

Abstract
The nmf193 mutant was generated by a large-scale ENU mutagenesis screen and originally described as having a dominantly inherited phenotype characterized by fundus abnormalities. We determined that nmf193 mice exhibit outer segment defects and progressive retinal degeneration. Clinical examination revealed retinal spotting apparent at 6 weeks of age. Histological analysis of homozygous mutant mice at 6 weeks indicated an absence of outer segments (OS) and a 50% reduction of photoreceptor cells which progressed to complete loss of photoreceptors by 10 months. Mice heterozygous for the nmf193 mutation had a less severe phenotype of shortened outer segments at 2 months with progressive loss of photoreceptor cells to 50% by 10 months. A positional cloning approach using a DNA pooling strategy was performed to identify the causative mutation in nmf193 mice. The nmf193 mutation linked to chromosome 17 and fine mapped to an interval containing the peripherin/rds (Prph2) gene. Mutation analysis identified a single base change in Prph2 that causes aberrant splicing between exon 1 and 2. Interestingly, a comparative histological analysis demonstrated that Prph2nmf193/+ mutants have similar photoreceptor degeneration to that of Nr2e3rd7/rd7. We show that Prph2 mRNA and protein levels are reduced in the Nr2e3rd7/rd7 mutant compared to control littermates. Further, chromatin immunoprecipitation analysis shows that Prph2 is a direct target of NR2E3. In addition, the down-regulation of Prph2 gene expression is similar in both the Nr2e3rd7/rd7 and Prph2nmf193/+ mutants suggesting that the reduction of Prph2 may contribute to the degenerative pathology seen in Nr2e3rd7/rd7.
Introduction
ENU mutagenesis screens in model organisms have yielded a wealth of information on gene function with particular relevance to disease. Several groups have performed large-scale mutagenesis screens using the mouse (Clark et al., 2004), including those focused on generating novel models of retinal disease (Baird et al., 2002; Jablonski et al., 2005; Pinto et al., 2004). The main advantage of this phenotype driven approach is that mutagenesis is random, thus allowing for the generation of an allelic series of mutations as well as mutations in genes that currently have no functional information or disease association (Justice et al., 1999). Allelic mutations, such as the one described in this study, provide valuable insight into protein function. Further, it becomes possible to generate a large number of unique single gene mutants with a particular phenotype, such as progressive retinal degeneration, and identify the causative mutations. The identification of these mutations associated with a group of mutants with similar phenotypic manifestations, ultimately provides a more complete understanding of pathological mechanisms that may be shared between genetically distinct diseases.
In this study, we aimed to identify the causative mutation in the novel mouse mutant nmf193. This mutant was originally described by the Neuroscience Mutagenesis Facility at the Jackson Laboratory as a dominant phenotype characterized by an abnormal fundus appearance by 8 weeks of age. We show that this phenotype is semi-dominant and has a slow progressive degeneration of the retina. We report the genetic mapping of nmf193 and identification of a causative mutation in the peripherin/rds (Prph2) gene. Prph2 is a tetraspanin protein that localizes the to the rims of the disc membranes (Travis et al., 1991). Prph2 is involved in the formation of outer segments and the maintenance of their morphology (Wrigley et al., 2000; Loewen et al., 2001). Additionally, Prph2 has been reported to promote fusion of opposing disk membranes in vitro (Boesze-Battaglia et al., 1998).
Prph2 was identified as the gene mutated in the retinal degeneration slow (rds) mutant (Travis et al., 1989; Connell et al., 1991). While the original rds mutant is due to an insertion that causes a null allele (Ma et al., 1995), subsequent mouse transgenic models have demonstrated that both dominant negative and haploinsufficient alleles cause disease, depending on the mutation that the transgene carries. Transgenic animals carrying common human mutations, R172W or P216L, exhibit reduced function of the photoreceptor on a wild-type background, which indicates a dominant negative effect of the allele (Kedzierski et al., 1997; Ding et al., 2004). The del307 mutation has a similar dominant negative effect, having a more severe phenotype then the null allele alone (McNally et al., 2002). In contrast, the C214S transgenic mutation does not affect the retina on a wild-type background or heterozygous mutant background, indicating that the mutation is a loss-of-function allele (Stricker et al., 2005). This is partly explained by the differential intracellular localization of allelic variants in the photoreceptor. The loss of function allele, C214S is not transported to the outer segment, while the dominant negative allele, P216L, forms a tetramer and is transported (Loewen et al., 2003), thereby, allowing the mutant allele to interfere in the structural organization of the outer segments. Several mutations in human Prph2 have been associated with autosomal dominant retinitis pigmentosa (Farrar et al., 1991; Keen et al., 1996) and various patterned macular dystrophies (Wells et al., 1993, Nichols et al., 1993; Kim et al., 1995; Felbor et al., 1997). Significant variation of the disease is seen with different mutations (Kohl et al., 1998), and also within a family segregating the same mutation (Weleber et al., 1993), suggesting the existence of genetic modifiers.
In the nmf193 mutant, the haploinsufficiency of Prph2 causes a slow progressive degeneration of the photoreceptors, grossly similar to that of the Nr2e3rd7/rd7 mutant (Akhmedov et al., 2000; Haider et al., 2001). The Nr2e3rd7/rd7 mouse has a developmental phenotype characterized by an excess of blue cone cells and whorls and rosettes in the retina caused by a mutation in the transcription factor NR2E3. Nr2e3rd7/rd7 mice also exhibit a delay in the formation of outer segments (Haider et al., 2006), when outer segments do form they are shortened, similar to the haploinsufficient Prph2nmf193/+ mouse. NR2E3 is involved in the regulation of several genes (Chen et al., 2005; Cheng et al., 2004; Corbo and Cepko, 2005). Since reduced levels of Prph2 are sufficient for photoreceptor loss, if a mutation in Nr2e3 either directly or indirectly reduced Prph2 levels then it would be expected to cause degeneration over a similar time course. In this study, we show that Prph2 is downregulated in Nr2e3rd7/rd7 mutants at the mRNA and protein level at a postnatal time-point prior to the onset of degeneration and after the morphogenesis of the outer segments should be complete. Further, chromatin immunoprecipitation data shows that NR2E3 binds to a recognition site upstream of Prph2. These data suggest that Prph2 is involved in the pathology of Nr2e3rd7/rd7, which is supportive of a common degenerative pathway.
Experimental Methods
Animal husbandry
All animals were bred and maintained under standard conditions at the UNMC research vivarium in accordance with IACUC standards. A mating cross of CAST/EiJ × C57BL6/Jnmf193/+ was established to generate F1 offspring. Affected offspring were backcrossed to CAST/EiJ to generate progeny for mapping. Mice were maintained under constant environmental conditions to ensure the disease course progressed naturally. Tissues were harvested from B6.Cg-Nr2e3rd7/rd7, C57BL/6J (B6), Prph2nmf193/+, Prph2nmf193/nmf193 at 6 weeks, 3 months, 6 months, and 10 months. A minimum of three mice were analyzed for each time point. Mice were genotyped for the Nr2e3rd7/rd7 mutation as previously described (Haider et al., 2001).
