between bioaldehydes and micronutrients/carbohydrates needs to be collected, in a manner similar to the research that established the formation of semialdehyde adducts with activated B6.24,44
Improved clinical assays
Better clinical tests need to be developed. Aldehydes are very reactive, and unless special care it taken, it is nearly impossible to detect their presence by any erythrocyte or urine assay. In SSADH deficiency (Fig. 4), for example, an excess of an organic acid, γ-hydroxybutyric acid (GHB), is detected, not succinic semialdehyde, the intermediate that accumulates as a result of a LoF mutation in ALDH5A1. Unfortunately, not all excess aldehydes convert to organic acids, the most common product considered in metabolic tests. In hyperprolinemia type II44 and other B6-dependent seizure disorders24,46,47 the intermediate aldehydes generated by LoF mutations in ALDH4A1 and ALDH7A1, respectively, form signature adducts with the activated form of B6. Other intermediate aldehydes might react with a plethora of large and small molecules, thus reducing the concentration of any single product, thereby increasing the difficulty of detection. Of cautionary note, several researchers have reported that the urinary aldehydes are unstable,148 likely prone to oxidation, so researchers may need to test very fresh urine or determine a method to stabilize the urine sample with a nonsulfur-containing antioxidant, such as ascorbic acid, as was done for the hydroxyhemopyrrolin-2-one analysis.149 The problems with clinical assays are not limited to aldehyde detection. As described, aldehydes react with a select set of micronutrients, inducing cell-localized deficiencies. Such deficiencies confined to the cells of the origination site of the aldehydes are unlikely to be detected in erythrocytes or urine assays. Fortunately, there are reasonable clinical tests to detect oxidative stress,139 but these tests do not determine the metabolic source of the oxidative stress. The central hypothesis, if correct, indicates the future direction needed to improve clinical assays.
Generalized treatment protocols
Should the accumulation of endogenous aldehydes prove to be a contributing source of ASD symptoms, then the most effective and safest treatment protocol needs to be ascertained. Until pharmacological therapeutics become available to target specific aldehydes, sulfur-containing antioxidants, such as NAC, taurine, GSH, and/or lipoic acid, are likely to form the core of any treatment protocol. Studies will need to be carried out to assess the most absorbable and effective aldehyde-neutralizing antioxidant(s), in either in vitro assays or in vivo animal models. In addition to one or more sulfur-containing antioxidants, broad-spectrum micronutrient supplementation, with an extra emphasis on nutrients rendered deficient by aldehyde toxicity, is likely to be required to rapidly halt the cascade of secondary metabolic disturbances. The most important micronutrient to emphasize is B6, given the proven reactivity with aldehydes, but Zn2+, Mg2+, B1, folate, essential fatty acids, and retinol in the form of cod liver oil should also be included. Theoretically, retinoic acid appears to be a better supplement than retinol to avoid any deleterious mutations in retinal-metabolizing enzymes, but studies would be needed to determine who should receive retinoic acid and how much, given detrimental effects at large doses.
Identification of pre- and postnatal markers
The current hypothesis, if proven correct, suggests possibilities to prevent autism, but additional studies will be needed to identify the earliest clinical symptoms of oxidative stress during pregnancy and after birth. Most important, rather than enlarging the ASD cohort for studies, it might be better to narrow the cohort group in order to correlate specific symptoms, such as head banging, with a cluster of early clinical manifestations. One such manifestation is mild-to-moderate neonatal jaundice, which is also associated with oxidative stress. Furthermore, prenatal supplements need to be examined more carefully. Most contain vitamins C and E, but not sulfur-containing antioxidants. Past studies suggest that prenatal supplementation with taurine143 or methionine144 might be harmful at elevated levels, but NAC appears to be safe and possibly beneficial for neuroprotection in cases of chorioamnionitis during pregnancy.145 Along with NAC, optimal levels of glutathione and lipoic acid need to be evaluated. Another early soft neurological symptom may be toe walking, which appears around 10–14 months. Lastly, if alcoholism is present in the first- and second-degree relatives, then extra precautions may need to be taken to prevent oxidative stress and aldehyde toxicity before, during, and after pregnancy.
Pairwise genetic analyses to identify genetic errors causing an accumulation of aldehydes
A generation or more will be needed to decipher the genomic studies for ASD in order to develop better targeted pharmacological interventions. To identify the sources of aldehyde accumulation, researchers can facilitate the process by conducting pairwise, not single gene, analyses of enzymes involved in the generation and clearance of aldehyde intermediates. Only pairwise analyses can determine whether an intermediate aldehyde is cleared quickly or accumulated. Moreover, such analyses will identify specific aldehydes involved in different subtypes of ASD so that pharmacological therapies can be better targeted to the aldehyde type and cellular location. In the interim, treating acute and chronic aldehyde toxicity with the available supplements describe herein may be the most effective approach to rescue some children and reduce the long-term social and financial costs of autism.
