

ABSTRACT
Cyclooxygenases (COX), commonly upregulated in numerous cancers, generate prostaglandin E2 (PGE2), which has been implicated in key aspects of malignant growth including proliferation, invasion and angiogenesis. Recently, we showed that production of PGE2 by cancer cells dominantly enables progressive tumor growth via immune escape and that cyclooxygenase inhibitors synergize with immunotherapy to enhance tumor eradication.
KEYWORDS: Cancer inflammation, cancer immunity, cancer immune evasion, COX-2, dendritic cells, immunotherapy
The remarkable recent success of certain type of immunotherapies such as those based on immune checkpoint blockade1 is testament to the notion that the immune system can act as a barrier to a wide range of malignancies. This has led, in turn, to a renewed interest in understanding the basis for cancer immune evasion. Identifying the mechanism/s underlying how cancer cells avoid immune control is fundamental for cancer treatment, especially as a marked fraction of patients fails to respond to immune-based therapies. In our recent study, “Cyclooxygenase-dependent tumor growth through evasion of immunity,”2 we demonstrate that COX-driven production of PGE2 by cancer cells enables immune escape and progressive tumor growth and show that this has therapeutic implications for cancer treatment.
Clinically apparent cancers are able to grow aggressively and invasively despite the presence of a seemingly functional immune system. The immunoediting theory posits that this characteristic is not due to an intrinsic inability of the immune system to detect oncogenically transformed cells but the result of a Darwinian process whereby the immune system selects for less immunogenic cancer cells.3 The immunogenic potential of tumor cells, as for any other entity, depends on antigen and adjuvant determinants.4 Low immunogenicity due to reduced antigenicity as a consequence of lack or loss of tumor antigens is a well-established mechanism of immunoediting.5-7 Adjuvanticity, that is, the ability of an antigenic stimulus to induce an immune response, has been less well studied in terms of cancer immunoediting. Adjuvant activity correlates with the ability to trigger innate immunity and induce activation of dendritic cells (DCs). Therefore, cancer cell traits that suppress or subvert DC activation are likely to contribute to decreased cancer immunogenicity and might constitute attractive therapeutic targets.
With this in mind, we set out to investigate the immunogenic properties of a cell line established from an autochthonous melanoma mouse model driven by the most common recurring mutation found in melanoma patients, a glutamic acid substitution for valine at position 600 of the B-RAF kinase (BrafV600E).8 We speculated that cancer cells isolated from a genetically engineered cancer-prone mouse with an intact immune system must have escaped immune surveillance and possess attributes that allowed them to avoid immune-mediated destruction. In agreement with our hypothesis, BrafV600E cells taken from cancer-bearing mice formed equally progressive tumors in T/B cell-sufficient or -deficient syngeneic mice underscoring their poor immunogenicity.
We went on to find that factors produced or released by BrafV600E cells had very potent immunomodulatory potential on mixed cultures of DCs and macrophages, which included the ability to directly induce the production of various tumor-promoting cytokines and chemokines such as IL-6, G-CSF or CXCL1. These effects could be attributed to PGE2 secretion by the melanoma cells following its synthesis through a canonical pathway dependent on the activity of COX and prostaglandin E synthases (PGES). Further in vitro and in vivo studies revealed that cancer cell-derived PGE2 was critical for the progressive growth of tumors formed by BrafV600E cells but also NrasG12D-driven melanoma, colorectal and breast cancer cell lines. Abrogation of the ability of cancer cells to produce PGE2 by means of genetic ablation of COX or PGES using CRISPR-Cas9 technology invariably and markedly impaired their potential for sustained growth in immunocompetent mice (Fig. 1). However, the COX- or PGES-deficient cells were able to grow in immunodeficient hosts indistinguishably from parental cells. This suggests that PGE2 production by tumor cells is primarily aimed at escaping adaptive immunity, which is unexpected given that it has been thought to be necessary for tumor cell proliferation, survival and angiogenesis.9 Finally, mice that rejected COX-deficient cancer cells were largely resistant to challenge with parental tumors indicating that PGE2 suppresses immunity rather than creating immune privilege.
Figure 1.