Fundus photography
Pupils were dilated with 1% atropine sulfate (Alcon, Forth Worth, TX) in a dark room. Fundus images were captured on a Kowa Genesis digital fundus camera (Kowa Company Ltd., Japan). A 90mm diopter lens (Volk, Mentor, OH) was held in place under the camera lens so that it filled the field of view (Hawes et al., 1999). Obligate wild-type, heterozygous and homozygous mutant mice, were imaged at 1.5, 3 and 9 months; n>3 for each genotype and age. Adult Nr2e3rd7/rd7 mice were phenotyped by indirect ophthalmoscopy for the characteristic panretinal spots prior to additional analysis.
Electroretinography (ERG)
ERG analysis was performed on wild-type, heterozygous and homozygous mutant mice at 2 and 10 months. Mice were dark adapted overnight and anesthetized with an intraperitoneal injection of normal saline solution containing ketamine (15 mg/g) and xylazine (7 mg/g body weight). ERG recordings were performed using the UTAS E4000 system (LKC Technologies Inc, Gaithersburg, MD). Pupils were dilated with 1% atropine sulfate and a silver coated nylon thread electrode was placed at the corneal surface of one eye for recording. A needle electrode placed under the skin of the forehead served as ground. A drop of 1% sodium carboxymethylcellulose was placed on the corneal surface in conjugation with a contact lens to ensure electrical contact and to maintain corneal integrity. Body temperature was maintained at a constant temperature of 38 °C using a Themipaq heating pad. All stimuli were presented in a Ganzfeld chamber/light emitter (LKC Technologies, Gaithersburg, MD) whose interior surface was painted with a highly reflective white matte paint (No. 6080; Eastman Kodak, Rochester, NY). Stimuli were generated with a Sunburst UTAS Visual Diagnostic system (LKC Technologies). Dark adapted responses were recorded to short wavelength (lmax = 470 nm; Wratten 47A filter) flashes of light over a 4.0 log unit range of intensities (0.3 log unit steps) up to the maximum allowable by the photic stimulator. Light adapted responses were obtained with white flashes (0.3 steps) on a rod saturated background after 10 min of exposure to the background light to allow complete light adaptation. Responses were amplified with an UBA-4200 series Universal DC Biomedical Amplifier (LKC Technologies) and digitized using an I/O board (model PCI-1200; National Instruments, Austin, TX) in a personal computer. Signal processing was performed with EM for Windows version 7.1.2. Signals were sampled every 0.8 ms over a response window of 200 ms. For each stimulus condition, responses were computer averaged with up to 50 records for the weakest signals. A signal rejection window could be adjusted during data collection to eliminate electrical artifacts.
Histological analysis
Eyes were collected from wild-type, homozygote and heterozygote mutants at 1.5, 6 and 10 months (n>3/sample and age). Tissues were marked with a cautery prior to enucleation to designate dorsal ventral orientation. Tissue was collected from age matched littermates and eyes were fixed in 3:1 methanol:acetic acid or 4% paraformaldehyde solution overnight, embedded in paraffin and sectioned at 5 μm. Sections covering an area of 200 microns, approximately 100 microns dorsal to the optic nerve head, were collected on vectabond coated slides. Hematoxylin and eosin staining and TUNEL analysis was performed as before (Haider et al., 2001; Haider et al. 2006). TUNEL positive cells were counted across the retina on sections totaling 100 microns from the central retina in 3 mice from each mutant. Sections were visualized on a Ziess AxioPlan 2 (Carl Ziess MicroImaging, Inc., Thornwood, NY) and imaged with an AxioCam C using Axiovision 4.3 software. Counts of existing photoreceptor nuclei were made at 10 months in Nr2e3rd7/rd7, Prph2nmf193/+ and control retinas (n=3) from hematoxylin and eosin stained sections. Two 50 micron fields were counted 400 microns on either side of the optic nerve head. The average number of photoreceptor nuclei were compared to wild-type to determine the percentage of lost photoreceptors.
Genetic mapping
DNA was isolated from tail tips excised from two week old pups. Genotyping for linkage analysis was performed using a screening set of 67 STRP markers. Equimolar amounts of DNA from 7 unaffected and 10 affected mice were pooled in two separate tubes. DNA was amplified by PCR using the following conditions: 40ng of DNA in an 10μl PCR reaction mixture containing 1.25 μl PCR buffer (100 mM Tris-HCl [pH 8.8], 500 mM KCl, 15 mM MgCl2, and 0.01% w/v gelatin), 200mM each dATP, dCTP, dGTP, and dTTP, 2.5 pmol of each forward and reverse primer and 0.25 U Taq polymerase. Reaction mixtures were subjected to 40 cycles of 94°C for 30s, 55°C for 30s and 72°C for 30s. Products were electrophoresed on a 4% agarose gel containing ethidium bromide, and visualized by UV transillumination.
Mutation analysis
Mutation analysis was performed by directly sequencing genomic DNA using primers that were designed flanking the exons of Prph2. DNA was amplified as above. RT-PCR was performed using primers specific to the Prph2 mRNA using the Titan-one RT-PCR kit (Roche, Basel, Switzerland) per manufacturer’s instructions. RNA was isolated from mutant and wild-type retinas as before (Haider et al., 2006). The appropriate bands were excised and DNA was recovered using the Qiagen Gel Extraction Kit (Qiagen Inc., Valencia, CA) per manufacturer’s instructions. Purified PCR and RT-PCR products were sequenced by the DNA Sequencing Core at UNMC. Nucleotide and amino acid sequences were analyzed using Sequencher 4.6 (Gene Codes Corp., Ann Arbor, MI).
Western blot analysis
Western blot analysis was performed using 20μg of protein per sample as before (Sachs et al., 2007). Briefly, control, Prph2nmf193/+, Prph2nmf193/nmf193 mutant retinas were dissected from 1.5 month old mice and lysed in RIPA buffer (1X Tris buffered saline (TBS), 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.04% sodium azide, 1mM PMSF). Samples were electrophoresed on 4–12% Tris-Bis NuPage gels (Invitrogen Corp., Carlsbad, CA). Proteins were transferred onto PVDF membranes and western blot analysis was performed using the Odyssey Infrared Imaging System (LiCOR Biosciences, Lincoln, NE) according to manufacturer’s recommendations. Blots were incubated in Odyssey blocking solution for one hour at room temperature. Primary antibody: anti-PRPH2 at 1:1000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or anti-b-actin at 1:1000 (Santa Cruz), was incubated in Odyssey blocking solution overnight at 4°C, then washed in PBS. Secondary antibody, anti-goat IgG was incubated at 1:20,000 for one hour on the blot at room temperature, and the blot washed in PBS (AlexaFluor680 (Invitrogen Corp.)). Images were detected with the Odyssey system using the proper infrared channels and visualized using Odyssey v.1.2 software.