Summary
The many symptoms and divergent theories of ASD are consistent with aldehyde toxicity in which reactive aldehydes accumulate as a consequence of errors associated with genes intended to oxidize, reduce, or otherwise neutralize aldehydes. No matter the source(s) of the accumulated aldehydes, all share a reactive chemical group, which inflicts similar types of intracellular damage, but also suggests that a common treatment plan will be beneficial to many with ASD. Aldehydes induce localized micronutrient deficiencies initiating a cascade of metabolic disturbances in hundreds of metabolic pathways, cause oxidative stress, inactivate proteins by adduct formation, and bind to DNA, ultimately causing mutations and strand breakage. These effects of aldehyde toxicity explain all of the current genetic and biochemical theories of ASD in the medical literature. In addition to damage done by lipid peroxidation-generated aldehydes, already recognized by some experts in ASD,84–87 the central hypothesis expands to other types of endogenous and exogenous aldehydes. The aldehyde toxicity hypothesis has profound implications for early clinical detection and treatment of ASD as well as its prevention. Due to the high risk of permanent, irreversible cellular damage, time is of the essence for treating aldehyde toxicity. Treatment for aldehyde toxicity is similar to nutritional plans already advocated in the orthomolecular community76 but with a few additional features: (1) one or more sulfur-containing antioxidants are essential to neutralize reactive aldehydes and (2) a broad spectrum of micronutrients, with a special emphasis on B6, Zn2+, B1, Mg2+, folate, and retinol, should be included, irrespective of results from currently available, but inadequate, clinical testing. Such a treatment plan appears to be the best option for treating ASD now, until the genetics of ASD is better understood, and targeted therapies can be implemented. The hypothesis likely applies to many other neurological disorders, such as schizophrenia, with the major differences among disorders being the source, concentration, and reactivity of the accumulated aldehydes.
Acknowledgments
The author thanks Dr. Guiseppe de Vito, Ph.D., for relevant mathematical discussions pertaining to the number of mutation combinations of an enzyme pair needed to accumulate an intermediate. The author also thanks Professor John Jay Gargus, M.D., Ph.D., for critical comments.
Abbreviations
- ADH
Alcohol Dehydrogenase
- ADHD
Attention Deficit Hyperactivity Disorder
- AGEs
Advanced Glycation End-Products
- AKR
Aldo-Keto Reductase
- ALDH
Aldehyde Dehydrogenase
- ALEs
Advanced Lipid Peroxidation End-Products
- ASD
Autism Spectrum Disorder
- AUTS2
Autism Susceptibility Gene 2
- B1
Thiamine
- B6
Pyridoxine
- B12
Cobalamin
- CAT
Catalase
- CYP2E1
Cytochrome P450 2E1
- dG
Deoxyguanine
- DHA
Docosohexaenoic Acid
- dnCNV
de novo Copy Number Variants
- DRD2
Dopamine D2 Receptor
- DRI
Dietary Reference Intake
- DSCAM
Down Syndrome Cell Adhesion Molecule
- EPA
Eicosapentaenoic Acid
- FASD
Fetal Alcohol Syndrome Disorder
- GABA
γ-Aminobutyric Acid
- GHB
γ-Hydroxybutyric Acid
- GSH
Reduced Glutathione
- GST
Glutathione-S-Transferase
- GWAS
Genome-Wide Association Studies
- 4-HHE
4-Hydroxyhexenal
- 4-HNE
4-Hydroxynonenal
- H2O2
Hydrogen peroxide
- IP3R
Inositol 1,4,5-Triphosphate Receptor
- LoF
Loss of Function
- MDA
Malondialdehyde
- MeCP2
Methyl-CpG-Binding Protein
- Mg2+
Magnesium ion
- NAC
N-Acetyl Cysteine
- •O2−
Superoxide anion radical
- OCD
Obsessive-Compulsive Disorder
- OH
An alcohol group
- •OH
Hydroxyl radical
- P5P
Pyridoxyl-5-Phosphate, activated form of vitamin B6, also called PLP
- P5CDH
Pyrroline-5-Carboxylate Dehydrogenase
- PKNOX2
PBX/Knotted 1 Homeobox 2
- PUFA
Polyunsaturated Fatty Acid
- RARE
Retinoic Acid Response Element
- ROS
Reactive Oxygen Species
- SDR
Short-chain Dehydrogenase/Reductase
- SHANK3
SH3 and Multiple Repeat Domain 3
- SLC6A4
Solute Carrier 6 (serotonin transporter) Member 4
- SNP
Single Nucleotide Polymorphism
- SSADH
Succinic Semialdehyde Dehydrogenase
- TPP
Thiamine Pyrophosphate, activated from of vitamin B1
- XDH
Xanthine Dehydrogenase or Xanthine Oxidase
- Zn2+
Bioactive Zinc ion
Footnotes
ACADEMIC EDITOR: Joseph Zhou, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 598 words, excluding any confidential comments to the academic editor.
FUNDING: Author discloses no external funding sources.
COMPETING INTERESTS: Author discloses no potential conflicts of interest.
