

ABSTRACT
Mice transgenic for the RET oncogene provide a remarkable model for investigating the mechanisms underlying the promotion and the development of melanoma. This model was established on the C57BL/6 genetic background. In the present study, we investigated an effect of the strongly proinflammatory and autoimmune genetic makeup of the non-obese diabetic (NOD) strain. We bred (NODxB6)F1 mice and backcrossed them with NOD mice. F1 mice and mice at subsequent generations of backcrossing showed marked acceleration of tumor development, in particular with a more frequent and earlier extension of the primary uveal melanoma. In close relation with this severe evolution, we observed a profound drop in Dectin-1 expression on CD11b+Ly6G+ granulocytic myeloid cells correlating with an expansion of CD4+Foxp3+ T regulatory cell and of interferon(IFN)γ-producing CD8+ T cell subsets in tumors. IFNγ is a major inducer of the type 2 nitric-oxide synthase (Nos2) gene whose products are known to be tumorigenic. Germline inactivation of the Nos2 gene was associated with a dramatically improved tumor prognosis and a restoration of Dectin-1 expression on myeloid cells. Moreover, in vivo treatment of (NODxB6)F1.RET+ mice with curdlan, a glucose polymer that binds Dectin-1, prevented tumor extension and was associated with marked reduction of the CD4+Foxp3+ T cell subset. These observations highlight the (NODxB6)F1.RET+ mice as a new model to investigate the role of the immune system in the host–tumor relationship and point to Dectin-1 and Nos2 as potentially promising therapeutic targets.
KEYWORDS: Dectin-1, myeloid cells, melanoma, NOD mouse, Nos2, RET
Introduction
The immune system and inflammation play a key role in the relationships between a tumor and its host.1 Recent studies established that, paradoxically, inflammation can promote tumor development rather than help mount an efficient response against the tumor cells.2 However, the parameters that tilt the balance toward a favorable, or unfavorable, outcome are still poorly understood. Genetic factors notably influence the risk to develop cancer.3 But, except for a minority of cases with simple genetics, the genetic susceptibility to cancer is largely multifactorial, involving multiple genes probably in combination with non-genetic factors,4 and therefore much remains to be elucidated.
In the era of high-throughput biology and mathematical modeling, the study of experimental models remains an essential component of cancer research.5 Animal models that spontaneously develop tumors are of particular interest as they provide a unique opportunity to dissect the pathogenic mechanisms in light of the tumor growth at the earliest stages and of its relationship with the host organism. In the RET transgenic mouse model, a TRIM27-RET fusion transgene, on the C57BL/6 (B6) genetic background, results in the spontaneous development of ocular melanoma that subsequently disseminates into metastases.6,7 This model thus allows to investigate the natural history of tumor development. It has provided a wealth of information on the mechanisms of tumor growth and its relationship to the host immune system.8-16 Genetic factors also appear to play a significant role since transgenic mice from the BALB/c background do not develop melanoma although the cellular mechanisms underlying this alteration are not known.17
The NOD mouse is a well-established model of autoimmune diabetes.18 It has been intensely investigated to characterize the genetic and immunologic mechanisms of the autoimmune response against insulin-producing β cells. Beyond type 1 diabetes, it has a general propensity to develop autoimmunity against a variety of target organs, either spontaneously or following immunization with self-antigens.19 It also displays a strong inflammatory background, notably leading to marked resistance to allograft or high delayed hypersensitivity response.20,21 In the present work, we intercrossed B6.RET+ transgenic mice with NOD mice. This resulted in a dramatically aggravated disease course. We also noted marked alterations of the immune cell populations, notably including a profound loss of Dectin-1 expression on a granulocytic myeloid cell subset. Moreover, tumor extension could be prevented by ligation of Dectin-1 by curdlan or by inactivation of the Nos2 gene.
Results
Acceleration of tumor development following intercross with NOD mice
An intercross of B6.RET+ transgenic mice with NOD mice resulted in an unexpected and marked acceleration of tumor extension. As seen in Fig. 1, whereas the course of tumor development was similar at the primary ocular site in B6.RET+ mice compared with (NODxB6) F1.RET+ mice and with mice of subsequent generation of backcross, including F2, F3 and F10 (Fig. 1A), the latter mice showed an earlier extension of tumors to the face (p = 2.3 × 10−8, Fig. 1B) and to the back (p = 2.5 × 10−17, Fig. 1C).
Figure 1.
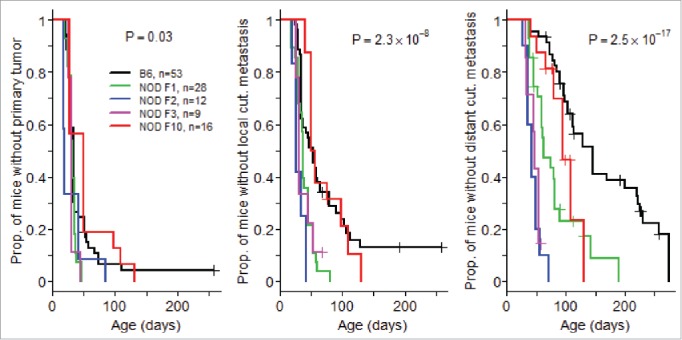
Acceleration of tumor development associated with NOD genetic background. Mice transgenic for the RET oncogene were monitored weekly for the onset of tumors at the eye, the primary site (A) and for local (B) and distant (C) cutaneous (cut.) metastases. The comparison included mice with a B6 background, F1 mice intercrossed with NOD mice, and these F1 mice backcrossed with NOD at the second (F2), third (F3) and tenth (F10) generation. Data are displayed using the Kaplan–Meier representation.
Alteration of lymphoid cell populations: expansion of CD4+Foxp3+ T cells in NOD.RET+ tumor correlates with increased expression of proinflammatory cytokines
Thus, the NOD genetic background markedly enhanced the impact of the RET transgene. The NOD strain is well known for its proinflammatory immune anomalies and we speculated that these played a role in acceleration of tumors. We therefore sought for alterations of the immune cells in NOD mice compared with B6 mice. Among the NOD group, we analyzed separately (NODxB6)F1 mice and mice after 5 to 10 generations of backcrossing onto the NOD background (NOD F5–F10) as the proportion of B6 alleles in their genome differs substantially, 50% for (NODxB6)F1 and in average less than 4% in NOD F5–F10. We focused our analyses on the lymphoid organs including the spleen, the inguinal and axillary lymph nodes, and the submandibular lymph nodes that drain the tumor of the eyes and the face. We also examined metastasis infiltrating immune cells. We accounted for the RET transgene effect by systematically including non-transgenic littermates.
Analysis of the lymphoid compartment revealed no quantitative change in the NK and γδ T cell compartments, but rather a marked expansion of the T cells resulting from an increased number of CD4+ T cells in the lymph nodes and the spleen of (NODxB6)F1 and NOD F5–F10 mice compared to the B6 controls (not shown). This CD4+ T-cell expansion was independent of the RET transgene and likely relates to the CD4+ T cell hyperplasia previously reported on the NOD background.22 In the tumors, we noted a substantially increased density of both CD4+ and CD8+ T cells in NOD mice compared with B6 (Fig. 2A). Beyond this global increase, we observed a specific increase of the CD4+Foxp3+ T regulatory cell subset in NOD tumors compared with lymphoid organs (p = 3.2 × 10−5 for comparison of tumor with lymph nodes), as previously reported in many tumors, and also with B6 tumors where no increase was observed indeed (p = 0.047 and p = 3.4 × 10−8 for comparison of (NODxB6)F1 and NOD F5–F10 with B6 mice, Fig. 2B). This was highly consistent with the more severe tumor progression seen on the NOD background. Most interestingly, this alteration was more pronounced in NOD F5–F10 than in (NODxB6)F1, reflecting the increase in the proportion of NOD alleles with the backcrossing.
Figure 2.