Expression analysis
Real-time RT-PCR was used to determine relative expression levels of Prph2 in Prph2nmf193/+, Prph2nmf193/nmf193 and control retinas, as previously described (Haider et al., 2006). Total RNA was isolated from retinas of P14 (n=3) mice. Primers spanning exons 1 and 2 of Prph2 were selected to amplify the wild-type message only (F, 5′-AGGTCAAAGATCGCATCAAG-3′ and R, 5′-CTGTAGTAATTCAGCAGAGC-3′) and total message, wild-type and mutant (F, 5′-TCCCTCCCACAAAGCACTATG-3′ and R, 5′-TGGTCTCAGATGGCCACAAC-3′). Reaction products were diluted 1 to 100 and 1μl was used as template for amplification using the following 10μl reaction mixture: 100 pmol each forward and reverse primer and 5μl Sybr green PCR master mix (Applied Biosystems, Foster City, CA). PCR was carried out on an ABI 7500 using the manufacturer’s recommended cycling parameters. Biological and technical triplicate reactions were performed. Expression of Prph2 was normalized to that of β-actin. Significance was calculated using student’s T test. Comparisons between mutant and wild-type controls were made for fold change estimation and relative expression using the following formula: 1000/2ΔCtbactin-ΔCttestgene. For comparison of Prph2 message level between Nr2e3rd7/rd7 and wild-type controls, the above protocol was repeated with P21 retinas (n=7).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using reagents from the ChIP kit (Upstate, Millipore) and a modified protocol. Briefly, retinas were dissected from P2 or P21 C57BL/6J eyes (8–10 eyes per ChIP; three independent assays per time point) and placed into 400 μl of PI buffer (1 tablet protease inhibitor cocktail, Roche) in 10ml phosphate buffered saline (PBS) on ice. Chilled retinas were dissociated and crosslinked in 37% formaldehyde (1% final concentration) for 20 minutes at room temperature on a rotating platform. One molar glycine to achieve a final concentration of 0.125mM and retinas were incubated for an additional 5 minutes and rinsed twice with PI buffer before lysis. Retinas were lysed on ice and sonicated on ice using a Misonix Sonicator 3000 (power 5, pulse 10×, 30 second pause between pulses; 150 total pulses). Retina lysates were pre-cleared using a salmon sperm DNA/protein A Agarose-50% slurry and immunoprecipitation was performed overnight with approximately one μg of NR2E3 antibody (Haider et al. 2006) at 4°C with rotation. Immunoprecipitated samples were eluted with 1%SDS, 0.1M NaHCO3 buffer and reverse cross-linked with NaCl (200 mM final concentration) and incubated with 10 mg RNAse A at 65°C for five hours. Input (control) samples were isolated similarly and incubated with pre-clear solution instead of antibody or with goat IgG antibody (negative control). PCR reactions were performed using 1 μl of 1:10 diluted Input or Experimental (NR2E3 precipitated) sample under standard PCR conditions as previously described (Haider et al. 2001) using an annealing temperature of 58°C and 35 cycles. PCR reactions were electrophoresed in a 2% agarose gel and visualized by ethidium bromide staining. Forward (GCTACACACTTCCTTGTTAAG) and reverse (CTGACCTGCTAGCACTTTGTTC) primers were chosen approximately 100 nucleotides 5′ and 3′, respectively, from a putative response element sequence to amplify approximately 200 base pair products (bp).
Immunohistochemistry
Immunohistochemical analysis was performed on 6 μm serial sections of B6 and Nr2e3rd7/rd7 eyes. Paraffin embedded eyes were fixed with either 4% PFA, Methanol/Acetic acid (3:1), or Z-fix (Anatech Ltd) and maintained at a dorsal/ventral orientation. Frozen sections were prepared from eyes enucleated and immediately placed in 30% sucrose overnight, followed by immersion in a 50:50 mixture of 30% sucrose and OCT for 8–12 hours. Specimens were subsequently embedded in OCT and sectioned on a cryostat. After blocking with 2% normal goat or horse serum (Vector, CA) in PBS, sections were incubated with primary PRPH2 antibody (goat, 1:200, Santa Cruz Biotechnology) at 4°C overnight. The next day, samples were rinsed and incubated in secondary antibody (anti-goat 488 AlexaFluor antibody by Molecular Probes, Invitrogen). Images from sections were collected on a Zeiss Axioplan II fluorescent microscope equipped with the appropriate bandpass filters for each flurochrome.
Results
Nmf193 maps to chromosome 17
A mapping cross was generated by backcrossing affected offspring from a C57BL6/Jnmf193/+ × CAST/EiJ cross to CAST/EiJ. Linkage was identified using a genome wide screen on pooled DNA samples. A single marker, D17Mit200, was identified as homozygous for the CAST/EiJ allele in the unaffected pool (Figure 1A,B). Individual DNA samples were genotyped with this marker and additional flanking markers. Linkage was observed with D17Mit200 and D17Mit28, while flanking markers, D17Mit46 and D17Mit86, had recombinant animals (Figure 1C). This narrowed the disease interval to approximately 19cM. This region is relatively large, however, upon examination of the genes within the interval, a strong candidate gene, Prph2, was found near the linked marker D17Mit200.
Figure 1. A genome wide screen using pooled samples shows that D17Mit200 is linked to the nmf193 phenotype.
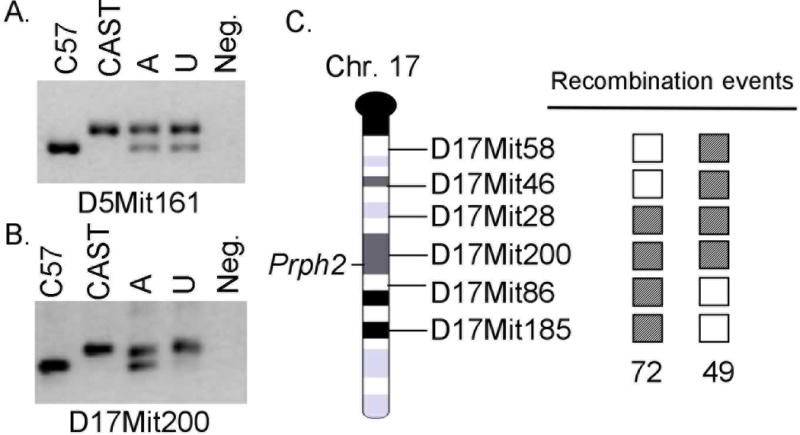
Panel A shows a typical unlinked marker, C57BL6/J and CAST/EiJ DNA were used as controls, the affected (A) and unaffected (U) pools show both alleles. In the linked marker (panel B) the unaffected pool does not have the C57BL6/J allele. Genotyping of individual samples identified two affected recombinant mice (panel C). Shaded box indicates a heterozygous genotype, and an open box is homozygous for the CAST/EiJ allele.