Paper subject to independent expert single-blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived the concepts: FJ. Analyzed the data: FJ. Wrote the first draft of the manuscript: FJ. Developed the structure and arguments for the paper: FJ. Made critical revisions: FJ. Author reviewed and approved of the final manuscript.
REFERENCES
- 1.Frye RE, Rossignol DA. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front Pediatr. 2014;2:66. doi: 10.3389/fped.2014.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LoPachin RM Gavin T. Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol. 2014;27(7):1081–1091. doi: 10.1021/tx5001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Brien P, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit Rev Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- 4.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283(32):21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pizzimenti S, Ciamporcero E, Daga M, et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front Physiol. 2013;00242:1–17. doi: 10.3389/fphys.2013.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner HW. Lipid hydroperoxide reactivity with proteins and amino acids: a review. J Agric Food Chem. 1979;27(2):220–229. [Google Scholar]
- 7.Voulgaridou G-P, Anestopoulos I, Franco R, Panayiotidis MI, Pappa A. DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat Res. 2011;711:13–27. doi: 10.1016/j.mrfmmm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153:6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5(1):194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juranek J, Ray R, Banach M, Rai V. Receptor for advanced glycation end-products in neurodegenerative diseases. Rev Neurosci. 2015;26(6):691–698. doi: 10.1515/revneuro-2015-0003. [DOI] [PubMed] [Google Scholar]
- 11.Kouidrat Y, Amad A, Arai M, et al. Advanced glycation end products and schizophrenia: a systematic review. J Psychiatr Res. 2015;6(6–67):112–117. doi: 10.1016/j.jpsychires.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 12.Barua M, Jenkins EC, Chen W, Kuizon S, Pullarkat RK, Junaid MA. Glyoxalase I polymorphism rs2736654 causing the Ala111Glu substitution modulates enzyme activity-implications for autism. Autism Res. 2011;4(4):262–270. doi: 10.1002/aur.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabriele S, Lombardi F, Sacco R, et al. The GLO1 C332 (Ala111) allele confers autism vulnerability; family-based genetic association and functional correlates. J Psychiatr Res. 2014;59:108–116. doi: 10.1016/j.jpsychires.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 14.Boleda MD, Saubi N, Farres J, Pares X. Physiological substrates for rat alcohol dehydrogenase classes: aldehydes of lipid peroxidation, ω-hydroxyfatty acids, and retinol. Arch Biochem Biophys. 1993;307:85–90. doi: 10.1006/abbi.1993.1564. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z-N, Davis GJ, Hurley TD, Stone CI, Li T-K, Bosron WF. Catalytic efficiency of human alcohol dehydrogenase for retinol oxidation and retinal reduction. Alcohol Clin Exp Res. 1994;18:587–591. doi: 10.1111/j.1530-0277.1994.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 16.Mårdh G, Dingley AL, Auld DS, Vallee BL. Human class II (π) alcohol dehydrogenase has a redox-specific function in norepinephrine metabolism. Proc Natl Acad Sci U S A. 1986;83:8908–8912. doi: 10.1073/pnas.83.23.8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Human Genome Organization (HUGO) Gene Nomenclature Committee Available at: http://www.genenames.org/genefamilies/ADH.
- 18.Vasiliou V, Bairoch A, Tipton KF, Nebert DW. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics. 1999;9:421–434. [PubMed] [Google Scholar]
- 19.Edenberg HJ, Xuei X, Chen H-J, et al. Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Hum Mol Genet. 2006;15(9):1539–1549. doi: 10.1093/hmg/ddl073. [DOI] [PubMed] [Google Scholar]
- 20.Jin S, Chen J, Chen L, et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesisProc Natl Acad Sci U S A 2015112299088–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann N Y Acad Sci. 2004;1011:36–44. doi: 10.1007/978-3-662-41088-2_4. [DOI] [PubMed] [Google Scholar]
- 22.Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chem Biol Interact. 2013;202:2–10. doi: 10.1016/j.cbi.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2(2):138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12(3):307–309. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 25.Kivuva EC, Parker MJ, Cohen MC, Wagner BE, Sobey G. De Barsy syndrome: a review of the phenotype. Clin Dysmorphol. 2008;17:99–107. doi: 10.1097/MCD.0b013e3282f4a964. [DOI] [PubMed] [Google Scholar]
- 26.Sass JO, Walter M, Shield JPH, et al. 3-hydroxyisobutyrate aciduria and mutations in the ALDH6A1 gene coding for methylmalonate semialdehyde dehydrogenase. J Inherit Metab Dis. 2012;35:437–442. doi: 10.1007/s10545-011-9381-x. [DOI] [PubMed] [Google Scholar]