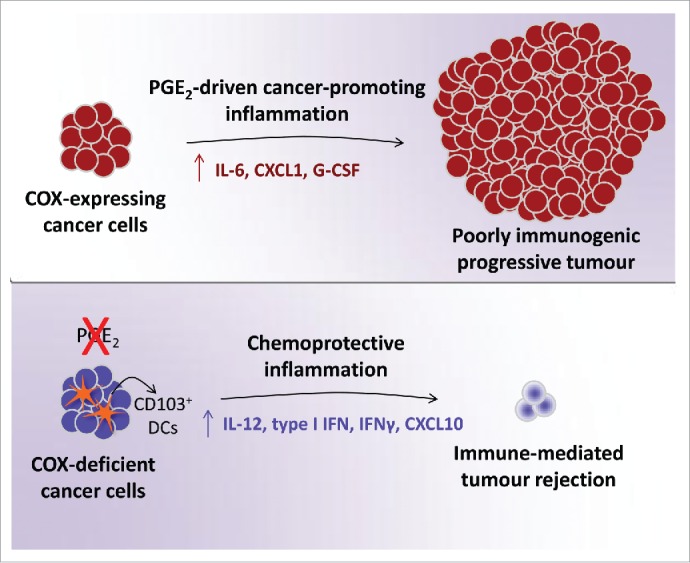
COX-driven PGE2 production by cancer cells fuels tumor-promoting inflammation and allows progressive tumor growth. Cancer cell-specific COX-deficiency alters the inflammatory profile at the tumor site increasing antitumor mediators and enabling immune-dependent tumor rejection.
COX deficiency in cancer cells was associated with a marked shift in the inflammatory signature at the tumor site characterized by lower expression of cancer-promoting factors and concomitant increase in several mediators typically associated with antitumor immunity. The latter included increased accumulation of IL-12-producing Batf3-dependent CD103+ DCs providing further evidence that PGE2 suppresses anticancer immunity (Fig. 1). Thus, a COX signature in tumor biopsies might constitute a biomarker of lessened immune control, which could perhaps be utilized to predict unresponsiveness to immunotherapy.
The drastic effects of genetically ablating PGE2 production by tumors prompted us to assess whether pharmacological intervention might achieve similar results. Given the genetic evidence indicating the necessity of completely blocking PGE2 synthesis before establishment of immune control could be observed, we did not expect COX inhibitors to be effective by themselves. However, even incomplete COX inhibition by drugs might be sufficient to potentiate immune-dependent control. Indeed, we found that giving mice COX-inhibitors, such as aspirin or celecoxib, together with anti-PD-1 blocking antibody synergistically promoted adaptive immune-mediated control of COX-competent cancers. These preclinical data suggest that COX-inhibitors could be useful to enhance the efficacy of anticancer immunotherapy. This prediction will now need to be tested in the context of a clinical trial and adds to the interest in assessing the efficacy of aspirin as a cancer chemopreventive agent.10
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015; 161:205-14; PMID:25860605; http://dx.doi.org/ 10.1016/j.cell.2015.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA et al.. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015; 162(6):1257-70; PMID:26343581; http://dx.doi.org/ 10.1016/j.cell.2015.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; http://dx.doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 4.Zelenay S, Reis e Sousa C. Adaptive immunity after cell death. Trends Immunol 2013; 34:329-35; PMID:23608152; http://dx.doi.org/ 10.1016/j.it.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 5.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y-S, Shea LK et al.. Cancer exome analysis reveals a T-cell-dependent mechanism of csancer immunoediting. Nature 2012; 482:400-4; PMID:22318521; http://dx.doi.org/25594174 10.1038/nature10755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012; 482:405-9; PMID:22318517; http://dx.doi.org/25594174 10.1038/nature10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160:48-61; PMID:25594174; http://dx.doi.org/ 10.1016/j.cell.2014.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell 2009; 15:294-303; PMID:19345328; http://dx.doi.org/ 10.1016/j.ccr.2009.02.022 [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Dubois RN. Eicosanoids and cancer. Nature Rev Cancer 2010; 10:181-93; PMID:20168319; http://dx.doi.org/ 10.1038/nrc2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. http://www.cancerresearchuk.org/about-us/cancer-news/press-release/2015-10-22-worlds-largest-clinical-trial-on-aspirin-to-stop-cancer-returning-launches-today [Google Scholar]