

ABSTRACT
Current subcutaneously (s.c.)-injected insulin (INS) products result in a hyperinsulin exposure to peripheral tissues (skeletal muscle and adipose) while INS hardly accesses to liver after injection. This unphysiological distribution raises risks of hypoglycemia episode and causes weight gain after long term treatment. An ideal INS replacement therapy requires the distribution or action of exogenous INS to more closely mimic physiological INS in terms of its preferential hepatic action. However, there are 2 factors that limit the ability of s.c. injected INS to restore the liver: peripheral gradient in INS deficient diabetes patients: (1) the transport of INS in capillary endothelium and peripheral tissues from the injection site; and (2) peripheral INS receptor (IR) mediated INS degradation. In this review, the tissue barriers against efficient liver targeting of s.c. injected INS are discussed and current advances in developing hepatoselective insulin therapeutics are introduced.
KEYWORDS: diabetes, hepatic-directed vesicle INS, hepatoselectivity, IR-B selective INS analog, Peglispro, proinsulin-transferrin, subcutaneously-injected insulin, thyroxyl-INS analog
Liver is the primary action site of physiologically secreted insulin (INS). INS secretion bursts are synchronous with blood glucose (BG) fluctuations and the oscillations of intracellular calcium levels.1 After exocytosis of secretory granules from pancreatic β cells, released INS is first delivered to the hepatic portal vein, where the concentration of INS fluctuates from 100 to 1000 pM, and then INS reaches the hepatic sinusoids. Hepatic sinusoids are unique capillary cells due to their open endothelial pores (50-300 nm diameter in humans) and discontinuous basal lamina lacking a diaphragm.2 These sinusoids function as a loose molecular sieve allowing substance exchange between the hepatic artery through the perisinusoidal space to the hepatocytes. In the lumen of hepatic sinusoids, INS freely diffuses through the highly permeable endothelium and is exposed to insulin receptors (IR) on hepatocytes. A substantial amount of INS is degraded in the liver after IR mediated endocytosis,3 with a hepatic clearance of 50~80%. The remaining INS is circulated systemically, where the range of INS is 10 to 30 pM at basal conditions,4 and then reaches peripheral tissues including skeletal muscle and adipose tissue. Different from the microvessels aligning hepatocytes, peripheral blood capillaries are considered as continuous and tight (fenestration 6-12 nm).5 The transendothelial movement of INS is non-receptor mediated and non-saturable. According to previous studies, the ratio of INS concentration in the plasma versus the interstitial fluid of skeletal muscles significantly decreased during INS infusion at higher pharmacological levels (1.37 ± 0.25) compared to lower physiological levels (1.98 ± 0.21).6 The higher amount of infused INS delivered to skeletal muscles at the ultraphysiological level suggests the transport efficiency and capacity increased under higher plasma INS concentrations.
Current INS therapeutics administered via the s.c. route are unable to recapitulate physiological INS distribution (Fig. 1). The connective tissue septa comprise the first physical and catabolism barriers against INS absorption. INS, which is below the upper limit of capillary uptake (16 kDa), primarily accesses systemic circulation via either passive diffusion through small pores (< 30 Å) or paracellular transport pathway in capillary endothelium.7 The overall influence of "first-pass catabolism" on protein biologics disposition is not conclusive. However, studies have shown that the bioavailability (BA) of s.c. injected INS in humans was as high as 84%, indicating minimal catabolic clearance.8 Even so, BA may vary on a case-dependent manner for individual INS analogs. When evaluating the influence of the s.c. catabolic barrier for each INS analog, factors including injection site (abdominal vs appendicular side), retention time at injection site (fast-acting INS vs depot effect of long-acting INS), as well as amino acid (AA) sequence (native INS vs INS analogs with AA substitution) should be considered.9,10,11 Once in systemic circulation, INS is almost equivocally distributed between liver and peripheral tissues. The plasma concentration of pharmacological INS is approximately 2.5-3.5 times of circulated INS in normal subjects.12 This higher pharmacological insulin concentration predisposes peripheral tissues into hyperinsulinemia and leaves the liver under-insulinized. Liver hypoinsulinemia was reported in studies on rats, where it was shown that when arterial INS levels were clamped at twofold above basal levels during peripheral INS infusion, the portal vein INS concentration was still marked as "deficient," or below the basal level.13 Peripheral IR mediated degradation and the short plasma T1/2 (5 min) of INS may presumably attribute to observations in those early studies, which showed that only 1% of exogenous INS was delivered to the liver.14 Therefore, in order to obtain sufficient INS concentrations in the liver, peripheral hyperinsulinemia is necessary. Moreover, s.c. INS administration shifts the main burden of INS metabolism from the liver to the kidney compared with endogenous INS. Subsequently, it also raises the hypoglycemia risk for renal impaired patients.3 Further, the higher amount of INS exposed to the peripheral tissues is also responsible for many of the adverse metabolic side effects and increased risk of mitogenicity associated with exogenous INS therapy.15
Figure 1.