- 27.Moreno-Ramos OA, Olivares AM, Haider NB, de Autismo LC, Lattig MC. Whole-exome sequencing in a South American cohort links ALDH1A3, FOXN1 and retinoic acid regulation pathways to autism spectrum disorders. PLoS One. 2015;10(9):e0135927. doi: 10.1371/journal.pone.0135927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A. Gut microbiota in autism and mood disorders. World J Gastroenterol. 2016;22(1):361–368. doi: 10.3748/wjg.v22.i1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell AK, Matthews SB, Vassel N, et al. Bacterial metabolic “toxins”: a new mechanism for lactose and food intolerance, and irritable bowel syndrome. Toxicology. 2010;278:268–276. doi: 10.1016/j.tox.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Kantarcioglu AS, Kiraz N, Avdin A. Microbiota-gut-brain axis: yeast species isolated from stool samples of children with suspected or diagnosed autism spectrum disorders and in vitro susceptibility against nystatin and fluconazole. Mycopathologia. 2016;181(1–2):1–7. doi: 10.1007/s11046-015-9949-3. [DOI] [PubMed] [Google Scholar]
- 32.Gainza-Cirauqui ML, Nieminen MT, Novak Frazer L, Aguirre-Urizar JM, Moragues MD, Rautemaa R. Production of carcinogenic acetaldehyde by Candida albicans from patients with potentially malignant oral mucosal disorders. J Oral Pathol Med. 2013;42(3):243–249. doi: 10.1111/j.1600-0714.2012.01203.x. [DOI] [PubMed] [Google Scholar]
- 33.Qiao Y, Sun J, Ding Y, Le G, Shi Y. Alterations of the gut microbiota in high-fat diet mice is strongly linked to oxidative stress. Appl Microbiol Biotechnol. 2013;97(4):1689–1697. doi: 10.1007/s00253-012-4323-6. [DOI] [PubMed] [Google Scholar]
- 34.Environmental Protection Agency Integrated Risk Information System for Acetaldehyde. Available at: http://cfpub.epa.gov/ncea/iris2/chemicalLanding.cfm?substance_nmbr=290.
- 35.Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29(4):245–254. [PMC free article] [PubMed] [Google Scholar]
- 36.Young JK, Giesbrecht HE, Eskin MN, Aliani M, Suh M. Nutrition implications for fetal alcohol spectrum disorder. Adv Nutr. 2014;5:675–692. doi: 10.3945/an.113.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16(4):667–685. doi: 10.1016/j.cld.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin PR, Singleton CK, Hiller-Sturmhöfel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27(2):134–142. [PMC free article] [PubMed] [Google Scholar]
- 39.Lumeng L, Li TK. Pyridoxal phosphate levels in plasma and the effects of acet-aldehyde on pyridoxal phosphate synthesis and degradation in human erythrocytes. J Clin Invest. 1974;53:693–704. doi: 10.1172/JCI107607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mieyal JJ, Bantle G, Votaw RG, Rosner IA, Sable HZ. Coenzyme interactions: V. The second carbanion in reactions catalyzed by thiamine. J Biol Chem. 1971;246(17):5213–5219. [PubMed] [Google Scholar]
- 41.Anni H, Pristatsky P, Israel Y. Binding of acetaldehyde to a glutathione metabolite: mass spectrometric characterization of an acetaldehyde-cysteinylglycine conjugate. Alcohol Clin Exp Res. 2003;10:1613–1621. doi: 10.1097/01.ALC.0000089958.65095.84. [DOI] [PubMed] [Google Scholar]
- 42.Ogasawara M, Nakamura T, Koyama I, Nemoto M, Yoshida T. Reactivity of taurine with aldehydes and its physiological role. Adv Exp Med Biol. 1994;359:71–78. doi: 10.1007/978-1-4899-1471-2_8. [DOI] [PubMed] [Google Scholar]
- 43.Shaw S, Jayatilleke E, Herbert V, Colman N. Cleavage of folates during ethanol metabolism. Biochem J. 1989;257:277–280. doi: 10.1042/bj2570277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farrant RD, Walker V, Mills GA, Mellor JM, Langley GJ. Pyridoxal phosphate de-activation by pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J Biol Chem. 2001;276(18):15107–15116. doi: 10.1074/jbc.M010860200. [DOI] [PubMed] [Google Scholar]
- 45.Geraghty MT, Vaughn D, Nicholson AJ, et al. Mutations in the delta-1-pyrroline 5-carboxylase dehydrogenase gene cause type II hyperprolinemia. Hum Mol Genet. 1998;7:1411–1415. doi: 10.1093/hmg/7.9.1411. [DOI] [PubMed] [Google Scholar]
- 46.Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency) Brain. 2010;133:2148–2159. doi: 10.1093/brain/awq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baumgart A, von Spiczak S, Verhoeven-Duif NM, et al. Atypical vitamin B6 deficiency: a rare cause of unexplained neonatal and infantile epilepsies. J Child Neurol. 2014;29(5):704–707. doi: 10.1177/0883073813505354. [DOI] [PubMed] [Google Scholar]
- 48.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 49.Urvalek A, Laursen KB, Gudas LJ. The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. Subcell Biochem. 2014;70:129–149. doi: 10.1007/978-94-017-9050-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deltour L, Ang HL, Duester G. Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J. 1996;10:1050–1057. [PubMed] [Google Scholar]