A point mutation causing aberrant splicing of Prph2 mRNA results in the nmf193 retinal degeneration
The Prph2 gene was screened for mutations by directly sequencing PCR products. Genomic DNA fragments were PCR amplified using primers flanking exons to amplify the coding region and splice sites of the gene. No coding region mutations were identified; however a T to A mutation was identified in the invariant T of the exon 1 splice donor (Figure 2). The Prph2 mRNA was amplified by RT-PCR. In the RNA from the mutant retina an aberrant sized transcript was identified indicating the use of an inappropriate downstream splice donor (Figure 3A). Further, using primers that were designed to span the exon 1 and 2 junction, and therefore amplify only wild-type message, a 2.8 fold reduction in the level of Prph2 mRNA was detected by quantitative RT-PCR from Prph2193/+ mutants (Figure 3B). In the homozygous mutants no wild-type message is detected and mutant message is present, but at 2.9 fold lower levels. The mutant and wild-type product were sequenced and the same T to A splice site mutation was identified in mutant transcripts causing the incorporation of 25 nucleotides (Figure 2D). The message then splices to the normal splice acceptor in exon 2 from a cryptic splice donor site. This mutation causes the incorporation of 49 missense amino acids before prematurely terminating the protein. Approximately, one half of the tetraspanin domain is altered or missing (Figure 4A). Western blot analysis showed that the wild-type protein is present in heterozygotes but absent from homozygotes, a truncated protein is not detected (Figure 4B). Consistent with quantitative RT-PCR results, the quantity of wild-type protein is visibly reduced in the heterozygote.
Figure 2. A splice site mutation that changes the invariant T in the splice donor to an A was identified in intron 1 of the Prph2 gene.
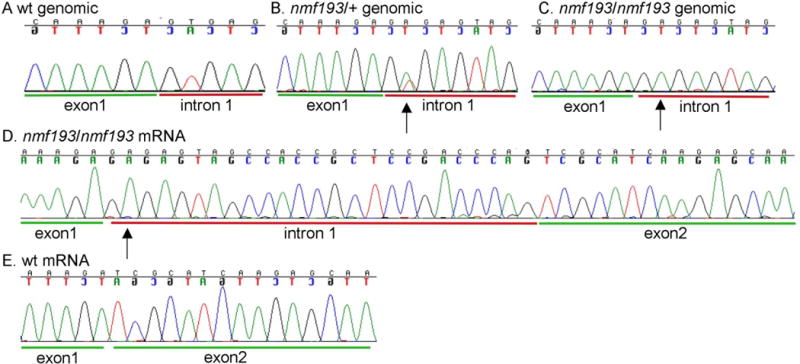
Panels A–C show sequence chromatograms from wild-type, heterozygous and homozygous mutant DNA, respectively. The mutant causes the use of a cryptic splice site downstream (panel D) and incorporates 25 extra base pairs into the mRNA, this is not detected in wild-type C57BL6/J mice (panel E).
Figure 3. The mutation causes a reduction of wild-type messages in mutant animals.
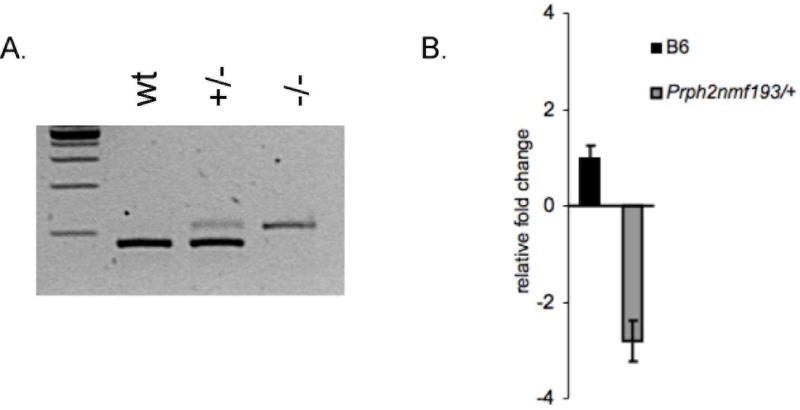
Panel A shows RT-PCR of RNA from C57BL/6J (wt), heterozygous (+/−) and homozygous mutants (−/−). The 25bp insertion into the message is seen at albeit reduced levels. Panel B depicts relative expression of Prph2 using a primer that detects only wild-type message in a quantitative RT-PCR assay. Expression is given as number of molecular per 1000 molecules of β-actin, genotype is indicated on the x-axis.
Figure 4. The mutation causes a frameshift and a nonsense codon that truncates the protein (panel A).

Shaded areas indicate the region of the protein that is equivalent to wild-type, the red underlined sequence indicates the tetraspanin domain. Western blot analysis (panel B) shows the reduction of wild-type protein in the heterozygous mutant (+/−) and its absence in the homozygous mutant (−/−), β-actin is shown as a loading control.
nmf193 mutant mice exhibit fundus spotting and hypopigmentation
In order to determine the pathophysiology of retinal degeneration in nmf193 mice, we tracked the clinical progression of the disease by indirect ophthalmoscopy. Wild-type, Prph2nmf193/+, and Prph2nmf193/nmf193 mice were evaluated for fundus abnormalities at 4–6 weeks, 3 months and 9 months. Prph2nmf193/nmf193 mice have severe degeneration characterized by large regions of hypopigmentation apparent at eye opening (data not shown) but progress to span the majority of the retina by three months (Figure 5B). In contrast, Prph2nmf193/+ mice show a progressive deterioration (Figure 5 C–E). Prph2nmf193/+ did not show fundal abnormalities until 1.5 months of age when a few light retinal spots were observed (Figure 5C). Interestingly, one week prior, these spots were not observed in the same animal, suggesting a key transition period in the retina at 4–5 weeks. The spots have an increased density at 3 months and attenuated blood vessels were commonly observed by this time (Figure 5D). At 9 months the fundus is severely hypopigmented, similar to a 3 month Prph2nmf193/nmf193 retina, and only remnants of blood vessels can be found (Figure 5E).
Figure 5. Abnormal appearance of the nmf193 fundus.
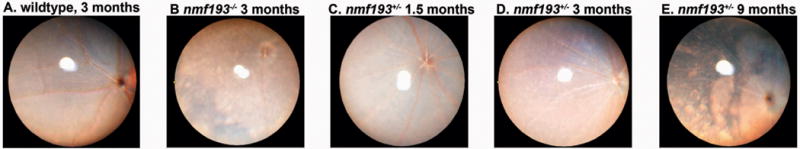
Fundus images of heterozygous mutants were taken at 1.5 (panel C), 3 (panel D), and 9 months (panel E). A 3 month old homozygous mutant (panel B) and wild-type control are shown for comparison (panel A).
Outer segment defects accompany photoreceptor loss in nmf193 mutant mice
To determine the morphological changes associated with the nmf193 mutation, retinal sections from wild-type, Prph2nmf193/+ and Prph2nmf193/nmf193 mutants were histologically examined by hematoxylin and eosin staining. Six week old heterozygous mutant mice had a normal retina with the exception that the outer segments are shorter in comparison to wild-type (Figure 6). By 6 months of age, Prph2nmf193/+ mutant mice had approximately 70% reduction of photoreceptor cells (outer nuclear layer, ONL) and many displaced nuclei were observed in the outer segments (OS). At 10 months of age the ONL was reduced to approximately 4 cells. Retinal degeneration is more pronounced in Prph2nmf193/nmf193 mutant mice where the ONL at one month is considerably thinner, similar to the 10 month old Prph2nmf193/+ mutants, with absent outer segments. Further degeneration was seen in Prph2nmf193/nmf193 mutants at 6 months (ONL is 2 cells thick) and by 10 months essentially no outer nuclear layer or outer segments were observed.