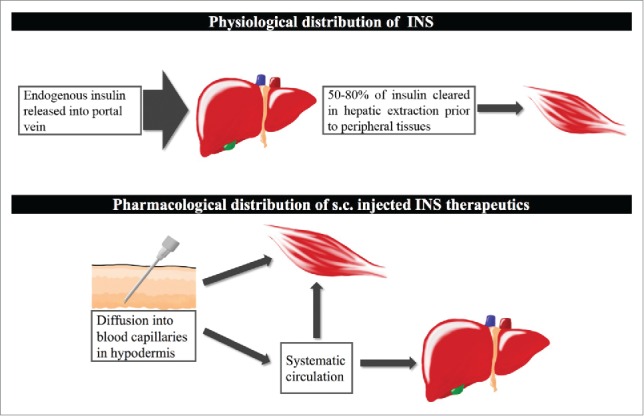
Comparison of physiological insulin distribution and exogenous insulin distribution. Under physiological conditions, insulin was secreted by pancreatic β cells and delivered to hepatic portal vein. 50-80% of insulin was cleared out in liver before its exposure upon peripheral tissues. 3 Exogenous insulin after s.c. injection appeared in blood capillaries through diffusion. It imposed hepatic hypoinsulinemia and peripheral hyperinsulinmemia compared to the physiological hepatic/peripheral insulin gradient.
Direct delivery of INS to the hepatic portal circulation allows efficient liver-targeting. According to empirical calculations, continuous intraportal INS infusion (0.35–0.56 U/kg/day) together with peripheral INS supplementation (0.08–0.11 U/kg/day) can reproduce the physiological distribution of INS in type 1 diabetic patients.1 However, such complicated delivery system is unfeasible and not suitable for self-administration by diabetic patients, where current administration via a s.c. bolus provides patient compliance and remains the main route for long-term INS treatment.16 As mentioned, the physiological barriers of peripheral tissues and liver when using a remote administration route (s.c.) hamper the ability of exogenous INS to mimic portal vein INS secretion. This review summarized current INS analogs and formulation refinements aimed at achieving liver targeting of s.c. injected INS (Fig. 2). Some examples are emerging novel concepts and some have been approved following clinical studies. By comparison and discussion of diverse targeting machineries, the aim of this review is to shed light on potential solutions to develop effective liver targeting INS therapy.
Figure 2.