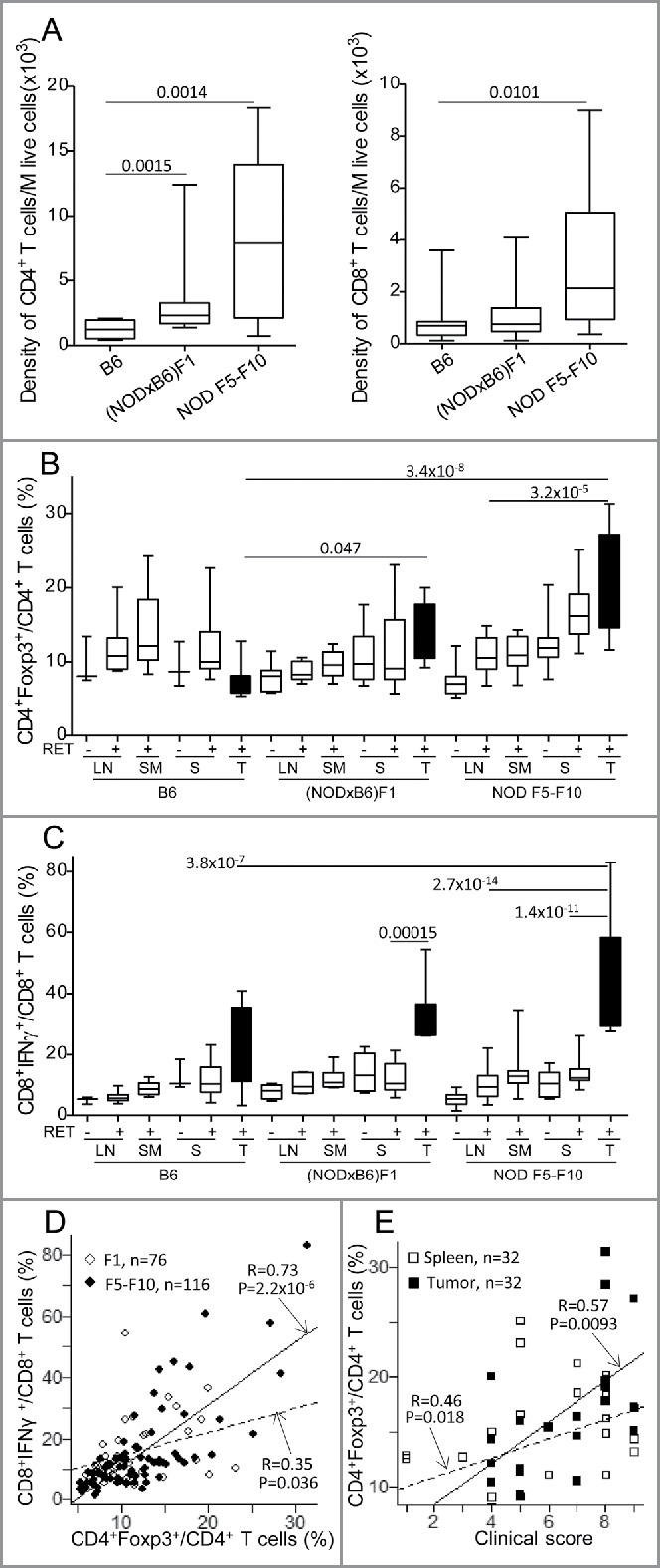
Alteration of T cell subsets in (NODxB6)F1 and NOD F5-F10 backcrossed mice carrying the RET transgene. (A) Density of CD4+ and CD8+ T cells in tumors. Boxplots show the mean and interquartile range (box) and the 5–95 percentiles (whiskers). B6, n = 10; (NODXB6)F1, n = 10; NOD F5-F10, n = 13. (B, C) The proportion of CD4+ T cells expressing Foxp3 (B), and of CD8+ T cells expressing IFNγ (C) was determined by intra-cellular immunolabeling and flow cytometry analysis in the peripheral (axillary and inguinal) lymph nodes (LN), submandibular lymph nodes (SM) draining the facial tumors, the spleen (S) and the tumor (T) of RET-transgenic mice and their non-transgenic littermates. Expression of IFNγ was assessed after stimulation with PMA and ionomycin for 4 h. The average proportions in the various conditions were compared with pairwise t tests. The resulting p values were systematically corrected for multiple testing using the Bonferroni method. For clarity, only relevant significant p values are reported. (D) Correlation between the proportions of CD4+Foxp3+ T cells and of CD8+ T cells expressing IFNγ in F1 hybrid (open symbols, dashed regression line) and F5–F10 backcrossed (closed symbols, solid regression line) mice. (E) Correlations of clinical score with proportion of CD4+Foxp3+ T cells in spleens (open symbols, dashed regression line) or in tumors (close symbols, solid regression line) in F1 and F5–F10 backcrossed mice. All correlations were tested using the non-parametric Spearman method. Number of samples in each group: B6.RET− LN, n = 3; B6.RET+ LN, n = 7; B6.RET+ SM, n = 5, B6.RET− S, n = 3; B6.RET+ S, n = 10; B6.RET+ T, n = 10; (NODxB6)F1.RET− LN, n = 7; (NODxB6)F1.RET+ LN, n = 6; (NODxB6)F1.RET+ SM, n = 6; (NODxB6)F1.RET− S, n = 10; (NODxB6)F1.RET+ S, n = 13; (NODxB6)F1.RET+ T, n = 9; NOD F5–F10.RET− LN, n = 22; NOD F5–F10.RET+ LN, n = 16; NOD F5–F10.RET+ SM, n = 16; NOD F5–F10.RET− S, n = 15; NOD F5–F10.RET+ S, n = 13; NOD F5–F10.RET+ T, n = 11.
We also observed a markedly increased expression of the proinflammatory cytokine IFNγ in CD8+ T cells in tumors compared to lymphoid tissues. As seen in Fig. 2C, the CD8+ T cells expressing IFNγ were dramatically expanded in NOD tumors compared with B6 tumors although this was significant only when comparing F5–F10 with B6 tumors (p = 3.8 × 10−7). Strikingly the proportion of CD8+IFNγ+ T cells was tightly correlated with that of CD4+Foxp3+ T regulatory cells (p = 2.2 × 10−6 in NOD F5–F10 mice and p = 0.036 in (NODxB6)F1 mice) (Fig. 2D).
We devised a clinical score reflecting the severity and the extent of the tumor process (see Materials and methods) to assess a quantitative correlation with the immunologic parameters. Mice were systematically scored at the time of the experiments. As seen in Fig. 2E, the disease severity was significantly correlated with the proportion of CD4+Foxp3+ T cells, in both spleens and tumors of F1 and F5–F10 backcrossed mice.
Marked alterations of the myeloid compartment
We then focused our analysis on myeloid cells in the spleen and the tumors where their presence was reliably detected in sufficient proportion. The CD11b+ cells are usefully split into two subsets using the Ly6G marker.23,24 As previously reported,12 there was a predominance of the CD11b+Ly6G− monocytic subset among CD45+ cells in the tumors of both B6 and NOD.RET+ mice and this was not influenced by the genetic background (Fig. 3A). The CD11b+Ly6G+ granulocytic compartment also was increased in tumors compared to spleen, although pairwise tests were significant only for comparison of spleen and tumors in NOD F5–F10 mice (Fig. 3B).
Figure 3.
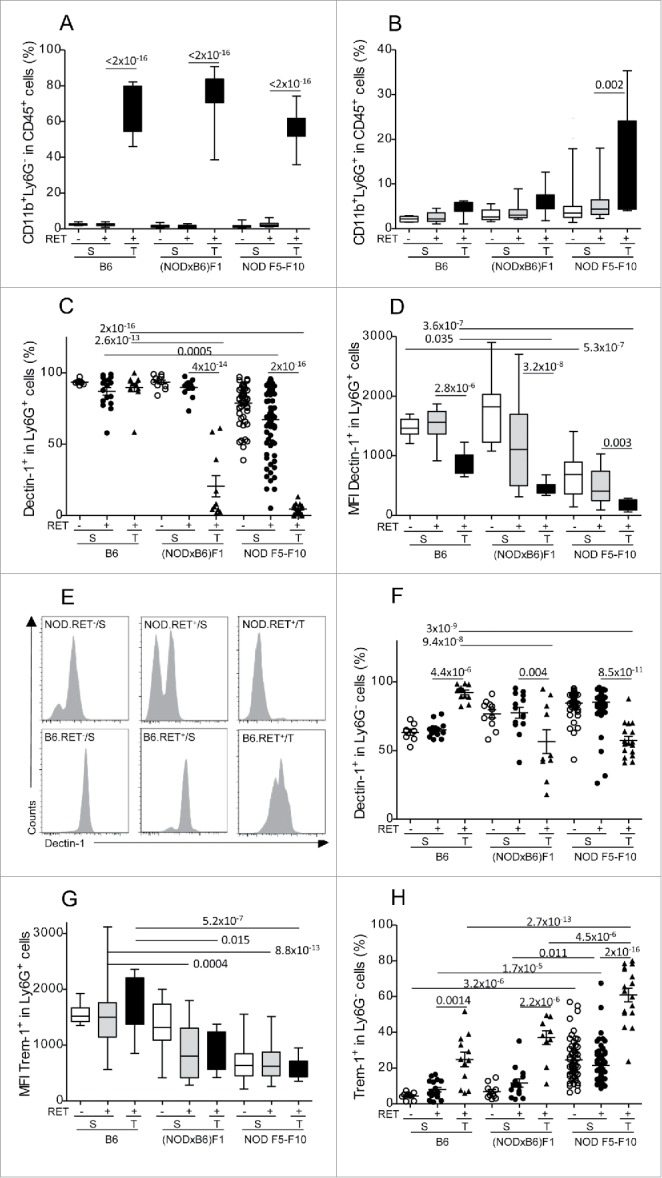
Marked alteration of Dectin-1 and Trem-1 expression on myeloid cells. Proportions of CD11b+Ly6G− (A) and CD11b+Ly6G+ (B) cells among CD45+ cells in spleens (S) and tumors (T) were determined by immunolabeling and flow cytometry. Expression of Dectin-1 (C–F) and Trem-1 (G, H) was determined on CD11b+Ly6G+ (C–E, G) and CD11b+Ly6G− (F, H). Data are depicted as the proportion of positively labeled cells (A–C, F–H) or using the mean fluorescence intensity (MFI) of the labeling (D, G). Representative histograms of fluorescence intensities are also shown for Dectin-1 on CD11b+Ly6G+ cells (E). Bars in scatterplots indicate the mean sample and its standard error. The sample means in the various conditions were compared with pairwise t tests. The resulting p values were corrected using the Bonferroni method. For clarity, only relevant significant p values are reported. Number of samples per group: B6.RET− S, n = 8; B6.RET+ S, n = 17; B6.RET+ T, n = 12; (NODxB6)F1.RET− S, n = 11; (NODxB6)F1.RET+ S, n = 16; (NODxB6)F1.RET+ T, n = 10; NOD F5–F10.RET− S, n = 55; NOD F5–F10.RET+ S, n = 67; NOD F5–F10.RET+ T, n = 17.
