

ABSTRACT
Several pattern recognition receptors including toll-like receptors and purinergic receptors are implicated in the anticancer immune response elicited by anthracyclines or oxaliplatin. Recently, formyl peptide receptor-1 (FPR1) has been involved in this response as well. FPR1 is required for the correct positioning of dendritic cells (DC) close to dying cancer cells. A genetic defect in FPR1 abrogates cross-presentation of tumor antigens by DC, thereby compromising therapy-elicited immunosurveillance.
KEYWORDS: Annexin A1, apoptosis, CTL, DAMPs, immunogenic cell death
Abbreviations
- ANXA1
annexin A1
- CALR
calreticulin
- DAMPS
damage-associated molecular pattern molecules
- DC
dendritic cell
- FPR1
formyl peptide receptor 1
- HMGB1
high mobility group box 1
- TLR
toll like receptor
Although it has been commonly conceived that chemotherapy can treat (and occasionally cure) cancer because its direct cytostatic and cytotoxic action on malignant cells, ever-more convincing evidences indicate that successful chemotherapies must stimulate an anticancer immune response to mediate long-term effects on patient survival.1-3 This may be achieved by two principal, mutually non-exclusive, mechanisms. First, chemotherapeutic agents may favor the immune response by direct effects on a diverse array of leukocyte subsets, for instance by eliminating or inhibiting myeloid-derived suppressor cells, M2 macrophages or regulatory T cells.2 Second, some chemotherapeutics (such as anthracyclines and oxaliplatin) possess the capacity to stress and kill cancer cells in a way that their agony is perceived as ‘dangerous’ and hence favors a cascade of events that leads to a long-term adaptive immune response against tumor-associated antigens: the attraction of myeloid cells including DC precursors into the tumor bed, the juxtaposition of differentiating DC to dying cells, the uptake of corpse material by DC, and the final cross-presentation of tumor antigens by DC to cytotoxic T lymphocytes, which are recruited into the tumor bed as well.4-6 At least in mouse models, the tumor growth-inhibiting immune response, induced by chemotherapy, can occur in the absence of draining lymph nodes,7 suggesting that all elements required for such a response may act in the tumor microenvironment, which after chemotherapy is converted into a fully functional lymphoid-like organ.
Over the past decade, a number of stress pathways, damage-associated molecular pattern molecules (DAMPs) and pattern recognition receptors have been identified as crucial players in the immune response triggered by anticancer therapy.2,8 For example, autophagy favors the lysosomal secretion of ATP, causing extracellular ATP to act on two different purinergic receptors, first, purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) for the recruitment of myeloid cells into the tumor microenvironment and, second, purinergic receptor P2X, ligand-gated ion channel, 7 (P2RX7) for the stimulation of the NLR family, pyrin domain containing three (NLRP3) inflammasome and consequent interleukin-1β secretion by DC. Endoplasmic reticulum stress culminates in the exposure of calreticulin (CALR) on the outer surface of the plasma membrane, and CALR then interacts with receptors (such as CD91) on the surface of DC, facilitating the phagocytic uptake of portions of the dying cancer cells. High mobility group box 1 (HMGB1) leaking out from the nuclei of dead cells can interact with toll-like receptor (TLR) four on DC, hence stimulating tumor antigen presentation. In addition, tumor cells recognize double-stranded RNA via TLR3 to mount a type-1 interferon (IFN) response, leading to autocrine and paracrine signaling via the type 1 interferon receptor (IFNAR), as well as to the release of the chemokine (C-X-C motif) ligand 10 (CXCL10), which favors the recruitment of effector CXCR3+ T cells into the tumor bed (Fig. 1).2,8,9
Figure 1.