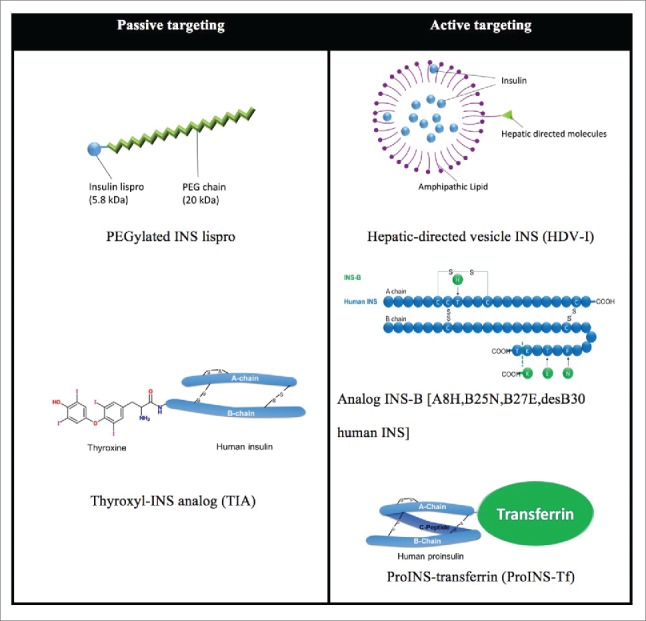
Two targeting approaches are exploited in the development of liver targeted INS therapy. Passive targeting by size increment of INS molecule (5.8 kDa) takes advantage of the different endothelial vascular sieves in peripheral tissues vs. the liver. Large molecules, which minimally diffuse through peripheral capillary walls, are freely filtered through fenestrated hepatic sinusoids. PEGylated INS lispro (Peglispro, LY2605541) is covalently coupled with a 20 kDa polyethylene glycol (PEG) and attains a hydrodynamic radius as large as a 71-98 kDa globular protein. Thyroxyl INS analog (TIA) is semi-synthesized by covalently linking thyronine to the ϵ-amino group of PheB1. In the active liver targeting area, hepatic-directed vesicle INS (HDV-I) encapsulates human INS in lipid vesicles (<150 nm in diameter) which have hepatocyte-targeting molecules (HTM) incorporated on the surface. HTMs include molecules targeting the asialoglycoprotein (galactose) receptor, hepatobiliary receptor or biotin receptor on hepatocytes. INS-B [A8H, B25N, B27E, desB30 human INS] is produced as an INS precursor in yeast with site directed mutagenesis, and the precursor is then enzymatically converted into 2-chain desB30 analogs. 30 ProINS-Tf is expressed in mammalian HEK293 cells by recombinant DNA techniques. MW increments of HDV-I and ProINS-Tf also presumably contribute to hepatoselectivity by passive targeting effect.
Current strategies in liver targeted insulin delivery
Current liver targeted INS analogs fall into either passive or active targeting mechanisms (Fig. 2). INS analogs, with increased size compared to INS (5.8 kDa), achieve hepatoselectivity by passive liver-targeting. These INS analogs exploit permeability differences between hepatic sinusoids and peripheral blood capillaries. Large molecules, which have restricted transport across peripheral capillaries, can easily diffuse through the open-pored hepatic sinusoids. Moreover, molecules with a molecular size above the renal filtration threshold (> 50-60 kDa) attain longer plasma T1/2 and protracted absorption as an s.c. depot. Thyroxyl insulin analogs 17 and insulin detemir 18 form larger complex via binding to plasma proteins: thyroxine-binding globulin (TBG; MW 54 kDa) and human serum albumin (HSA; 65 kDa), respectively. Another example is Peglispro (LY2605541, Eli Lilly and Company), which is composed of a 20 kDa PEG attached to lysine B28 on INS lispro. Each PEG monomer binds 3 molcules of water, resulting in a hydrodynamic radius as large as 71-98 kDa for Peglispro.19 The preferential hepatic action of Peglispro has been substantiated by several studies by comparing the ratio of the peripheral glucose disposal (PGD) rate, Rd, to the hepatic glucose production (HGP) rate, Ra. In euglycemic clamp studies on conscious dogs, the change from baseline in glucose Rd-to-Ra ratio was 0.5-0.6 in the Peglispro infusion group compared with 1.4 ± 0.3 in the INS group.20 It is believed that the large hydrodynamic radius of Peglispro impedes its transport across peripheral blood capillaries to the interstitial fluid of skeletal muscles and adipose, which leaves more Peglispro available for filtration through hepatic sinusoids 19 In addition, the reduced binding affinity of Peglispro to IR (less than 6% of INS lispro) possibly decreases peripheral IR-mediated clearance and contributes to a longer residence time in liver microcirculation.21 However, hepato-preference of Peglispro was prominent only below a certain dose. When the amount of infused Peglispro increased, PGD also increased indicating decreased hepatoselectivity.20