Figure 6. Histological analysis shows degeneration of the photoreceptors in mutant mice.
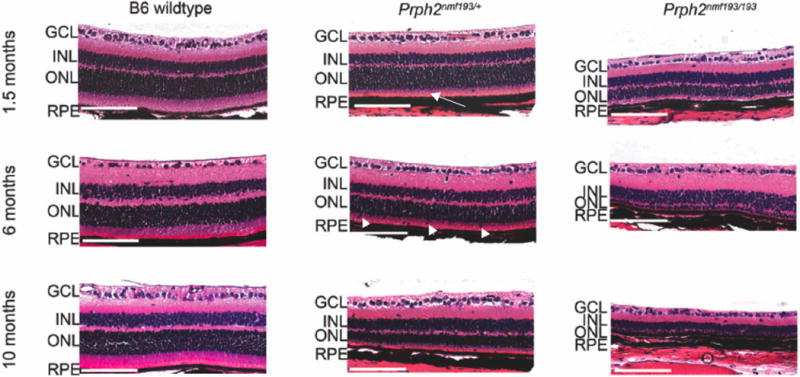
Wild-type, heterozygous and homozygous mutant retinas are shown at the indicated time-points. Note the shortened outer segments (OS) in the mutants (arrow). Displaced nuclei are frequently seen in the OS of heterozygous mutants at 6 and 10 months (arrowhead). A marked reduction of the outer nuclear layer (ONL) in the heterozygote and its absence in the homozygous mutants is observed at 10 months. Scale bar indicates 100mm.
ERG analysis reveals loss of photoreceptor function in nmf193 mutant mice
To determine the effect of outer segment defects and loss of photoreceptor cells on rod and cone photoreceptor function, electroretinogram analyses were performed on Prph2nmf193/+ and Prph2nmf193/nmf193 mutants and controls at 2 and 10 months of age. Consistent with clinical and histological observations, Prph2nmf193/+ mutants have progressive loss of rod function and cone function and Prph2nmf193/nmf193 mutants exhibit a more severe phenotype (Figure 7). As observed for other mutants involving Prph2, rod degeneration precedes cone degeneration (Cheng et al., 1997; Farjo et al., 2006). Prph2nmf193/+ mice showed 68.4% rod function (dark adapted) and 84.6% cone function (light adapted) at 2 months compared to normal. By 10 months, Prph2nmf193/+ mutants had approximately 43.6% of normal rod function and 63.9% of normal cone function. In contrast, Prph2nmf193/nmf193 mutants show negligible rod function compared to normal, even at 2 months, while cone function was approximately 31.6% of normal at 2 months. A complete loss of rod and cone function was observed in homozygous mutants by 10 months.
Figure 7. ERG analysis shows a diminished response in mutant animals.
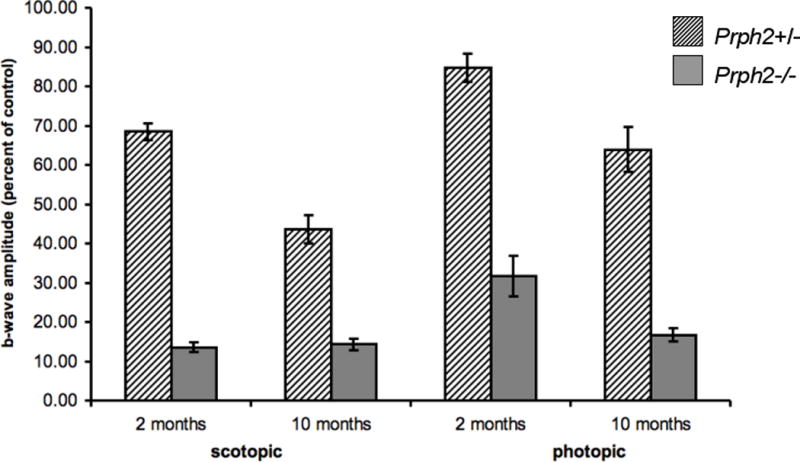
Heterozygous and homozygous mutants at 2 and 10 months were compared to age matched wild-type controls. Average b-wave amplitude from mice of each genotype is given as a percentage of wild-type control.
Similar degeneration observed in photoreceptor degeneration in Prph2nmf193/+ and Nr2e3rd7/rd7 mutants, direct targeting of Prph2 by Nr2e3
Prph2nmf193/+ and Nr2e3rd7/rd7 both have slow degeneration of the photoreceptor layer. To assess whether Prph2nmf193/+ and Nr2e3rd7/rd7 mice have a similar rate of retinal degeneration, 10 month old Prph2nmf193/+ and Nr2e3rd7/rd7 mice were histologically compared. A similar reduction in the ONL between Prph2nmf193/+ and Nr2e3rd7/rd7 mice was observed (Figure 8A,B). Remaining photoreceptor nuclei were counted in 50mm fields, 400mm on either side of the optic nerve head in both mutants compared to age-matched wild-type controls (n=3). A 32.6% reduction in photoreceptor nuclei was observed in Nr2e3rd7/rd7 mice, while Prph2nmf193/+ showed a 37.2% reduction. Both Prph2rds and Nr2e3rd7/rd7 mutant photoreceptors are known to undergo apoptosis (Chang et al., 1993; Haider et al., 2006). TUNEL analysis was used to identify apoptotic neurons in the ONL of 1.5 month and 10 month Prph2193/+ and Nr2e3rd7/rd7 mutant mice. Approximately 23 TUNEL positive neurons per 5μm retinal section were observed in both mutants indicating a comparable degeneration at 1.5 months old, while at 10 months an average of 5 TUNEL positive neurons were observed in the thinned ONL (Figure 8C–E).
Figure 8. Histological analysis shows similarities in the degeneration of the photoreceptor between Prph2nmf193/+ and Nr2e3rd7/rd7 mutants.