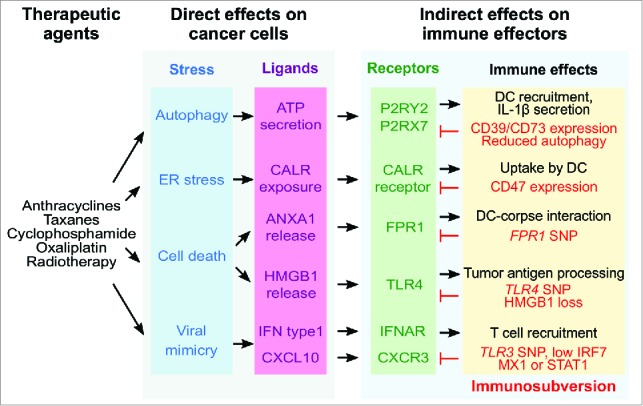
Pattern recognition receptors in the anticancer immune response elicited by immunogenic chemotherapy. Immunogenic cell death inducers act on cancer cells to stimulate a series of stress and death pathways including the toll-like receptor-3 (TLR3)-dependent induction of a type 1 interferon response. In this context, TLR3 is acting at the level of the malignant cells. Ligands released from stressed/dying malignant cells or exposed on their surface then add on a series of receptors on the surface of DC and their precursors, thus stimulating a cascade of events that is indispensable for stimulating an anticancer immune response. For example, annexin A1 (ANXA1) liberated from malignant cells can stimulate formyl peptide receptor-1 (FPR1) on DC to stimulate their positioning close to, and interaction with, dead cancer cells. A series of aberrations (labeled in red) has been shown to negatively affect breast cancer patients, likely because they compromise natural or therapy-induced immunosurveillance. Abbreviations: ANXA1, annexin A1; CALR, calreticulin; CXCL10, chemokine (C-X-C motif) ligand 10; CXCR3, chemokine (C-X-C motif) receptor 3; DC, dendritic cell; ER, endoplasmic reticulum; FPR1, formyl peptide receptor 1; HMGB1, high mobility group box 1; IFN, interferon; IFNAR, type 1 interferon receptor; IL, interleukin; IRF7, interferon regulatory factor 7; MX1, MX dynamin-like GTPase 1; P2RX7, purinergic receptor P2X, ligand-gated ion channel, 7; P2RY2, purinergic receptor P2Y, G-protein coupled, 2; SNP, single nucleotide polymorphism; STAT1, signal transducer and activator of transcription 1; TLR, toll like receptor.
Recently, we found yet another player in the intratumoral anticancer immune response. We observed that mice lacking FPR1 are unable to mount an immune response against dying cancer cells.10 Chemotherapy with anthracyclines failed to reduce tumor growth in such mice, correlating with a reduced or absent anticancer T cell response. Within the tumor bed, post-chemotherapy, FPR1 is mostly expressed by DC progenitor CD11b+Ly6ChiCD11c+ cells and neutrophil granulocytes.10 Given that the latter are dispensable for chemotherapeutic anticancer immune responses, while the former are essential,7 we focused on the phenotypic characterization of FPR1-deficient DC. Such DC failed to elongate within the tumor bed post-chemotherapy, suggesting a migration defect, and actually mislocalized within the tumor bed. Whereas normal DCs were systematically located in closer proximity of corpses (activated Caspase 3+) than to live cancer cells, FPR1-negative DC failed to manifest such a preference for interactions with dead cells. Experiments performed in a microfluidic device, designed to study the interaction between anthracycline-killed cancer cells and leukocytes allowed to differentiate into DC in situ, confirmed that FPR1 is required for immune cells to migrate toward dead cells and to engage in stable interactions with them.10
FPR1 received its name due to its capacity to interact with formyl peptides from bacteria and mitochondria. However, the deletion of (i) cathepsin G (Ctsg), (ii) family with sequence similarity 19 (chemokine (C-C motif)-like), member A4 (Fam19a4) or (iii) mitochondrial methionyl-tRNA formyltransferase (Mtfmt), an enzyme that catalyzes formylation reactions in mitochondria failed to interfere with the capacity of dying cancer cells to stimulate anticancer immune responses. In contrast, knockout of the gene coding for another known ligand of Fpr1, annexin A1 (Anxa1) abolished the capacity of dying cancer cells to do so. Moreover, ANXA1-negative cancers became refractory to tumor growth inhibition by treatment with anthracyclines, phenocopying the failure of chemotherapy responses observed for ANXA1-positive cancers growing in hosts lacking Fpr1. Cancer cells, treated with chemotherapy in vitro release ANXA1, which normally is contained in their cytosol, into the supernatant.10 The exact mechanisms, accounting for ANXA1 release, remain to be elucidated. Nonetheless, it appears plausible that ANXA1 is the DAMP that activates FPR1 in the context of chemotherapy (Fig. 1).