In active liver-targeting, one approach is to preferentially target IR isoform B (IR-B), which is the dominant isoform expressed on human liver compared to the other isoform of IR, IR-A. The INS analog INS-B [A8H, B25N, B27E, desB30 human insulin] 22 displays a 2 to 4-fold higher binding affinity toward IR-B than IR-A. In ex vivo rodent studies, INS-B exhibited higher potency in hepatic glycogen accumulation (75% potency of human INS) than glycogen synthesis in skeletal muscles (45% potency of human INS). The hepatoselectivity of INS-B largely depends on tissue-specific expression of IR isoforms. However, IR-B is also prominently expressed on skeletal muscles in humans.23 This discrepancy obligates further investigation of INS-B's liver-targeting in alternate animal models which are more human-relevant. Another example in actively targeting INS to the liver is hepatic-directed vesicle INS (HDV-I). HDV-I entraps INS inside vesicles, along with hepatocyte-targeting molecules (HTM) attached to the amphipathic lipid layers. After absorption and vesicle release, free INS transits to systemic circulation and triggers peripheral IR activation. Meanwhile, lipid layer-associated INS is driven to hepatocytes by virtue of the HTM/hepatic receptor mediation. INS is then dissociated from the lipid complex over time and exerts INS action in the liver.24,25 HTM is composed of molecules targeted either the asialoglycoprotein (galactose) receptor, hepatobiliary receptor or biotin receptor on hepatocytes. The amount of INS delivered to liver has been enhanced in the HDV-I formulation. Still, the vast majority of HDV-I was the free form encapsulated inside of vesicles (99% free INS).25 In addition, vesicles with increased diameter over 90 nm exhibit increased uptake by nonparenchymal cells which decreases the specificity to hepatocytes.26 The final example of active liver targeting is Proinsulin-transferrin (ProINS-Tf) fusion protein, which consists of the inactive human INS precursor, ProINS, and Tf. In cultured adipocytes and after short-term incubation in hepatocytes, ProINS-Tf itself displayed weak INS potency, similar to ProINS. However, following prolonged incubation with hepatocytes, ProINS-Tf was converted to immuno-reactive INS-transferrin (irINS-Tf). The resultant irINS-Tf acquired enhanced INS potency and was recognized by human INS specific radioimmunoassay.27 When evaluated in a type 1 diabetes mouse model (streptozotocin-treated mice), ProINS-Tf exerted a delayed but prolonged BG lowering effect in vivo.28 The delayed effect was attributed to the requirement of conversion from ProINS-Tf to irINS-Tf to achieve bioactivity, while the prolonged effect was attributed to Tf, a stable protein established for its ability to prolong the T1/2 of fused proteins.29 Compared with native INS, ProINS-Tf led to a higher ratio of hepatic/peripheral IR activation and enhanced hepatic glycogen accumulation after an s.c. bolus injection. Uniquely, ProINS-Tf is the first described liver targeted INS prodrug with low stimulation of PGD.27 In contrast, the aforementioned INS analogs and formulation refinements still bring circulated active INS to peripheral tissues. Consequently, the efficiency of their hepatopreference may be attenuated during dosage escalation (smaller Ra to Rd ratio), which is a potential drawback. However, compared with most INS analogs listed in this review, the anti-diabetic effect of ProINS-Tf has only been tested on rodent models so far. Therefore, its hepatic action on human and liver toxicity needs further exploration.
Conclusion
INS therapy with improved glycemic control without compromising safety issues remains a critical challenge for chronic treatment in diabetes. Currently applied s.c. INS analogs or formulations work mostly to achieve an anti-hyperglycemic effect via promoting PGD, and reverse the physiological portal: peripheral INS gradient. Liver targeted INS mimicking endogenous INS distribution may be able to restore normal glucose control. The use of liver targeted INS also reduces the required peripheral INS dose, thereby reducing the risk of subsequent hypoglycemia. Liver-targeted INS analogs may serve as the next generation of INS therapy in diabetes treatment with improved safety.