We next analyzed the expression of two major surface receptors of CD11b+ cells involved in their response to inflammatory stimuli, including Dectin-1 and Trem-1 (triggering receptor expressed on myeloid cells 1). Of note, our unpublished data indicated that expression of both these receptors was altered in NOD mice. As seen in Fig. 3C–E, basal expression of Dectin-1 on granulocytic cells from spleen of non-transgenic mice was decreased in NOD F5–F10 compared to B6 mice (Fig. 3D, p = 5.3 × 10−7). Moreover (Fig. 3C), there was a profound drop of Dectin-1 expression on CD11b+Ly6G+ cells in tumors of NOD mice compared to B6 (p = 2.6 × 10−13 for F1 vs. B6 and p = 2 × 10−16 for F5–F10 vs. B6) and also compared to spleen (for tumor vs. spleen: p = 4 × 10−14 in F1 and 2 × 10−16 in F5–F10). Dectin-1 was also significantly lower in the spleen of F5–F10 mice compared with B6 (p = 0.0005). In other words, Dectin-1 was expressed at the membrane of nearly all CD11b+Ly6G+ cells in B6 tumors and was virtually absent from CD11b+Ly6G+ cells in NOD tumors. Interestingly, assessment of the mean fluorescence intensities revealed that expression of Dectin-1 was quantitatively decreased at the surface of CD11b+Ly6G+ cells in tumors compared to spleen even in B6 mice (p = 2.8 × 10−6, Fig. 3D; see Fig. 3E for representative histograms). However, the level in B6 tumors remained significantly above that measured in NOD mice (p = 0.035 for comparison with F1 and 3.6 × 10−7 with F5–F10).
The pattern of expression of Dectin-1 on CD11b+Ly6G− cells was more complex though also quite significant (Fig. 3F). In B6 mice, it was increased in tumors compared with spleens (p = 4.4 × 10−6). The opposite pattern (decreased in tumors vs. spleens) was observed in F1 and F5–F10 NOD mice (p = 0.004 in F1 and p = 8.5 × 10−11 in F5–F10). This resulted in a decreased expression of Dectin-1 on CD11b+Ly6G− cells in tumors of NOD mice compared with B6 (p = 9 × 10−8 for F1 vs. B6 and p = 3 × 10−9 for F5–F10 vs. B6).
Regarding expression of Trem-1 on CD11b+Ly6G+ cells, it was also decreased in NOD F1 and F5–F10 mice compared with B6, and this was seen in both the tumors and the spleen (Fig. 3G). In marked contrast, expression of Trem-1 on CD11b+Ly6G− cells was increased in tumors compared with spleen in the three groups of mice (Fig. 3H) but this increase in tumors was much stronger in NOD F5–F10 than in NOD F1 and B6 mice (p = 2.7 × 10−13 for F5–F10 vs. B6 and p = 4.5 × 10−6 for F5–F10 vs. F1). This effect was tightly dependent on the genetic background as it was also seen in the spleen of non-transgenic mice (p = 3.2 × 10−6 for F5–F10 against B6).
Remarkably, downregulation of Dectin-1 expression on Ly6G+ cells was correlated with the increase of both CD4+Foxp3+ T cells and CD8+IFNγ+ T cells in tumors of F1 and F5–F10 NOD mice (Fig. 4A–B). In addition, expression of Trem-1 on Ly6G− cells was positively correlated with an expansion of both of these lymphoid cell subsets (Fig. 4C–D). These observations suggested a coordinate mechanism underlying the changes in these lymphoid and myeloid cell populations.
Figure 4.
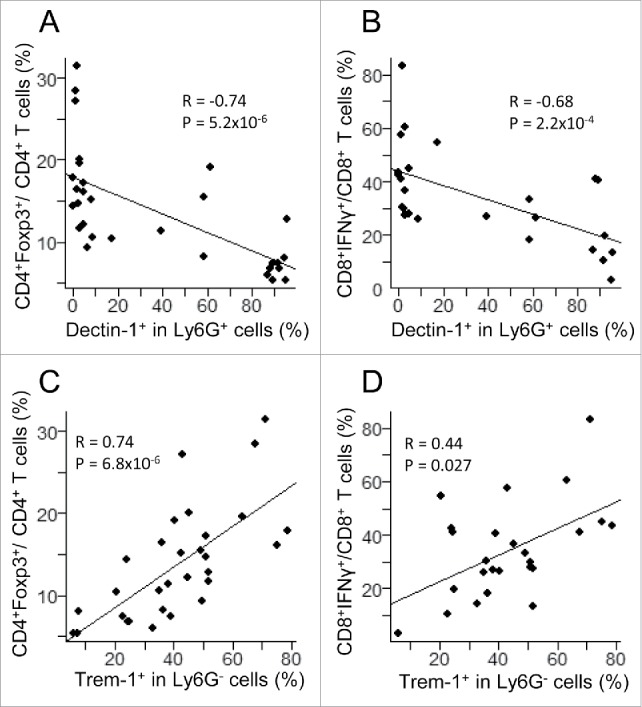
Correlation between myeloid subsets, including CD11b+Ly6G+Dectin-1+ (upper panels) and CD11b+Ly6G−Trem-1+ (lower panels) with T lymphoid subsets, including CD4+Foxp3+ (left panels) and CD8+IFNγ+ (right panels) in 36 tumors from (NODxB6)F1 and NOD F5–F10 mice. Significance of correlations was assessed with the non-parametric method of Spearman.
Dectin-1 and Trem-1 are thus functionally important receptors that are both expressed on CD11b+ cell subsets. We examined whether their expression on these cells was independent or not and how they combined at the surface of CD11b+ cells under the influence of the genetic background and the localization. In the spleen of non-transgenic B6 mice (the baseline), all CD11b+Ly6G+ cells express both markers (Fig. 5A, top left pie chart). On the B6 background in the presence of the RET transgene, the Dectin-1+Trem-1+ double positive population remained largely predominant and the Trem-1+ and Dectin-1+ single positives and the double negative populations were seen in small proportions (top center and top right pie charts). The introduction of the NOD genetic background was associated with the massive loss of Dectin-1 expression (compare top left and bottom right pie charts). Consequently, in tumors of NOD F1 and even more so of NOD F5–F10 RET+ mice, CD11b+Ly6G+ cells expressed little Dectin-1 and were in majority (˜73%) Trem-1 single positives; a significant proportion of cells also lost Trem-1 and therefore were double negatives (˜20%).
Figure 5.