- 51.Marrs JA, Clendonen SG, Ratcliffe DR, Fielding SM, Liu Q, Bosron WF. Zebrafish fetal alcohol syndrome model: effects of ethanol are rescued by retinoic acid supplement. Alcohol. 2010;44:707–715. doi: 10.1016/j.alcohol.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miles JH. Autism spectrum disorders—a genetics review. Genet Med. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 53.Duignan E, Kenna P, Watson R, Fitzsimon S, Brosnahan D. Ophthalmic manifestations of vitamin A and D deficiency in two autistic teenagers: case reports and a review of the literature. Case Rep Ophthalmol. 2015;6(1):24–29. doi: 10.1159/000373921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Megson MN. Is autism a G-alpha protein defect reversible with natural vitamin A? Med Hypotheses. 2000;54(6):979–983. doi: 10.1054/mehy.1999.0999. [DOI] [PubMed] [Google Scholar]
- 55.Eriksson CJ. The role of acetaldehyde in the actions of alcohol. Alcohol Clin Exp Res. 2001;25(5 suppl ISBRA):15S–32S. doi: 10.1097/00000374-200105051-00005. [DOI] [PubMed] [Google Scholar]
- 56.Niemelä O. Acetaldehyde adducts in circulation. Novartis Found Symp. 2007;286:183–192. doi: 10.1002/9780470511848.ch13. [DOI] [PubMed] [Google Scholar]
- 57.Zeng J, Davies MJ. Evidence for the formation of adducts and S-(carboxymethyl) cysteine on reaction of α-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem Res Toxicol. 2005;18:1232–1241. doi: 10.1021/tx050074u. [DOI] [PubMed] [Google Scholar]
- 58.Tuma DJ, Casey CA. Dangerous byproducts of alcohol breakdown-focus on adducts. Alcohol Res Health. 2003;27(4):285–290. [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Q, Maret W. Aldehydes release zinc from proteins. A pathway from oxidative stress/lipid peroxidation to cellular functions of zinc. FEBS J. 2006;273:4300–4310. doi: 10.1111/j.1742-4658.2006.05428.x. [DOI] [PubMed] [Google Scholar]
- 60.Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol. 2005;166(6):1681–1690. doi: 10.1016/S0002-9440(10)62478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aldeyra Therapeutics clinical trial information. Available at: http://www.aldeyra.com/development-status/
- 62.Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29(2):212–216. doi: 10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- 63.Saronwala A, Tournay A, Gargus JJ. Genetic inborn error of metabolism provides a unique window into molecular mechanisms underlying taurine therapy. In: Huxtable RJ, Michalk D, editors. Taurine in Health and Disease. New York, NY: Springer Science & Business Media; 2012. pp. 357–369. [Google Scholar]
- 64.Oliveira MWS, Minotto JB, de Oliveira MR, et al. Scavenging and antioxidant potential of physiological taurine concentrations against different reactive oxygen/nitrogen species. Pharmacol Rep. 2010;62:185–193. doi: 10.1016/s1734-1140(10)70256-5. [DOI] [PubMed] [Google Scholar]
- 65.Deepmala D, Slattery J, Kumar N, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. 2015;55:294–321. doi: 10.1016/j.neubiorev.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 66.Shahripour RB, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4(2):108–122. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167–177. doi: 10.1016/j.tips.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 68.Koop DR. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992;6(2):724–730. doi: 10.1096/fasebj.6.2.1537462. [DOI] [PubMed] [Google Scholar]
- 69.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, super-oxide, and hypochlorous acid. Free Radic Biol Med. 1989;6(6):593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 70.Sprince H, Parker CM, Smith GG, Gonzales LJ. Protective action of ascorbic and sulfur compounds against aldehyde toxicity: implications in alcoholism and smoking. Agents Actions. 1975;5(2):164–173. doi: 10.1007/BF02027359. [DOI] [PubMed] [Google Scholar]
- 71.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11(1):81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 72.Abdul HM, Butterfield DA. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by co-treatment of acetyl- l-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Free Radic Biol Med. 2007;42(3):371–384. doi: 10.1016/j.freeradbiomed.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM-4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 74.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013. [Google Scholar]
- 75.Herbert MR. Autism: a brain disorder, or a disorder that affects the brain? Clin Neuropsychol. 2005;2:354–379. [Google Scholar]
- 76.Walsh W. Nutrient Power: Heal Your Biochemistry and Heal Your Brain. New York, NY: Skyhorse Publishing; 2012. pp. 91–112. [Google Scholar]