Abbreviations
- Rd
peripheral glucose disposal rate
- Ra
hepatic glucose production rate
- PGD
peripheral glucose disposal
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Studies of ProINS-Tf in this review were supported by NIH grant GM063647 and USC Bensussen Innovative Challenge Grant.
References
- [1].Ferrannini E. Physiology of glucose homeostasis and insulin therapy in type 1 and type 2 diabetes. Endocrinol Metab Clin North Am 2012; 41:25-39; PMID:22575405; http://dx.doi.org/ 10.1016/j.ecl.2012.01.003 [DOI] [PubMed] [Google Scholar]
- [2].Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol 2002; 1:1; PMID:12437787; http://dx.doi.org/ 10.1186/1476-5926-1-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev 1998; 19:608-24; PMID:9793760; doi: 10.1210/edrv.19.5.0349 [DOI] [PubMed] [Google Scholar]
- [4].Matteucci E, Giampietro O, Covolan V, Giustarini D, Fanti P, Rossi R. Insulin administration: present strategies and future directions for a noninvasive (possibly more physiological) delivery. Drug Des Devel Ther 2015; 9:3109-18; PMID:26124635; http://dx.doi.org/ 10.2147/DDDT.S79322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res 2010; 2:14; PMID:20701757; http://dx.doi.org/ 10.1186/2040-2384-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Steil GM, Ader M, Moore DM, Rebrin K, Bergman RN. Transendothelial insulin transport is not saturable in vivo. No evidence for a receptor-mediated process. J Clin Invest 1996; 97:1497-503; PMID:8617883; http://dx.doi.org/ 10.1172/JCI118572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McDonald TA, Zepeda ML, Tomlinson MJ, Bee WH, Ivens IA. Subcutaneous administration of biotherapeutics: current experience in animal models. Curr Opin Mol Ther 2010; 12:461-70; PMID:20677097 [PubMed] [Google Scholar]
- [8].Hoffman A, Ziv E. Pharmacokinetic considerations of new insulin formulations and routes of administration. Clin Pharmacokinet 1997; 33:285-301; PMID:9342504; http://dx.doi.org/ 10.2165/00003088-199733040-00004 [DOI] [PubMed] [Google Scholar]
- [9].ter Braak EW, Woodworth JR, Bianchi R, Cerimele B, Erkelens DW, Thijssen JH, Kurtz D. Injection site effects on the pharmacokinetics and glucodynamics of insulin lispro and regular insulin. Diabetes Care 1996; 19:1437-40; PMID:8941480; http://dx.doi.org/ 10.2337/diacare.19.12.1437 [DOI] [PubMed] [Google Scholar]
- [10].Jockel JP, Roebrock P, Shergold OA. Insulin depot formation in subcutaneoue tissue. J Diabetes Sci Technol 2013; 7:227-37; PMID:23439181; http://dx.doi.org/ 10.1177/193229681300700128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kuerzel GU, Shukla U, Scholtz HE, Pretorius SG, Wessels DH, Venter C, Potgieter MA, Lang AM, Koose T, Bernhardt E. Biotransformation of insulin glargine after subcutaneous injection in healthy subjects. Curr Med Res Opin 2003; 19:34-40; PMID:12661778; http://dx.doi.org/ 10.1185/030079902125001416 [DOI] [PubMed] [Google Scholar]
- [12].Hayford JT, Thompson RG. Free and total insulin integrated concentrations in insulin dependent diabetes. Metabolism 1982; 31:387-97; PMID:7043174; http://dx.doi.org/ 10.1016/0026-0495(82)90116-0 [DOI] [PubMed] [Google Scholar]