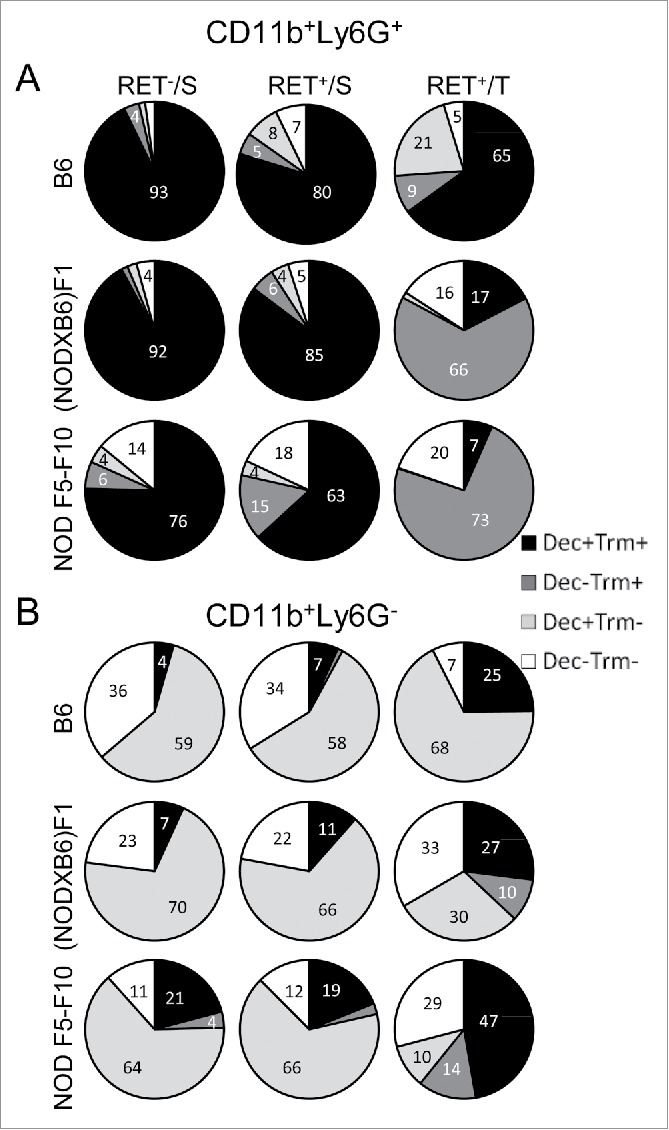
Major changes in subsets of myeloid cells are associated with the NOD genetic background. Spleen (S) and tumor (T) CD11b+Ly6G+ (A) and CD11b+Ly6G− (B) cells were gated electronically and the percentages of cell subsets defined by Dectin-1/Trem-1 double labeling were determined and are depicted using pie charts. Dec, Dectin-1; Trm, Trem-1.
In contrast, CD11b+Ly6G− cells exhibited a different pattern characterized by the absence of expression of Trem-1 at the baseline (spleen of non-transgenic B6 mice) and therefore were either Dectin-1 single positive (˜59%) or double negative (˜36%) (Fig. 5B, top left pie chart). In the B6 tumors, the majority of CD11b+Ly6G− cells were either Dectin-1 single positive or double positive (top right pie chart). The introduction of the NOD genetic background was characterized by a shrinkage of the Dectin-1 single positive cell subset (down to 10% from 68% in B6 tumors) whereas expression of Trem-1 was increased, resulting in an expansion of the double positive and of the Trem-1+ single positive cell populations (bottom right pie chart).
Protection against tumor by curdlan
To investigate the relevance of the loss of Dectin-1 expression at the tumor sites in our model, we treated the (NODxB6)F1.RET+ mice with curdlan, a 1,3-β glucose polymer that cross-links Dectin-1 and activates its signaling. 6-to-7 week-old mice, all with tumor score of one, were injected i.p. with 100 µg curdlan per gram of body weight or with PBS. They received a second injection one week later and were monitored weekly for tumor progression for 15 weeks in total. As seen in Fig. 6A, curdlan-injected mice were dramatically protected against tumor development whereas the number of tumors increased markedly in vehicle-injected mice (p = 2 × 10−6). Accordingly, in the few tumors available for analysis, the proportion of CD4+Foxp3+ T cells in CD4+ T cells was significantly reduced (p = 0.006, Fig. 6B).
Figure 6.
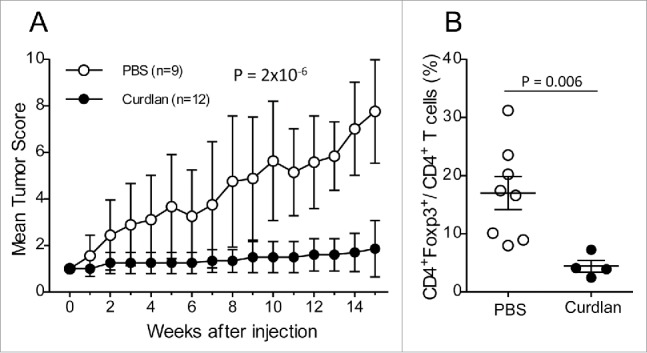
Protection against tumor development by curdlan treatment. (A) (NODxB6)F1.RET+ transgenic mice (6–7 weeks of age) were injected intraperitoneally twice with curdlan (100 µg/g of body weight, n = 12) or with PBS (n = 9) with a one-week interval and were monitored weekly for tumor progression. The data are pooled from two experiments conducted at an interval of 6 mo. The mean tumor score and its standard deviation are shown at each timepoint. The difference of tumor scores between both groups was tested using a linear model with mixed effects. (B) Reduction of CD4+Foxp3+ T cells in tumors of curdlan-treated mice.
Protection against tumor progression in Nos2−/− mice
The Nos2 products appear to play a major aggravating role in progression of various models of melanoma and also in many tumors. In order to investigate their role in the (NODxB6)F1.RET+ mouse model, we crossed B6.RET+.Nos2−/− mice with NOD.Nos2−/− mice and investigated the F1.RET+ mice that were therefore also Nos2−/− homozygous. As seen in Fig. 7A, their clinical score was significantly lower than that of F1.RET+.Nos2+/+ at the same age (p = 3.3 × 10−7) that exhibited exceptional extension of the tumors to the back and the viscera. Only four of the 27 analyzed mice developed one tumor distantly from the primary ocular site. Of note, B6.RET+.Nos2−/− mice were investigated in parallel and were also strongly protected as they displayed no metastasis (not shown). In what follows, we provided their relevant data, and also those of B6.RET+.Nos2+/+ shown just above, as references for discriminating the effects of the strain genetic background and of the Nos2 gene inactivation.
Figure 7.
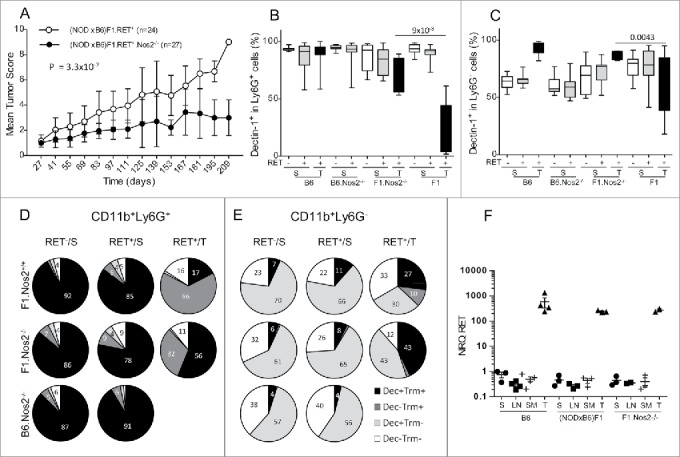
Less severe tumor evolution and upregulation of Dectin-1 expression in (NODxB6)F1.RET+.Nos2−/− mice. Hybrid (NODxB6)F1.RET+ transgenic mice proficient (open circles, n = 24) or deficient (dark circles, n = 27) for the Nos2 gene were monitored for tumor development every other week for 7 mo. The mean tumor score and its standard deviation are shown at each timepoint. The difference of tumor scores between both groups was tested using a linear model with mixed effects. (B, C) Upregulation of Dectin-1 expression on CD11b+Ly6G+ (B) and CD11b+Ly6G− (C) myeloid cells in spleen (S) and tumors (T) of (NODxB6)F1 and B6.RET+ mice deficient for Nos2 compared with their Nos2+/+ counterparts. (D, E) Dectin-1/Trem-1 double labeling of myeloid subsets in spleens and tumors. CD11b+Ly6G+ (D) and CD11b+Ly6G− (E) cells were gated electronically and the percentages of cell subsets defined by Dectin-1/Trem-1 double labeling were determined in mice with indicated genetic background and are depicted using pie charts. Dec, Dectin-1; Trm, Trem-1. (F) Quantification by qPCR of RET transgene expression in tumors (T) and in lymphoid organs, including spleen (S), peripheral lymph nodes (LN) and submandibular lymph nodes (SM), of RET+ mice with indicated genetic background. Normalized relative quantities (NRQ) of RET messages were determined as described in Materials and Methods. Number of samples per group: B6.RET− S, n = 8; B6.RET+ S, n = 17; B6.RET+ T, n = 12; B6.Nos2−/−.RET− S, n = 7; B6.Nos2−/−.RET+ S, n = 12; F1.Nos2−/−.RET− S, n = 17; F1.Nos2−/−.RET+ S, n = 16; F1.Nos2−/−.RET+ T, n = 4; F1.RET− S, n = 11; F1.RET+ S, n = 14; F1.RET+ T, n = 10.