- 77.Frye RE, Rose S, Slattery J, MacFabe DF. Gastrointestinal dysfunction in autism spectrum disorder; the role of the mitochondria and the enteric microbiome. Microb Ecol Health Dis. 2015;26:27458. doi: 10.3402/mehd.v26.27458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry. 2014;4:1–23. doi: 10.1038/tp.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 80.Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders; immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17:389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007;17(4):434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics. 2007;38(6):276–281. doi: 10.1055/s-2008-1065354. [DOI] [PubMed] [Google Scholar]
- 83.González-Fraguela ME, Diaz Hung M-L, Vera H, et al. Oxidative stress markers in children with autism spectrum disorders. Br J Med Med Res. 2013;3(2):307–317. [Google Scholar]
- 84.Damodaran LPM, Arumugam G. Urinary oxidative stress markers in children with autism. Redox Rep. 2011;16(5):216–222. doi: 10.1179/1351000211Y.0000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meguid NA, Dardir AA, Abdel-Raouf ER, Hashish A. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biol Trace Elem Res. 2011;143:58–65. doi: 10.1007/s12011-010-8840-9. [DOI] [PubMed] [Google Scholar]
- 86.Chauhan A, Chauhan V, Brown WT, Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin-the antioxidant proteins. Life Sci. 2004;75(21):2539–2549. doi: 10.1016/j.lfs.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 87.Signorini C, DeFelice C, Durand T, et al. Isoprostanes and 4-hydroxy-2-nonenal: markers or mediators of disease? Focus on Rett syndrome as a model of autism spectrum disorder. Oxid Med Cell Longev. 2013;2013:343824. doi: 10.1155/2013/343824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roos L, Fang M, Dali C, et al. A homozygous mutation in a consanguineous family consolidates the role of ALDH1A3 in autosomal recessive microphthalmia. Clin Genet. 2014;86:276–281. doi: 10.1111/cge.12277. [DOI] [PubMed] [Google Scholar]
- 89.Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. J Am Med Assoc. 2014;311(7):1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robinson EB, Neale BM, Hyman SE. Genetic research in autism spectrum disorders. Curr Opin Pediatr. 2015;27(6):685–691. doi: 10.1097/MOP.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gaugler T, Klei L, Sanders SJ, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Warrier V, Chee V, Smith P, Chakrabarti B, Baron-Cohen S. A comprehensive meta-analysis of common genetic variants in autism spectrum conditions. Mol Autism. 2015;6:49. doi: 10.1186/s13229-015-0041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Roak BJ, Stessman HA, Boyle EA, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iossifov I, Levy D, Allen J, et al. Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci U S A. 2015;112(41):E5600–E5607. doi: 10.1073/pnas.1516376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanders SJ, He X, Willsey AJ, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70(5):898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DeLong GR, Dwyer JT. Correlation of family history with specific autistic subgroups: Asperger’s syndrome and bipolar affective disease. J Autism Dev Disord. 1988;18(4):593–600. doi: 10.1007/BF02211877. [DOI] [PubMed] [Google Scholar]
- 101.Smalley SL, McCracken J, Tanguay P. Autism, affective disorders, and social phobia. Am J Med Genet. 1995;60(1):19–26. doi: 10.1002/ajmg.1320600105. [DOI] [PubMed] [Google Scholar]
- 102.Miles JH, Takahashi TN, Haber A, Hadden L. Autism families with a high incidence of alcoholism. J Autism Dev Disord. 2003;33(4):403–415. doi: 10.1023/a:1025010828304. [DOI] [PubMed] [Google Scholar]
- 103.Sundquist J, Sundquist K, Jianguang J. Autism and attention-deficit/hyperactivity disorder among individuals with a family history of alcohol use disorders. eLife. 2014;3:e02917. doi: 10.7554/eLife.02917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuo L, Wang K, Zhang X-Y, et al. Association between common alcohol dehydrogenase gene (ADH) variants and schizophrenia and autism. Hum Genet. 2013;132(7):735–743. doi: 10.1007/s00439-013-1277-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kanduri C, Kantojärvi K, Salo PM, et al. The landscape of copy number variations in Finnish families with autism spectrum disorders. Autism Res. 2016;9(1):9–16. doi: 10.1002/aur.1502. [DOI] [PubMed] [Google Scholar]