There are numerous clinical observations suggesting that the aforementioned pathway of immunogenic cell death recognition is clinically important. If we focus on breast cancer, which is commonly treated with anthracyclines, it has been shown that enhanced expression of CD47 by cancer cells (that can antagonize CALR because it serves as a ‘don't eat-me’ signal) or that of CD39 and CD73 (which act as ecto-enzymes to degrade ATP) has a negative prognostic impact. Similarly, lack of autophagy (which reduces ATP release) or HMGB1 expression correlate with poor survival in breast cancer patients treated with adjuvant chemotherapy. Low expression of interferon regulatory factor 7 (IFR7), MX dynamin-like GTPase 1 (MX1) or signal transducer and activator of transcription 1 (STAT1), which indicate the absence of type 1 IFN response also indicate poor prognosis.8 Finally, breast cancer patients bearing loss-of-function mutations in TLR3, TLR4 or FPR1 exhibit a shorter metastasis-free and overall survival than patients bearing functional variants of these pattern recognition receptors.8,10 Importantly, TLR3 and TLR4 are epistatic to FPR1, indicating that all these receptors act on a similar, presumable immune-related pathway.10 Hence, clinical evidence supports the idea that FPR1 contributes to anticancer immunosurveillance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
G.K. and L.Z. are supported by the Ligue Nationale contre le Cancer (Equipes labelisées), Sites de Recherche Intégrées sur le Cancer (SIRIC) Socrates and Carpem, the ISREC Foundation, Agence Nationale pour la Recherche (AUTOPH, Emergence), Cancéropôle Ile-de-France, European Commission (ArtForce), European Research Council Advanced Investigator Grant (to G.K.), Fondation pour la Recherche Médicale, Fondation de France, the LabEx Immuno-Oncology, Institut National du Cancer (INCa), and the Paris Alliance of Cancer Research Institutes. Y.M. is supported by the LabEx Immuno-Oncologie and the Chinese National Thousand Talents Program.
References
- 1.Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M et al.. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012; 337:1678-1684; PMID:23019653; http://dx.doi.org/ 10.1126/science.1224922 [DOI] [PubMed] [Google Scholar]
- 2.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013; 39:74-88; PMID:23890065; http://dx.doi.org/ 10.1016/j.immuni.2013.06.014 [DOI] [PubMed] [Google Scholar]
- 3.Stoll G, Enot D, Mlecnik B, Galon J, Zitvogel L, Kroemer G. Immune-related gene signatures predict the outcome of neoadjuvant chemotherapy. Oncoimmunology 2014; 3:e27884; PMID:24790795; http://dx.doi.org/ 10.4161/onci.27884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N et al.. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3:e955691; PMID:25941621; http://dx.doi.org/ 10.4161/21624011.2014.955691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, Baracco EE, Galluzzi L, Zitvogel L, Kepp O et al.. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014; 3:e28473; PMID:25050214; http://dx.doi.org/ 10.4161/onci.28473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vacchelli E, Aranda F, Eggermont A, Galon J, Sautès-Fridman C, Cremer I, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014; 3:e27878; PMID:24800173; http://dx.doi.org/ 10.4161/onci.27878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K et al.. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013; 38:729-741; PMID:23562161; http://dx.doi.org/ 10.1016/j.immuni.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 8.Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nature Medicine 2015; 21(10):1128-38; PMID:26444637; http://dx.doi.org/ 10.1038/nm.3944 [DOI] [PubMed] [Google Scholar]
- 9.Michaud M, Xie X, Bravo-San Pedro JM, Zitvogel L, White E, Kroemer G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology 2014; 3:e944047; PMID:25610726; http://dx.doi.org/ 10.4161/21624011.2014.944047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M et al.. Chemotherapy-induced anti-tumor immunity requires formyl peptide receptor 1 Science 2015; PMID:26516201; http://dx.doi.org/ 10.1126/science.aad0779 [DOI] [PubMed] [Google Scholar]