Analysis of immune cell markers revealed striking changes in myeloid cells in relation with the inactivation of the Nos2 locus. Expression of Dectin-1 on CD11b+Ly6G+ cells was massively up-regulated (p = 9 × 10−8 for comparison of F1.Nos2+/+ with F1.Nos2−/−, Fig. 7B) and almost up to normal levels. In contrast, Trem-1 labeling was not influenced by the Nos2 locus (not shown). Consequently, in the CD11b+Ly6G+, the proportion of double positive Dectin-1+Trem-1+ (˜90%) was less decreased in F1.Nos2−/− (56%) compared to F1.Nos2+/+ (17%) tumors (Fig. 7D, compare top and middle right pie charts), whereas the Trem-1+ single positive cell subset was less increased.
Likewise, on CD11b+Ly6G− cells in F1 tumors, Dectin-1 expression was upregulated in Nos2−/− mice (Fig. 7C, p = 0.0043 for comparison of Nos2+/+ and Nos2−/−), while Trem-1 expression was not altered (data not shown). Examination of double labeling indicated that Dectin-1−Trem-1− cells were decreased in tumors of F1.Nos2−/− compared with F1.Nos2+/+, whereas Dectin-1+Trem-1− and Dectin-1+Trem-1+ cells were expanded compared to their Nos2+/+ counterpart (Fig. 7E, compare top and middle right pie charts).
Of substantial interest, expression of the RET transgene was not influenced by the Nos2 gene. As seen in Fig. 7F, the transgene was expressed at much higher levels, by 3 orders of magnitude, in tumors compared to spleen and lymph nodes, but not differently in B6, (NODxB6)F1 and NOD.Nos2−/− mice.
Discussion
The RET transgenic mouse provides a remarkable model for investigating the molecular and cellular mechanisms of spontaneous tumor growth and dissemination.6,7,25-27 Its study has also revealed a wealth of information on tumor interaction with the immune system and how the tumor potentially deviates the immune response and escapes the immune surveillance.8-16 These findings were obtained essentially using the RET transgene on the C57BL/6 genetic background. In this model, the tumor appears in average at the age of one month at the primary site and subsequently extends locally to the face and more distantly. Expression of the transgene, however, appears to be influenced by the genetic background. A cross with BALB/c mice revealed protective alleles.17 The mechanisms of this protection, however, were not studied. Here, on the contrary, using the NOD strain, we observed an aggravated phenotype, with much more frequent and earlier metastasis to the rest of the face, to the back and to the viscera than on the B6 background. This was seen as soon as the first generation of intercross suggesting that either dominantly predisposing NOD alleles or recessively protective B6 alleles are involved. This more severe phenotype was confirmed at successive generation of backcrossing. However, the F10 mice were more fragile and underweighted relative to their non-transgenic littermates suggesting additional complex effects of the fusion transgene on metabolism and development depending on the NOD genetic background (data not shown). The aggravating effect of NOD alleles is unlikely to be exerted at the level of the RET transgene itself as its expression was not quantitatively altered by the mouse genetic makeup. The endogenous Ret gene expression also was not different in B6 and NOD mice (not shown). This was important as RET has been shown to be expressed in peripheral blood cells and its loss of function in Hirschsprung patients associated with inflammatory response.28
Because the NOD strain is well known for its immune alterations,29 we speculated that these might play a primary role in the NOD phenotype. Indeed, we observed highly significant changes in key parameters of both the innate and the adaptive immune system that are overall consistent with the aggravated phenotype of RET-transgenic mice carrying NOD alleles. In the first line, we observed a marked increase of the CD4+Foxp3+ T cell subset, assumed to bear immunoregulatory function, in tumors from (NODxB6)F1 and NOD F5–F10 mice compared to their B6 counterpart. This immunoregulatory subset is known to be expanded in many malignancies where it antagonizes the effector T cells and is currently a major target for therapeutic research.30,31 In RET+ mice on B6 background, regulatory CD4+ T cells accumulate in the spleen and tumor-draining lymph nodes and inhibit the antitumor properties of inflammatory monocytes.15 Our study revealed a direct correlation between the amplification of this subset within the spleen and the tumor microenvironment and tumor extension (Fig. 2E). Besides, we noted a dramatic expansion of another lymphoid subset, this from the CD8+ T cell population. It was characterized by a high proportion of IFNγ-producing cells in tumors from (NODxB6)F1 and NOD F5–F10 mice. Remarkably, it was tightly correlated with the CD4+Foxp3+ T cell population (Fig. 2D). Moreover, whereas IFNγ is well known for its pro-inflammatory effects, it also exerts immunomodulatory functions through various mechanisms, notably including stimulation of IL1 receptor antagonist and IL18 binding protein expression.32,33 Another obvious mechanism through which IFNγ might facilitate the tumor growth is the generation of highly tumorigenic NOS2 products (see below) as IFNγ is a major inducer of the Nos2 gene transcription.34 How this CD8+ T cell population relates to the functional CD8+ T cells described previously in a slightly different model8 is presently unclear. It is possible that the population of CD8+ T cells expanded in tumors from the NOD model rather belong the kind of so-called regulatory CD8+ T cells reported to infiltrate tumors.35,36 Their phenotype, however, is quite variable between the various studies. In our model, they were negative for Foxp3 and expressed no IL10 (not shown). Their detailed phenotypic and functional characterization nonetheless remains to be done. The (NODxB6)F1.RET+ mice thus provide a powerful model to investigate the mechanisms underlying development and functions of this cell population.
Myeloid cells play a key role in metastasis development37 and displayed also major changes in our model. These included a defective expression of Dectin-1, most pronounced on CD11b+Ly6G+ granulocytic myeloid cells but also significant on CD11b+Ly6G− monocytic myeloid cells (Fig. 3). Indeed, Dectin-1 expression was virtually abolished from the surface of granulocytic myeloid cells in tumors of NOD mice compared with B6. It was also markedly decreased in the spleen of transgenic NOD mice and also, very interestingly, of non-transgenic NOD mice. This strongly suggests that there are two components in the Dectin-1 decreased expression in NOD, determined one by the genetic background, the other by an interaction between the genetic background and the tumor, likely reflecting immunoediting by the tumor cells. Dectin-1, a C-type lectin that is a membrane receptor for 1,3 β-glucan, is a key molecule in activating inflammatory response to a number of infectious agents, notably including bacteria and fungi.38 In contrast, much less is known about its potential role in tumor–host relationship. Recently, mice with a germline-inactivated Dectin-1 gene were reported to fail to control development and metastasis of transplanted B16 melanoma cells.39 In addition, studies using transplanted tumor cells revealed an antitumor effect of treatment with curdlan, a ligand of Dectin-1.40,41 Our study, however, is the first that demonstrates a strongly protective effect of curdlan in a model of spontaneously growing tumor. Remarkably, the protection afforded by curdlan was correlated with a marked decrease of the proportion of CD4+Foxp3+ T regulatory cell population in the few available tumors of curdlan-treated mice (Fig. 6B). This is quite consistent with the correlation observed between Dectin-1 expression level and the proportion of CD4+Foxp3+ T cells and CD8+IFNγ+ T cells in untreated NOD.RET+ mice (Fig. 4A–B). Finally, the restoration of Dectin-1 expression on myeloid cells in NOD.RET+.Nos2−/− mice along with their strong protection against tumor development (Fig. 7B–C), further strengthens the hypothesis of a critical role of Dectin-1 in the control of tumor expansion and in mounting an efficient host response to a tumor challenge. Whereas the cellular and molecular mechanisms linking NOS2, Dectin-1 and the immune control of tumor progression undoubtedly remain to be characterized, activation of Dectin-1 by appropriate ligands already appears as a potentially promising therapeutic strategy in cancer immunotherapy.