- 106.Schumann G, Coin LJ, Lourdusamy A, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci U S A. 2011;108(17):7119–7124. doi: 10.1073/pnas.1017288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarachana T, Hu VW. Genome-wide-identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. 2013;4(1):14. doi: 10.1186/2040-2392-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Riebold M, Mankuta D, Lerer E, et al. All-trans retinoic acid upregulates reduced CD38 transcription in lymphoblastoid cell lines from autism spectrum disorder. Mol Med. 2011;17(7–8):799–806. doi: 10.2119/molmed.2011.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Diss JK, Calissano M, Gascoyne D, Djamgoz MB, Latchman DS. Identification and characterization of the promoter region of the Nav1.7 voltage-gated sodium channel gene (SCN9A) Mol Cell Neurosci. 2008;37(3):537–547. doi: 10.1016/j.mcn.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 110.El Andaloussi-Lilja J, Lundqvist J, Forsby A. TRPV1 expression and activity during retinoic acid-induced neuronal differentiation. Neurochem Int. 2009;55(8):768–774. doi: 10.1016/j.neuint.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 111.Rhinn M, Dollé P. Retinoic acid signaling during development. Development. 2012;139:843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- 112.Tanoury ZA, Piskunov A, Andriamoratsiresy D, et al. Genes involved in cell adhesion and signaling: a new repertoire of retinoic acid receptor target genes in mouse embryonic fibroblasts. J Cell Sci. 2014;127:521–533. doi: 10.1242/jcs.131946. [DOI] [PubMed] [Google Scholar]
- 113.Anitha A, Thanseem I, Nakamura K, et al. Zinc finger protein 804A (ZNF804A) and verbal deficits in individuals with autism. J Psychiatry Neurosci. 2014;39(5):294–303. doi: 10.1503/jpn.130126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ji N, Findling RL. An update on pharmacotherapy for autism spectrum disorder in children and adolescents. Curr Opin Psychiatry. 2015;28(2):91–101. doi: 10.1097/YCO.0000000000000132. [DOI] [PubMed] [Google Scholar]
- 115.Ghanizadeh A, Moghimi-Sarani E. A randomized double blind placebo controlled clinical trial of N-acetylcysteine added to risperidone for treating autistic disorders. BMC Psychiatry. 2013;13:196–202. doi: 10.1186/1471-244X-13-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nikoo M, Radnia H, Farokhnia M, Mohammadi M-R, Akhondzadeh S. N-acetylcysteine as an adjunctive therapy to risperidone for treatment of irritability in autism: a randomized, double-blind, placebo-controlled clinical trial of efficacy and safety. Clin Neuropharmacol. 2015;38(1):11–17. doi: 10.1097/WNF.0000000000000063. [DOI] [PubMed] [Google Scholar]
- 117.Reichow B, Barton EE, Boyd BA, Hume K. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD) Cochrane Database Syst Rev. 2012;10:CD009260. doi: 10.1002/14651858.CD009260.pub2. [DOI] [PubMed] [Google Scholar]
- 118.Geraghty ME, Depasquale GM, Lane AE. Nutritional intake and therapies in autism. Infant Child Adolesc Nutr. 2010;2(1):62–69. [Google Scholar]
- 119.Frustaci A, Neri M, Cesario A, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med. 2012;52:2128–2141. doi: 10.1016/j.freeradbiomed.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 120.Filipek PA, Juranek J, Nguyen MT, Cummings C, Gargus JJ. Relative carnitine deficiency in autism. J Autism Dev Disord. 2004;34(6):615–623. doi: 10.1007/s10803-004-5283-1. [DOI] [PubMed] [Google Scholar]
- 121.Adams JB, Audhya T, McDonough-Means S, et al. Nutritional and metabolic status of children with autism vs neurotypical children, and the association with autism severity. Nutr Metab. 2011;8:1–34. doi: 10.1186/1743-7075-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vancassel S, Durand G, Barthélémy C, et al. Plasma fatty acid levels in autistic children. Prostaglandins Leukot Essent Fatty Acids. 2001;65(1):1–7. doi: 10.1054/plef.2001.0281. [DOI] [PubMed] [Google Scholar]
- 123.Gvozdjáková A, Kucharská J, Ostatníková D, Babinská K, Nakládal D, Crane FL. Ubiquinol improves symptoms in children with autism. Oxid Med Cell Longev. 2014;2014:798957. doi: 10.1155/2014/798957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hardan AY, Fung LK, Libove RA, et al. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol Psychiatry. 2012;71:956–961. doi: 10.1016/j.biopsych.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McGinnis WR. Oxidative stress in autism. Altern Ther Health Med. 2004;10(6):22–36. [PubMed] [Google Scholar]
- 126.Obrenovich ME, Shamberger RJ, Lonsdale D. Altered heavy metals and transketolase found in autistic spectrum disorder. Biol Trace Elem Res. 2011;144:475–486. doi: 10.1007/s12011-011-9146-2. [DOI] [PubMed] [Google Scholar]
- 127.Murza KA, Pavelko SL, Malani MD, Nye C. Vitamin B6-magnesium treatment for autism: the current status of the research. Magnes Res. 2010;23(2):115–117. doi: 10.1684/mrh.2010.0209. [DOI] [PubMed] [Google Scholar]
- 128.Mehl-Madrona L, Leung B, Kennedy C, Paul S, Kaplan BJ. Micronutrients versus standard medication management in autism: a naturalistic case-control study. J Child Adolesc Psychopharmacol. 2010;20(2):95–103. doi: 10.1089/cap.2009.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Raskin NH. Alcoholism or acetaldehydism? N Engl J Med. 1975;292(8):422–423. doi: 10.1056/NEJM197502202920811. [DOI] [PubMed] [Google Scholar]
- 130.Edenberg HJ, Foroud T. Genetics and alcoholism. Nat Rev Gastroenterol Hepatol. 2013;10:487–494. doi: 10.1038/nrgastro.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Edenberg HJ. The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res Health. 2007;30(1):5–13. [PMC free article] [PubMed] [Google Scholar]