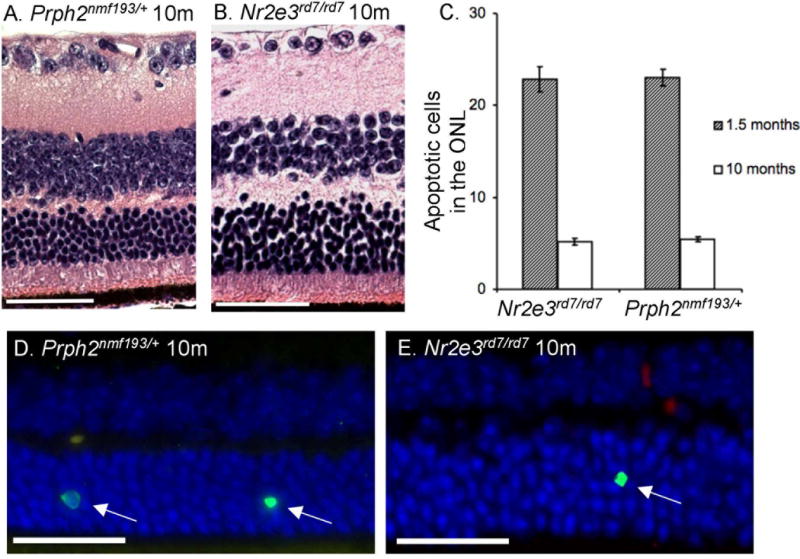
Hematoxylin and eosin staining shows outer nuclear layers reduced to 5–7 cells thick in the central retina of both mutants (panels A, B). Scale bar indicates 50 mm. TUNEL staining was used to identify apoptotic nuclei in the ONL per 5mm section in both mutants at 1.5 and 10 months (panels C–E). Wild-type controls showed negligible TUNEL staining at both time points. A total of 100 microns of sections were counted per mouse.
Reduced levels of Prph2 are sufficient for photoreceptor loss, therefore to examine the possibility that Prph2 is involved in the photoreceptor degeneration observed in Nr2e3rd7/rd7 mice the levels of Prph2 were measured at the mRNA and protein level in Nr2e3rd7/rd7 mutants relative to wild-type controls. Quantitative RT-PCR revealed 3.2 fold reduction (p=0.03) of Prph2 expression in Nr2e3rd7/rd7 mutants relative to wild-type controls (n=7) (Figure 9A). This reduction is far greater than that seen for expression of other rod genes such as rhodopsin, which is only reduced by 1–1.4 fold in Nr2e3rd7/rd7 mice (Corbo and Cepko 2005; Peng et al., 2005; Haider et al., 2006). examining approximately 30 kb of 5′ sequence in the Prph2 gene, we identified a single NR2E3 response element site and used chromatin immunoprecipitation to evaluate the ability of NR2E3 to bind this site in vivo. These data demonstrate that Prph2 gene expression is directly regulated by NR2E3 (Figure 9B,C). In addition, consistent with the message level, immunohistochemical analysis showed reduced expression of PRPH2 protein in NR2e3rd7/rd7 retinas compared to normal controls (Figure 9D,E).
Figure 9. Similar degeneration observed in photoreceptor degeneration in Prph2nmf193/+ and Nr2e3rd7/rd7 mutants, and direct targeting of Prph2 by Nr2e3.
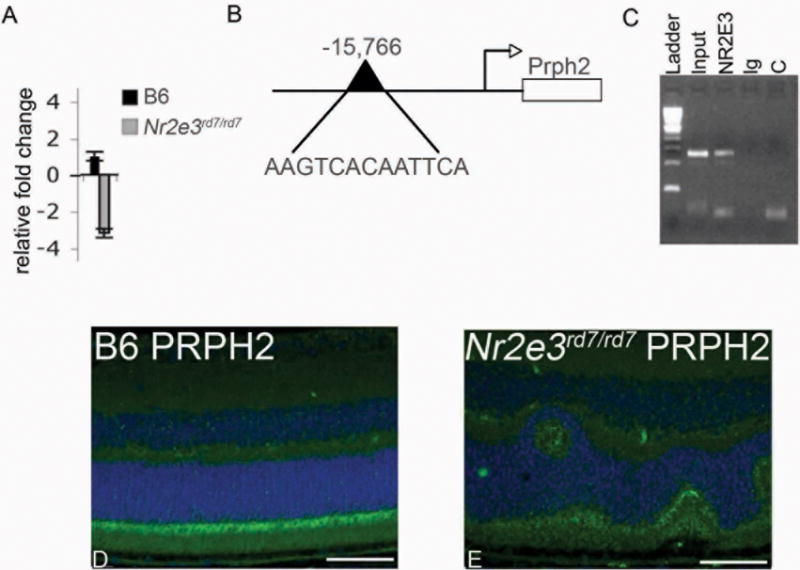
A. Quantitative real time PCR analysis showing 3.2 fold reduced expression of Prph2 in P21 Nr2e3rd7/rd7 relative to control. B. Schematic representation of Prph2 genomic sequence indicating location and sequence of NR2E3 binding response element (RE). C. Chromatin immunoprecipitation assay showing direct binding of NR2E3 to Prph2 RE sequence. L-ladder, I-input (no Antibody, positive control); NR2E3 (NR2E3 antibody precipitated sample); Ig (IgG antibody precipitated, negative control); C (no template negative control). D (B6 control), E (Nr2e3rd7/rd7) immunohistochemisty of P30 retina sections. Photoreceptor outer segments labeled with PRPH2 (green) and DAPI (blue nuclear stain) show reduced expression in Nr2e3rd7/rd7 retinas.
Discussion
In this report, we identified a novel mutation in the Prph2 gene that causes photoreceptor degeneration in the nmf193 mouse mutant. The temporal and spatial pattern of degeneration is interesting due to its similarity to another mouse mutant Nr2e3rd7/rd7. Nmf193 is inherited in a dominant fashion and linked to chromosome 17. Fine mapping localized the mutation to a region containing Prph2. The similarity between the nmf193 phenotype and that of rds both in the heterozygous and homozygous state indicated that Prph2 was a strong candidate gene. We identified a splice mutation altering the invariable T in the splice donor. Sequencing RNA confirmed the use of a cryptic splice site and a resulting frame shift. This mutation is a T:A transversion, which is the most common change induced by ENU seen in the mouse (Barbaric et al., 2007). This nucleotide change was not detected in the strain of origin. The mutation reduced mRNA levels significantly in the heterozygous mutant mouse, where mutant message is barely detectable, and wild-type message is estimated at slightly less than 50% of normal. This probably reflects an early onset of abnormalities in the retina, rather than an adverse effect on expression that the mutant allele has on the wild-type allele. The mutation has a drastic effect on the protein, significantly altering the second intradiskal loop and deleting the fourth transmembrane region and a known fusion domain (Boesze-Battaglia et al., 2000, Ritter et al., 2004). The second intradiskal loop is essential for the formation of homo- and heterocomplexes with ROM-1 (Ding et al., 2005). However, very low levels of mutant message are detected; therefore, this allele would likely result in a loss-of-function.
Prph2 is essential for the development and maintenance of photoreceptor outer segments. It appears that the protein is required for both rods and cones; however, the mutant protein affects the two photoreceptors differently. Electroretinography studies showed that haploinsufficiency of Prph2 has a greater disruptive effect on the rods than it does on cones (Cheng et al., 1997). Using the cone enriched Nrl−/− mutant, it was shown that cone outer segments are abnormal in a compound mutant; however, they are present and phototransduction competent, in contrast to rod outer segments (Boesze-Battaglia et al., 2000; Farjo et al., 2007). Further, there are differences in degeneration, early onset apoptosis is seen in rods but has not been observed in cones.