Upregulation of Trem-1 on monocytic (CD11b+Ly6G−) myeloid cells of F1 and F5–F10 NOD mice was another major alteration of myeloid cells, seen in tumors but also in spleen, including in non-transgenic littermates. Thus, the NOD genetic background, the RET transgene and the tumor location concur to increase the expression level of Trem-1 in our model. Moreover, this was quantitatively correlated with the lymphoid cell changes discussed just above (Fig. 4C–D). This inflammatory receptor has long been investigated predominantly in the context of inflammatory responses to infectious agents.42 Recently, however, reports have associated its increased expression with tumor recurrence and poor survival rate, consistent with the findings in our model.43,44
The NOS2 products are established as tumor promoters in many, though not all, cancer models, notably including melanoma.45 The tumorigenic effect of NO and of its oxidation products is thought to occur through multiple mechanisms, including alteration of signal transduction and cell metabolism, DNA damage, epigenetic modifications, stimulation of angiogenesis and immune suppression. The dramatically improved prognosis of NOD mice with an inactivated Nos2 gene along with the changes in their immune system configuration suggests that additional immunologic mechanism might be involved in the Nos2 gene effect on tumor growth. The Nos2 gene is primarily expressed in CD11b+Ly6G− monocytic myeloid cells.46 Our working hypothesis, based on ongoing experiments in our lab (L.K. et al., manuscript in preparation), is that the CD11b+Ly6G+Dectin-1+ granulocytic population is highly sensitive to NOS2 products that must be released in massive amounts in tumors by CD11b+Ly6G− monocytic myeloid cells exposed to high levels of IFNγ, in our model expressed by CD8+ T cells. Loss of Dectin-1 expression has then important consequences as Dectin-1 signaling was shown to impair CD4+CD25+ T cell regulatory function,47 to activate NK cells,39 and to promote IL12 expression by dendritic cells.48 The inverse correlation between Dectin-1 expression and CD4+Foxp3+ T cell numbers we reported is thus consistent with previous reports. In contrast, however, we would have expected a positive correlation of Dectin-1 expression with IFNγ-producing CD8+ T cell population. Instead, we noted also a negative correlation (Fig. 4B). To account for this discrepancy, we assume that other mechanisms are involved. Thus, NOD mouse macrophages were consistently reported to spontaneously express high amounts of IL12.21,49 This mechanism might bypass the modulation resulting from loss of Dectin-1 and explains the upregulation of IFNγ expression. In addition, the constitutive elevation of Trem-1 expression in NOD macrophages, which we report here for the first time, further documents the broad dysregulation of innate immune cells in the NOD strain and might also contribute to exacerbate the inflammatory adaptive response in NOD mice. Trem receptors, however, have pleiotropic effects.42 The molecular mechanisms underlying the significant correlations we noted between Trem-1 expression on macrophages and lymphoid cell populations (Fig. 4C–D) need to be analyzed specifically in our model.
In summary, our study highlights the NOD mouse as a new model to investigate the complex host–tumor relationship in a setting of rapidly disseminating tumor. It also emphasizes the Dectin-1 and the Nos2 as important players in this model. Whether these observations are relevant to human melanoma and also can be extended to other experimental models of tumors now deserves to be investigated.
Materials and methods
Mouse strains and breeding
C57BL/6 mice transgenic for a human TRIM27/RET fusion gene (in short, B6.RET+ mice) were described previously.6 NOD mice (Leiter substrain) were obtained from the Jackson Institute. C57BL/6 mice with a germline inactivated Nos2 gene as described elsewhere.50 were purchased from Taconics. The inactivated Nos2 locus was iteratively backcrossed for 20 generations on the NOD background in our animal facility to generate the NOD.Nos2−/− mice used in our study.
B6.RET+ mice were intercrossed with NOD mice to produce the (NODxB6)F1.RET+ mice and the transgene was then transferred on the NOD background by backcross for 10 generations. At each generation, transgenic mice were selected at weaning on their phenotype, i.e. dark pigmentation, which was confirmed by genotyping with no discrepancy. B6.RET+.Nos2−/− mice were obtained following an F2 cross between B6.RET+ and B6.Nos2−/− mice. (NODxB6)F1.RET+.Nos2−/− mice were F1 hybrids between B6.RET+.Nos2−/− and NOD.Nos2−/− mice. All mice were maintained in our pathogen-free animal facility. All experiments were performed in compliance with French Ministry of Agriculture regulations for animal experimentation.
Tumor monitoring
Tumor evolution was first assessed using the time to tumor onset at a given site (eye for primary tumors, face for local cutaneous metastases or back or limbs for distant cutaneous metastases) after clinical examination every other week. Alternatively, a tumor score was devised for a finer appreciation of tumor severity. Thus, we assigned a value to each mouse. This was equal to one for exophthalmos, much invaded eyes and face tumor. A dorsal tumor was scored 2 and a visceral metastasis was scored 3. Additional dorsal tumors were scored one. Weight loss greater than 15% was also scored one. These scores were added together to produce a composite score with maximum 10.
Antibodies and cell staining
Anti-CD45-PE-Cy7, anti-CD11b-APC or anti-CD11b-PercpCy5.5, anti-Ly6G-V450, anti-CD4-PerCP, anti-CD8-PE or anti-CD8-FITC or anti-CD8-V450, anti-FoxP3-PE, anti-IFNγ-PE antibodies were from BD PharMingen, anti-TREM1-PE antibody was from R&D Systems, anti-Dectin-1-FITC was obtained from AbD serotec and their corresponding isotypes from the same manufacturers.
Single cell suspensions of spleens (S) and lymph nodes (LN) were obtained by mechanical dissociation, followed by filtration through a 100 µm Cell Strainer (BD Falcon), washing and resuspension in PBS 1X, 2% FCS (Fetal Calf Serum). Cutaneous tumor masses were dissected from transgenic mice and were mechanically dissociated and then digested with Collagenase A (1 mg/mL; Roche) and DNase I (0.1 mg/mL; Roche) for 20 min at 37°C. Single cell suspensions were filtered on 100 µm Cell Strainer washed in PBS 1X, 2% FCS, and resuspended in RPMI 1640 medium. Cells were incubated with Live/Dead Fixable Aqua Dead Cell stain (Invitrogen), according to the manufacturer's protocol. Then, after blocking with anti-FcγR antibodies, cell samples were surface-stained with specific antibodies on ice for 30 min.
For intracellular staining, cell suspensions were stimulated 4 h with PMA (Phorbol 12-myristate 13-acetate) (500 ng/mL) (Sigma-Aldrich), Ionomycin (500 ng/mL) (Sigma-Aldrich) and Brefeldin A (10 µg/mL) (Sigma-Aldrich). Next, cells were surface-stained as explained previously and fixed with 100 µL of Cytofix (Becton Dickinson Biosciences) for 20 min at room temperature or with 50 µL paraformaldehyde for 15 min at room temperature. Intracellular staining was performed by permeabilizing cells with permeabilization buffer (Saponin 10 mg/mL PBS with 2% FCS) and incubating them with anti-IFNγ antibodies. The Foxp3 intracellular staining was performed following the manufacturer's instruction (eBiosciences) after surface staining as above. Flow cytometry data were acquired on the FacsCantoII cytofluorometer (BD Biosciences) and analyzed using FlowJo software (version 7.6.4). Data acquisition was performed at the Cochin Cytometry and Immunobiology facility. Fig. 8 shows an example of gating strategy for Dectin-1/Trem-1 double labeling of CD11b+Ly6G+ and CD11b+Ly6G− myeloid cells.
Figure 8.
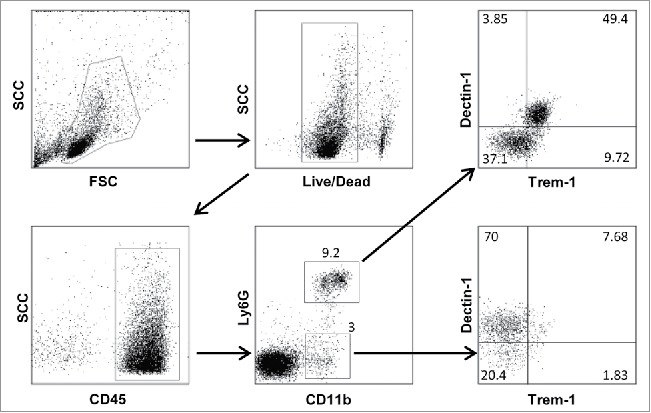
Gating strategy for flow cytometry labeling of spleen myeloid cells from an NOD F5–F10 RET+ mouse. A sample with a decreased level of Dectin-1 expression was selected. Figures indicate the percentages of events in the gates or in each quadrant.
RNA preparation and quantification by real-time PCR
Total RNA was extracted with the Qiagen RNeasy extraction kit. Briefly, spleen, lymph nodes and tumor cells in suspension were mixed with the RLT buffer. RNA was then isolated using RNeasy columns and treated with DNaseI to avoid genomic DNA contamination. RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Scientific). RNA integrity was assessed using the Bioanalyzer (Agilent). We consistently obtained RIN above 8. RNA (200 ng) was reverse-transcribed with SuperScriptM II reverse transcriptase (Invitrogen Life Technologies) using random hexamer primers (Applied Biosystems). Real-time quantitative PCR was performed using SYBR Green Master Mix (Bio-Rad) and a real time PCR system (Bio-Rad CFX96). Primer sequences are listed in Table 1.