- 132.Janowska B, Komisarski M, Prorok P, et al. Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli. Int J Biol Sci. 2009;5(6):611–620. doi: 10.7150/ijbs.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rossignol DA, Bradstreet JJ. Evidence of mitochondrial dysfunction in autism and implications for treatment. Am J Biochem Biotechnol. 2008;4(2):208–217. [Google Scholar]
- 134.Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate. Mayo Clin Proc. 2013;88(10):1127–1140. doi: 10.1016/j.mayocp.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kloubert V, Rink L. Zinc as a micronutrient and its preventive role of oxidative damage in cells. Food Funct. 2015;6:3195–3204. doi: 10.1039/c5fo00630a. [DOI] [PubMed] [Google Scholar]
- 136.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 137.Ho E, Galougahi KK, Liu C-C, Bhindi R, Figtree GA. Biological markers of oxidative stress: applications to cardiovascular research and practice. Redox Biol. 2013;1:483–491. doi: 10.1016/j.redox.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jangaard KA, Fell DB, Dodds L, Allen AC. Outcomes in a population of healthy term and near-term infants with serum bilirubin levels of ≥325 μmol/L (≥19 mg/dL) who were born in Nova Scotia, Canada, between 1994 and 2000. Pediatrics. 2008;122(1):119–124. doi: 10.1542/peds.2007-0967. [DOI] [PubMed] [Google Scholar]
- 139.Ayyappan S, Philip S, Bharathy N, et al. Antioxidant status in neonatal jaundice before and after phototherapy. J Pharm Bioallied Sci. 2015;7(suppl 1):S16–S21. doi: 10.4103/0975-7406.155766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Davutoglu M, Guler E, Olgar S, et al. Oxidative stress and antioxidant status in neonatal hyperbilirubinemia. Saudi Med J. 2008;29(12):1743–1748. [PubMed] [Google Scholar]
- 141.Gitto E, Pellegrino S, Gitto P, Barberi I, Reiter RJ. Oxidative stress of the newborn in the pre- and postnatal period and the clinical utility of melatonin. J Pineal Res. 2009;46:128–139. doi: 10.1111/j.1600-079X.2008.00649.x. [DOI] [PubMed] [Google Scholar]
- 142.Maimburg RD, Bech BH, Væth M, Møller-Madsen B, Olson J. Neonatal jaundice, autism, and other disorders of psychological development. Pediatrics. 2010;126(5):872–878. doi: 10.1542/peds.2010-0052. [DOI] [PubMed] [Google Scholar]
- 143.Aerts L, Van Assche FA. Taurine and taurine-deficiency in the perinatal period. J Perinat Med. 2002;30(4):281–286. doi: 10.1515/JPM.2002.040. [DOI] [PubMed] [Google Scholar]
- 144.Rees WD, Wilson FA, Maloney CA. Sulfur amino acid metabolism in pregnancy: the impact of methionine in the maternal diet. J Nutr. 2006;136(6 suppl):1701S–1705S. doi: 10.1093/jn/136.6.1701S. [DOI] [PubMed] [Google Scholar]
- 145.Jenkins DD, Wiest DB, Mulvihill DM, et al. Fetal and neonatal effects of N-acetylcysteine when used for neuroprotection in maternal chorioamnionitis. J Pediatr. 2016;168:67–76. doi: 10.1016/j.jpeds.2015.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lawrence RA. The risks of not breastfeeding: new associations. Breastfeeding Medicine. 2014;9(5):237–236. doi: 10.1089/bfm.2014.9985. [DOI] [PubMed] [Google Scholar]
- 147.Said HM. Intestinal absorption of water-soluble vitamins in health and diseases. Biochem J. 2011;437(3):357–372. doi: 10.1042/BJ20110326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marcadier JL, Smith AM, Pohl D, et al. Mutations in ALDH6A1 encoding methylmalonate semialdehyde dehydrogenase are associated with dysmyelination and transient methylmalonic aciduria. Orphanet J Rare Dis. 2013;8:98. doi: 10.1186/1750-1172-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McGinnis WR, Audhya T, Walsh WJ, et al. Discerning the mauve factor, part I. Altern Ther Health Med. 2008;14(2):40–50. [PubMed] [Google Scholar]
- 150.Zuo L, Lu L, Tan Y, et al. Genome-wide association discoveries of alcohol dependence. Am J Addict. 2014;23(6):526–539. doi: 10.1111/j.1521-0391.2014.12147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schmunk G, Boubion BJ, Smith IF, Parker I, Gargus JJ. Shared functional defect in IP3R-mediated calcium signaling in diverse monogenic autism syndromes. Transl Psychiatry. 2015;5:e643. doi: 10.1038/tp.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang M, Tsuang J, Wan YJ. A halotype analysis of CYP2E1 polymorphisms in relation to alcoholic phenotypes in Mexican Americans. Alcohol Clin Exp Res. 2007;31(12):1991–2000. doi: 10.1111/j.1530-0277.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- 153.Kang TS, Woo SW, Park HJ, Lee Y, Roh J. Comparison of genetic polymorphisms of CYP2E1, ADH2, and ALDH2 genes involved in alcohol metabolism in Koreans and four other ethnic groups. J Clin Pharm Ther. 2009;34(2):225–230. doi: 10.1111/j.1365-2710.2008.00986.x. [DOI] [PubMed] [Google Scholar]
- 154.OMIM Available at: http://www.ncbi.nlm.nih.gov/omim.