The nmf193 mutant was generated by ENU mutagenesis, which is a powerful phenotypic driven approach to define gene function relevant to disease pathology. It is noteworthy that many of the novel mutants generated are new alleles of genes that had already been associated with a particular phenotype (Jablonski et al., 2005; Sachs et al., 2007; Kiernan et al., 2002; Pinto et al., 2005). This effect could be due to the combination of selection bias, early embryonic lethality and the large number of knockout and spontaneous mouse mutants that have been generated and identified over the years. In a recent mapping study, nearly half of the genes in a 1Mb interval on chromosome 6 had already been associated with a phenotype (Nystuen et al., 2007). Thus, it appears that saturation of the mouse genome with mutations might come sooner rather than later.
We determined that Prph2 is a direct target of the nuclear hormone receptor gene Nr2e3. Mutations in mouse Nr2e3 (Nr2e3rd7/rd7) result in a slow progressive retinal degeneration (Akhmedov et al., 2000, Haider et al., 2006) while mutations in human Nr2e3 are associated with Enhanced S Cone syndrome and more recently, autosomal dominant retinitis pigmentosa (Haider et al., 2000; Lam et al., 2007; Coppieters et al., 2007). While the major phenotype of Nr2e3rd7/rd7 mice is a superfluous production of blue cone cells, they also exhibit outer segment defects. Reduced Prph2 message and protein levels were observed in Nr2e3rd7/rd7 mutant retinas, comparable to the reduction we observed in Prph2nmf193/+. Since it is well established by the rds mutant that the deficiency of Prph2 is sufficient for photoreceptor degeneration, it is of interest when reduced levels are seen in other mutants. While degenerating photoreceptors in mature Nr2e3rd7/rd7 retinas may cause some reduction of Prph2 expression, it is more difficult to explain how Prph2 this expression would be altered simply as a secondary effect given that expression of other rod genes were found to be very minimally reduced (Corbo and Cepko 2005; Peng et al., 2005; Haider et al. 2006). A comparison of the phenotypes of the two mutants revealed that similarities exist between them in the severity and progression of photoreceptor degeneration. It is an intriguing possibility that Prph2 has a role in the degeneration observed in the Nr2e3rd7/rd7 mutants and in the related human retinal diseases. The decreased levels of Prph2 are either indicative that Nr2e3 is involved in the regulation of Prph2 and may also be an effect of photoreceptor degeneration in the Nr2e3rd7/rd7 retina. While the data we have shown suggests that Nr2e3 is involved in the regulation of Prph2, gene transfer experiments to over-express Prph2 in an Nr2e3rd7/rd7 retina will be required to show that reduction of Prph2 in the Nr2e3rd7/rd7 retina is an integral part of the degenerative process.
Acknowledgments
This study was supported by the Center for Biomedical Excellence Award through the National Center for Research Resources, NIH (NIH 5 P20 RRO18788-02 (NBH), and the Nebraska Tobacco Settlement Biomedical Research Development (AMN, NBH), and Hope for Vision (NBH). The DNA Sequencing Core at UNMC receives partial support from the NIH grant number P20 RR016469 from the INBRE Program of the National Center for Research Resources.
References
- Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, Danciger M, Davisson MT, Farber DB. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci U S A. 2000;97:5551–5556. doi: 10.1073/pnas.97.10.5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird PN, Guymer RH, Chiu D, Vincent AL, Alexander WS, Foote SJ, Hilton DJ. Generating mouse models of retinal disease using ENU mutagenesis. Vision Res. 2002;42:479–485. doi: 10.1016/s0042-6989(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Barbaric I, Wells S, Russ A, Dear TN. Spectrum of ENU-induced mutations in phenotype-driven and gene-driven screens in the mouse. Environ Mol Mutagen. 2007;48:124–142. doi: 10.1002/em.20286. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Lamba OP, Napoli AA, Jr, Sinha S, Guo Y. Fusion between retinal rod outer segment membranes and model membranes: a role for photoreceptor peripherin/rds. Biochemistry. 1998;37:9477–9487. doi: 10.1021/bi980173p. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP, Fenner M, Napoli AA., Jr A peptide analogue to a fusion domain within photoreceptor peripherin/rds promotes membrane adhesion and depolarization. Biochim Biophys Acta. 2000;1463:343–354. doi: 10.1016/s0005-2736(99)00226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Chen J, Rattner A, Nathans J. The rod photoreceptor-specific nuclear receptor Nr2e3 suppresses transcription of multiple cone-specific genes. J Neurosc. 2005;25:118–129. doi: 10.1523/JNEUROSCI.3571-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Khanna H, Oh EC, Hicks D, Mitton KP, Swaroop A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum Mol Genet. 2004;13:1563–1575. doi: 10.1093/hmg/ddh173. [DOI] [PubMed] [Google Scholar]
- Cheng T, Peachey NS, Li S, Goto Y, Cao Y, Naash MI. The effect of peripherin/rds haploinsufficiency on rod and cone photoreceptors. J Neurosci. 1997;17:8118–8128. doi: 10.1523/JNEUROSCI.17-21-08118.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AT, Goldowitz D, Takahashi JS, Vitaterna MH, Siepka SM, Peters LL, Frankel WN, Carlson GA, Rossant J, Nadeau JH, Justice MJ. Implementing large-scale ENU mutagenesis screens in North America. Genetica. 2004;122:51–64. doi: 10.1007/s10709-004-1436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell G, Bascom R, Molday L, Reid D, McInnes RR, Molday RS. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A. 1991;88:723–726. doi: 10.1073/pnas.88.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters F, Leroy BP, Beysen D, Hellemans J, De Bosscher K, Haegeman G, Robberecht K, Wuyts W, Coucke PJ, De Baere E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007;81:147–57. doi: 10.1086/518426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Cepko CL. A Hybrid Photoreceptor Expressing Both Rod and Cone Genes in a Mouse Model of Enhanced S-Cone Syndrome. PLoS Genet. 2005;5:140–153. doi: 10.1371/journal.pgen.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, Naash MI. The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet. 2004;13:2075–2087. doi: 10.1093/hmg/ddh211. [DOI] [PubMed] [Google Scholar]
- Ding XQ, Stricker HM, Naash MI. Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry. 2005;44:4897–4904. doi: 10.1021/bi048414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjo R, Fliesler SJ, Naash MI. Effect of Rds abundance on cone outer segment morphogenesis, photoreceptor gene expression, and outer limiting membrane integrity. J Comp Neurol. 2007;504:619–630. doi: 10.1002/cne.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjo R, Skaggs JS, Nagel BA, Quiambao AB, Nash ZA, Fliesler SJ, Naash MI. Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J Cell Biol. 2006;173:59–68. doi: 10.1083/jcb.200509036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, Kenna P, Jordan SA, Kumar-Singh R, Humphries MM, Sharp EM, Sheils DM, Humphries P. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature. 1991;354:478–480. doi: 10.1038/354478a0. [DOI] [PubMed] [Google Scholar]
- Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Haider NB, Demarco P, Nystuen AM, Huang X, Smith RS, McCall MA, Naggert JK, Nishina PM. The transcription factor Nr2e3 functions in retinal progenitors to suppress cone cell generation. Vis Neurosci. 2006;23:917–929. doi: 10.1017/S095252380623027X. [DOI] [PubMed] [Google Scholar]
- Haider NB, Jacobson SG, Cideciyan AV, Swiderski R, Streb LM, Searby C, Beck G, Hockey R, Hanna DB, Gorman S, Duhl D, Carmi R, Bennett J, Weleber RG, Fishman GA, Wright AF, Stone EM, Sheffield VC. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24:127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- Haider NB, Naggert JK, Nishina PM. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet. 2001;10:1619–1626. doi: 10.1093/hmg/10.16.1619. [DOI] [PubMed] [Google Scholar]
- Hawes NL, Smith RS, Chang B, Davisson M, Heckenlively JR, John SW. Mouse fundus photography and angiography: a catalogue of normal and mutant phenotypes. Mol Vis. 1999;5:22. [PubMed] [Google Scholar]
- Jablonski MM, Wang X, Lu L, Miller DR, Rinchik EM, Williams RW, Goldowitz D. The Tennessee Mouse Genome Consortium: identification of ocular mutants. Vis Neurosci. 2005;22:595–604. doi: 10.1017/S0952523805225087. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8:1955–1963. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Lloyd M, Birch DG, Bok D, Travis GH. Generation and analysis of transgenic mice expressing P216L-substituted rds/peripherin in rod photoreceptors. Invest Ophthalmol Vis Sci. 1997;38:498–509. [PubMed] [Google Scholar]
- Keen TJ, Inglehearn CF. Mutations and polymorphisms in the human peripherin-RDS gene and their involvement in inherited retinal degeneration. Hum Mutat. 1996;8:297–303. doi: 10.1002/(SICI)1098-1004(1996)8:4<297::AID-HUMU1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Erven A, Voegeling S, Peters J, Nolan P, Hunter J, Bacon Y, Steel KP, Brown SD, Guenet JL. ENU mutagenesis reveals a highly mutable locus on mouse Chromosome 4 that affects ear morphogenesis. Mamm Genome. 2002;13:142–148. doi: 10.1007/BF02684018. [DOI] [PubMed] [Google Scholar]
- Kim RY, Dollfus H, Keen TJ, Fitzke FW, Arden GB, Bhattacharya SS, Bird AC. Autosomal dominant pattern dystrophy of the retina associated with a 4-base pair insertion at codon 140 in the peripherin/RDS gene. Arch Ophthalmol. 1995;113:451–455. doi: 10.1001/archopht.1995.01100040067029. [DOI] [PubMed] [Google Scholar]
- Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B. The role of the peripherin/RDS gene in retinal dystrophies. Acta Anat (Basel) 1998;162:75–84. doi: 10.1159/000046471. [DOI] [PubMed] [Google Scholar]
- Lam BL, Goldberg JL, Hartley KL, Stone EM, Liu M. Atypical mild enhanced S-cone syndrome with novel compound heterozygosity of the NR2E3 gene. Am J Ophthalmol. 2007;144:157–159. doi: 10.1016/j.ajo.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Moritz OL, Molday RS. Molecular characterization of peripherin-2 and rom-1 mutants responsible for digenic retinitis pigmentosa. J Biol Chem. 2001;276:22388–22396. doi: 10.1074/jbc.M011710200. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Moritz OL, Tam BM, Papermaster DS, Molday RS. The role of subunit assembly in peripherin-2 targeting to rod photoreceptor disk membranes and retinitis pigmentosa. Mol Biol Cell. 2003;14:3400–3413. doi: 10.1091/mbc.E03-02-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Norton JC, Allen AC, Burns JB, Hasel KW, Burns JL, Sutcliffe JG, Travis GH. Retinal degeneration slow (rds) in mouse results from simple insertion of a t haplotype-specific element into protein-coding exon II. Genomics. 1995;28:212–219. doi: 10.1006/geno.1995.1133. [DOI] [PubMed] [Google Scholar]
- McNally N, Kenna PF, Rancourt D, Ahmed T, Stitt A, Colledge WH, Lloyd DG, Palfi A, O’Neill B, Humphries MM, Humphries P, Farrar GJ. Murine model of autosomal dominant retinitis pigmentosa generated by targeted deletion at codon 307 of the rds-peripherin gene. Hum Mol Genet. 2002;11:1005–1016. doi: 10.1093/hmg/11.9.1005. [DOI] [PubMed] [Google Scholar]
- Nichols BE, Sheffield VC, Vandenburgh K, Drack AV, Kimura AE, Stone EM. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat Genet. 1993;3:202–207. doi: 10.1038/ng0393-202. [DOI] [PubMed] [Google Scholar]
- Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. 2007;8:1–10. doi: 10.1007/s10048-006-0068-7. [DOI] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, McDearmon EL, Fenner D, Lumayag SL, Omura C, Andrews AW, Baker M, Invergo BM, Olvera MA, Heffron E, Mullins RF, Sheffield VC, Stone EM, Takahashi JS. Generation, characterization, and molecular cloning of the Noerg-1 mutation of rhodopsin in the mouse. Vis Neurosci. 2005;22:619–629. doi: 10.1017/S0952523805225117. [DOI] [PubMed] [Google Scholar]
- Peng HP, Ahmad O, Ahmad F, Liu J, Chen S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum Mol Genet. 2005;14:747–764. doi: 10.1093/hmg/ddi070. [DOI] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Siepka SM, Shimomura K, Lumayag S, Baker M, Fenner D, Mullins RF, Sheffield VC, Stone EM, Heffron E, Takahashi JS. Results from screening over 9000 mutation-bearing mice for defects in the electroretinogram and appearance of the fundus. Vision Res. 2004;44:3335–3345. doi: 10.1016/j.visres.2004.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter LM, Boesze-Battaglia K, Tam BM, Moritz OL, Khattree N, Chen SC, Goldberg AF. Uncoupling of photoreceptor peripherin/rds fusogenic activity from biosynthesis, subunit assembly, and targeting: a potential mechanism for pathogenic effects. J Biol Chem. 2004;279:39958–39967. doi: 10.1074/jbc.M403943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AJ, Schwendinger JK, Yang AW, Haider NB, Nystuen AM. The mouse mutants recoil wobbler and nmf373 represent a series of Grm1 mutations. Mamm Genome. 2007;18:749–756. doi: 10.1007/s00335-007-9064-y. [DOI] [PubMed] [Google Scholar]
- Stricker HM, Ding XQ, Quiambao A, Fliesler SJ, Naash MI. The Cys214–>Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem J. 2005;388:605–613. doi: 10.1042/BJ20041960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds) Nature. 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, Bok D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- Weleber RG, Carr RE, Murphey WH, Sheffield VC, Stone EM. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111(11):1531–1542. doi: 10.1001/archopht.1993.01090110097033. [DOI] [PubMed] [Google Scholar]
- Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- Wrigley JD, Ahmed T, Nevett CL, Findlay JB. Peripherin/rds influences membrane vesicle morphology. Implications for retinopathies. J Biol Chem. 2000;275:13191–13194. doi: 10.1074/jbc.c900853199. [DOI] [PubMed] [Google Scholar]