Table 1.
Primer pairs used for real-time PCR experiments.
| Target cDNA | Forward/Reverse | Sequences (5′ to 3′) |
|---|---|---|
| Human TRIM27-RET fusion transgene | FR | TCC AAG AAT TAA GAG AGG CTCAGCTT CTC CTC CTC AGG GAA GC |
| Mouse gene = Ret | F | AAT CAG AGC CTG GAC CAC AG |
| R | TGG CAT TCT CCC TCT CTC TG | |
| GAPDH | F | TGT GTC CGT CGT GGA TCT GA |
| R | TTG CTG TTG AAG TCG CAG GAG | |
| β-Actin | F | GTG GCA TCC ATG AAA CTA CT |
| R | GGC ATA GAG GTC TTT ACG G |
Gene expression was normalized to GAPDH and β-Actin expression. Expression of GAPDH and β-Actin was used as endogenous reference, and expression of the RET target gene in each sample was normalized to the average (mean) of all Cq (Cq Calib). The relative gene expression level was calculated as the normalized relative quantity:
Curdlan treatment
6-to-7 week old (NODxB6)F1.RET+ mice were injected intraperitoneally twice with an interval of one week with either 100 µg/g body weight of curdlan (InvivoGen) or an equivalent volume of PBS (100 µL/10 g body weight). Weight, onset of ocular melanoma and metastasis were monitored weekly and blindly to the injected material.
Statistical analyses
Difference in time to onset of tumors in various groups of mice was tested using survival models with the R survival package. An influence of experimental factors, including RET transgene, localization, and genetic background on the clinical and immunological parameters was tested with a linear model, after adjustment for covariates, including sex, age and experiment. Pairwise comparisons to identify the relevant significant differences were done using t tests and Bonferroni correction for the multiple tests was systematically applied. Correlations were tested using the non-parametric test of Spearman. Longitudinal data (tumor scores) were compared between groups of mice using a linear mixed model as implemented in the lme4 R package. Effect of the grouping factor (curdlan treatment or Nos2 gene inactivation) was assessed by comparing models with and without the grouping factor with the anova function.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Karine Labroquère, Muriel Andrieu (Cochin Immunobiology facility) and Agnès Lebon (Cochin Animal Facility) for their invaluable help. We thank Maxime Breban and Gilles Chiocchia (Inserm U1173) for support throughout this project. ED was a recipient of a fellowship from the Center National de la Recherche Scientifique (PhD-engineer program) and the RAMSES association.
References
- 1.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007; 117:1137-46; PMID:17476343; http://dx.doi.org/ 10.1172/JCI31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrand-Rosenberg S. Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev 2008; 18:11-8; PMID:18308558; http://dx.doi.org/ 10.1016/j.gde.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pomerantz MM, Freedman ML. The Genetics of Cancer Risk. Cancer J 2011; 17:416-22; PMID:22157285; http://dx.doi.org/ 10.1097/PPO.0b013e31823e5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galvan A, Ioannidis JPA, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet 2010; 26:132-41; PMID:20106545; http://dx.doi.org/ 10.1016/j.tig.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrne HM. Dissecting cancer through mathematics: from the cell to the animal model. Nat Rev Cancer 2010; 10:221-30; PMID:20179714; http://dx.doi.org/ 10.1038/nrc2808 [DOI] [PubMed] [Google Scholar]
- 6.Kato M, Takahashi M, Akhand AA, Liu W, Dai Y, Shimizu S, Iwamoto T, Suzuki H, Nakashima I. Transgenic mouse model for skin malignant melanoma. Oncogene 1998; 17:1885-8; PMID:9778055; http://dx.doi.org/ 10.1038/sj.onc.1202077 [DOI] [PubMed] [Google Scholar]
- 7.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y et al.. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 2010; 120:2030-9; PMID:20501944; http://dx.doi.org/ 10.1172/JCI42002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lengagne R, Graff-Dubois S, Garcette M, Renia L, Kato M, Guillet JG, Engelhard VH, Avril MF, Abastado JP, Prevost-Blondel A. Distinct role for CD8 T cells toward cutaneous tumors and visceral metastases. J Immunol 2008; 180:130-7; PMID:18097012; http://dx.doi.org/ 10.4049/jimmunol.180.1.130 [DOI] [PubMed] [Google Scholar]
- 9.Umansky V, Abschuetz O, Osen W, Ramacher M, Zhao F, Kato M, Schadendorf D. Melanoma-specific memory T cells are functionally active in Ret transgenic mice without macroscopic tumors. Cancer Res 2008; 68:9451-8; PMID:19010920; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1464 [DOI] [PubMed] [Google Scholar]
- 10.Kimpfler S, Sevko A, Ring S, Falk C, Osen W, Frank K, Kato M, Mahnke K, Schadendorf D, Umansky V. Skin melanoma development in ret transgenic mice despite the depletion of CD25+Foxp3+ regulatory T cells in lymphoid organs. J Immunol 2009; 183:6330-7; PMID:19841169; http://dx.doi.org/ 10.4049/jimmunol.0900609 [DOI] [PubMed] [Google Scholar]
- 11.Zhao F, Falk C, Osen W, Kato M, Schadendorf D, Umansky V. Activation of p38 Mitogen-Activated Protein Kinase Drives Dendritic Cells to Become Tolerogenic in Ret Transgenic Mice Spontaneously Developing Melanoma. Clin Cancer Res 2009; 15:4382-90; PMID:19549770; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0399 [DOI] [PubMed] [Google Scholar]
- 12.Lengagne R, Pommier A, Caron J, Douguet L, Garcette M, Kato M, Avril MF, Abastado JP, Bercovici N, Lucas B et al.. T Cells Contribute to Tumor Progression by Favoring Pro-Tumoral Properties of Intra-Tumoral Myeloid Cells in a Mouse Model for Spontaneous Melanoma. PLoS One 2011; 6:e20235; PMID:21633700; http://dx.doi.org/ 10.1371/journal.pone.0020235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, Kato M, Prevost-Blondel A, Thiery JP, Abastado JP. Mesenchymal Transition and Dissemination of Cancer Cells Is Driven by Myeloid-Derived Suppressor Cells Infiltrating the Primary Tumor. PLoS Biol 2011; 9:e1001162; PMID:21980263; http://dx.doi.org/ 10.1371/journal.pbio.1001162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer Biol 2012; 22:319-26; PMID:22349515; http://dx.doi.org/ 10.1016/j.semcancer.2012.02.003 [DOI] [PubMed] [Google Scholar]
- 15.Pommier A, Audemard A, Durand A, Lengagne R, Delpoux A, Martin B, Douguet L, Le Campion A, Kato M, Avril M-F et al.. Inflammatory monocytes are potent antitumor effectors controlled by regulatory CD4+ T cells. Proc Natl Acad Sci U S A 2013; 110:13085-90; PMID:23878221; http://dx.doi.org/ 10.1073/pnas.1300314110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tham M, Tan KW, Keeble J, Wang X, Hubert S, Barron L, Tan NS, Kato M, Prevost-Blondel A, Angeli V et al.. Melanoma-initiating cells exploit M2 macrophage TGFβ and arginase pathway for survival and proliferation. Oncotarget 2014; 5:12027-42; PMID:25294815; http://dx.doi.org/ 10.18632/oncotarget.2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragani TA, Peissel B, Zanesi N, Aloisi A, Dai Y, Kato M, Suzuki H, Nakashima I. Mapping of melanoma modifier loci in RET transgenic mice. Jpn J Cancer Res 2000; 91:1142-7; PMID:11092979; http://dx.doi.org/ 10.1111/j.1349-7006.2000.tb00897.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999; 5:601-4; PMID:10371488; http://dx.doi.org/ 10.1038/9442 [DOI] [PubMed] [Google Scholar]
- 19.Bernard NF, Ertug F, Margolese H. High incidence of thyroiditis and anti-thyroid autoantibodies in NOD mice. Diabetes 1992; 41:40-6; PMID:1727738; http://dx.doi.org/ 10.2337/diab.41.1.40 [DOI] [PubMed] [Google Scholar]
- 20.Markees TG, Serreze DV, Phillips NE, Sorli CH, Gordon EJ, Shultz LD, Noelle RJ, Woda BA, Greiner DL, Mordes JP et al.. NOD mice have a generalized defect in their response to transplantation tolerance induction. Diabetes 1999; 48:967-74; PMID:10331399; http://dx.doi.org/ 10.2337/diabetes.48.5.967 [DOI] [PubMed] [Google Scholar]
- 21.Alleva DG, Johnson EB, Wilson J, Beller DI, Conlon PJ. SJL and NOD macrophages are uniquely characterized by genetically programmed, elevated expression of the IL-12(p40) gene, suggesting a conserved pathway for the induction of organ-specific autoimmunity. J Leukoc Biol 2001; 69:440-8; PMID:11261792 [PubMed] [Google Scholar]
- 22.Prochazka M, Leiter EH, Serreze DV, Coleman DL. Three recessive loci required for insulin-dependent diabetes in nonobese diabetic mice. Science 1987; 237:286-9; PMID:2885918; http://dx.doi.org/ 10.1126/science.2885918 [DOI] [PubMed] [Google Scholar]
- 23.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13:739-52; PMID:24060865; http://dx.doi.org/ 10.1038/nrc3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Damuzzo V, Pinton L, Desantis G, Solito S, Marigo I, Bronte V, Mandruzzato S. Complexity and challenges in defining myeloid-derived suppressor cells: Defining Myeloid-Derived Suppressor Cells. Cytometry B Clin Cytom 2015; 88:77-91; PMID:25504825; http://dx.doi.org/ 10.1002/cyto.b.21206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato M, Liu W, Akhand AA, Dai Y, Ohbayashi M, Tuzuki T, Suzuki H, Isobe K, Takahashi M, Nakashima I. Linkage between melanocytic tumor development and early burst of Ret protein expression for tolerance induction in metallothionein-I/ret transgenic mouse lines. Oncogene 1999; 18:837-42; PMID:9989837; http://dx.doi.org/ 10.1038/sj.onc.1202329 [DOI] [PubMed] [Google Scholar]
- 26.Kato M, Liu W, Akhand AA, Hossain K, Takeda K, Takahashi M, Nakashima I. Ultraviolet radiation induces both full activation of ret kinase and malignant melanocytic tumor promotion in RFP-RET-transgenic mice. J Invest Dermatol 2000; 115:1157-8; PMID:11121157; http://dx.doi.org/ 10.1046/j.1523-1747.2000.0202a-2.x [DOI] [PubMed] [Google Scholar]
- 27.Kato M, Takeda K, Kawamoto Y, Tsuzuki T, Hossain K, Tamakoshi A, Kunisada T, Kambayashi Y, Ogino K, Suzuki H et al.. c-Kit-targeting immunotherapy for hereditary melanoma in a mouse model. Cancer Res 2004; 64:801-6; PMID:14871802; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-2532 [DOI] [PubMed] [Google Scholar]
- 28.Rusmini M, Griseri P, Lantieri F, Matera I, Hudspeth KL, Roberto A, Mikulak J, Avanzini S, Rossi V, Mattioli G et al.. Induction of RET Dependent and Independent Pro-Inflammatory Programs in Human Peripheral Blood Mononuclear Cells from Hirschsprung Patients. PLoS One 2013; 8:e59066; PMID:23527089; http://dx.doi.org/ 10.1371/journal.pone.0059066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005; 23:447-85; PMID:15771578; http://dx.doi.org/ 10.1146/annurev.immunol.23.021704.115643 [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014; 27:1-7; PMID:24413387; http://dx.doi.org/ 10.1016/j.coi.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 31.Roychoudhuri R, Eil RL, Restifo NP. The interplay of effector and regulatory T cells in cancer. Curr Opin Immunol 2015; 33:101-11; PMID:25728990; http://dx.doi.org/ 10.1016/j.coi.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 32.Muhl H, Pfeilschifter J. Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int Immunopharmacol 2003; 3:1247-55; PMID:12890422; http://dx.doi.org/ 10.1016/S1567-5769(03)00131-0 [DOI] [PubMed] [Google Scholar]
- 33.Kelchtermans H, Billiau A, Matthys P. How interferon-gamma keeps autoimmune diseases in check. Trends Immunol 2008; 29:479-86; PMID:18775671; http://dx.doi.org/ 10.1016/j.it.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 34.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol 1997; 15:323-50; PMID:9143691; http://dx.doi.org/ 10.1146/annurev.immunol.15.1.323 [DOI] [PubMed] [Google Scholar]
- 35.Niederkorn JY. Emerging concepts in CD8+ T regulatory cells. Curr Opin Immunol 2008; 20:327-31; PMID:18406591; http://dx.doi.org/ 10.1016/j.coi.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parodi A., Battaglia FA, Kalli F, Ferrera F, Conteduca G, Tardito S, Stringara S, Ivaldi F, Negrini S, Borgonovo G et al.. CD39 is highly involved in mediating the suppression activity of tumor-infiltrating CD8+ T regulatory lymphocytes. Cancer Immunol Immunother 2013; 62:851-62; PMID:23359087; http://dx.doi.org/ 10.1007/s00262-013-1392-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu Rev Med 2015; 66:97-110; PMID:25341012; http://dx.doi.org/ 10.1146/annurev-med-051013-052304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol 2015; 32:21-7; PMID:25553393; http://dx.doi.org/ 10.1016/j.coi.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, Nishio J, Negishi H, Tamura T, Saijo S et al.. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. ELife 2014; 3:e04177; PMID:25149452; http://dx.doi.org/ 10.7554/eLife.04177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masuda Y, Inoue H, Ohta H, Miyake A, Konishi M, Nanba H. Oral administration of soluble β-glucans extracted from Grifola frondosa induces systemic antitumor immune response and decreases immunosuppression in tumor-bearing mice. Int J Cancer 2013; 133:108-19; PMID:23280601; http://dx.doi.org/ 10.1002/ijc.27999 [DOI] [PubMed] [Google Scholar]
- 41.Tian J, Ma J, Ma K, Guo H, Baidoo SE, Zhang Y, Yan J, Lu L, Xu H, Wang S. β-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells: Cellular immune response. Eur J Immunol 2013; 43:1220-30; PMID:23424024; http://dx.doi.org/ 10.1002/eji.201242841 [DOI] [PubMed] [Google Scholar]
- 42.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol 2009; 21:38-46; PMID:19230638; http://dx.doi.org/ 10.1016/j.coi.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho CC, Liao WY, Wang CY, Lu YH, Huang HY, Chen HY, Chan WK, Chen HW, Yang PC. TREM-1 Expression in Tumor-associated Macrophages and Clinical Outcome in Lung Cancer. Am J Respir Crit Care Med 2008; 177:763-70; PMID:18096709; http://dx.doi.org/ 10.1164/rccm.200704-641OC [DOI] [PubMed] [Google Scholar]
- 44.Li J J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res 2012; 72:3977-86; PMID:22719066; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer 2006; 6:521-34; PMID:16794635; http://dx.doi.org/ 10.1038/nrc1910 [DOI] [PubMed] [Google Scholar]
- 46.Bogdan C. Nitric oxide and the immune response. Nat Immunol 2001; 2:907-16; PMID:11577346; http://dx.doi.org/ 10.1038/ni1001-907 [DOI] [PubMed] [Google Scholar]
- 47.Tian J, Ma J, Wang S, Yan J, Chen J, Tong J, Wu C, Liu Y, Ma B, Mao C et al.. Increased expression of mGITRL on D2SC/1 cells by particulate β-glucan impairs the suppressive effect of CD4+CD25+ regulatory T cells and enhances the effector T cell proliferation. Cell Immunol 2011; 270:183-7; PMID:21636079; http://dx.doi.org/ 10.1016/j.cellimm.2011.05.003 [DOI] [PubMed] [Google Scholar]
- 48.Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, Sher A. Dectin-1 Interaction with Mycobacterium tuberculosis Leads to Enhanced IL-12p40 Production by Splenic Dendritic Cells. J Immunol 2007; 179:3463-71; PMID:17785780; http://dx.doi.org/ 10.4049/jimmunol.179.6.3463 [DOI] [PubMed] [Google Scholar]
- 49.Simpson PB, Mistry MS, Maki RA, Yang W, Schwarz DA, Johnson EB, Lio FM, Alleva DG. Cutting Edge: Diabetes-Associated Quantitative Trait Locus, Idd4, Is Responsible for the IL-12p40 Overexpression Defect in Nonobese Diabetic (NOD) Mice. J Immunol 2003; 171:3333-7; PMID:14500624; http://dx.doi.org/ 10.4049/jimmunol.171.7.3333 [DOI] [PubMed] [Google Scholar]
- 50.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N et al.. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995; 81:641-50; PMID:7538909; http://dx.doi.org/ 10.1016/0092-8674(95)90085-3 [DOI] [PubMed] [Google Scholar]