- [13].Farmer TD, Jenkins EC, O'Brien TP, McCoy GA, Havlik AE, Nass ER, Nicholson WE, Printz RL, Shiota M. Comparison of the physiological relevance of systemic vs. portal insulin delivery to evaluate whole body glucose flux during an insulin clamp. Am J Physiol Endocrinol Metab 2015; 308:E206-222; PMID:25516552; http://dx.doi.org/ 10.1152/ajpendo.00406.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Canfield RE, Kaye GI, West SB. The preparation and evaluation of tritiated polyalanyl insulin derivatives. Endocrinology 1972; 90:112-22; PMID:5009052; http://dx.doi.org/ 10.1210/endo-90-1-112 [DOI] [PubMed] [Google Scholar]
- [15].Stout RW. Insulin as a mitogenic factor: role in the pathogenesis of cardiovascular disease. Am J Med 1991; 90:62S-5S; PMID:1994720; http://dx.doi.org/ 10.1016/0002-9343(91)90041-U [DOI] [PubMed] [Google Scholar]
- [16].Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos 2014; 42:1881-9; PMID:25100673; http://dx.doi.org/ 10.1124/dmd.114.059238 [DOI] [PubMed] [Google Scholar]
- [17].Shojaee-Moradie F, Powrie JK, Sundermann E, Spring MW, Schüttler A, Sönksen PH, Brandenburg D, Jones RH. Novel hepatoselective insulin analog: studies with a covalently linked thyroxyl-insulin complex in humans. Diabetes Care 2000; 23:1124-9; PMID:10937509; http://dx.doi.org/ 10.2337/diacare.23.8.1124 [DOI] [PubMed] [Google Scholar]
- [18].Hordern SV, Wright JE, Umpleby AM, Shojaee-Moradie F, Amiss J, Russell-Jones DL. Comparison of the effects on glucose and lipid metabolism of equipotent doses of insulin detemir and NPH insulin with a 16-h euglycaemic clamp. Diabetologia 2005; 48:420-6; PMID:15729576; http://dx.doi.org/ 10.1007/s00125-005-1670-1 [DOI] [PubMed] [Google Scholar]
- [19].Madsbad S. LY2605541–a preferential hepato-specific insulin analogue. Diabetes 2014; 63:390-2; PMID:24464715; http://dx.doi.org/ 10.2337/db13-1646 [DOI] [PubMed] [Google Scholar]
- [20].Moore MC, Smith MS, Sinha VP, Beals JM, Michael MD, Jacober SJ, Cherrington AD. Novel PEGylated basal insulin LY2605541 has a preferential hepatic effect on glucose metabolism. Diabetes 2014; 63:494-504; PMID:24089512; http://dx.doi.org/ 10.2337/db13-0826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pandyarajan V, Weiss MA. Design of non-standard insulin analogs for the treatment of diabetes mellitus. Curr Diab Rep 2012; 12:697-704; PMID:22983891; http://dx.doi.org/ 10.1007/s11892-012-0318-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vienberg SG, Bouman SD, Sørensen H, Stidsen CE, Kjeldsen T, Glendorf T, Sørensen AR, Olsen GS, Andersen B, Nishimura E. Receptor-isoform-selective insulin analogues give tissue-preferential effects. Biochem J 2011; 440:301-8; PMID:21851336; http://dx.doi.org/ 10.1042/BJ20110880 [DOI] [PubMed] [Google Scholar]
- [23].Anderson CM, Henry RR, Knudson PE, Olefsky JM, Webster NJ. Relative expression of insulin receptor isoforms does not differ in lean, obese, and noninsulin-dependent diabetes mellitus subjects. J Clin Endocrinol Metab 1993; 76:1380-2; PMID:7684396 [DOI] [PubMed] [Google Scholar]
- [24].Lau JR, Geho WB (Google Patents, 2013). [Google Scholar]
- [25].Geho WB, Geho HC, Lau JR, Gana TJ. Hepatic-directed vesicle insulin: a review of formulation development and preclinical evaluation. J Diabetes Sci Technol 2009; 3:1451-9; PMID:20144401; http://dx.doi.org/ 10.1177/193229680900300627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harashima H, Kiwada H. Liposomal targeting and drug delivery: Kinetic consideration. Adv Drug Deliver Rev 1996; 19:425-44; http://dx.doi.org/ 10.1016/0169-409X(96)00012-9 [DOI] [Google Scholar]
- [27].Wang Y, Chen YS, Zaro JL, Shen WC. Receptor-mediated activation of a proinsulin-transferrin fusion protein in hepatoma cells. J Control Release 2011; 155:386-92; PMID:21756950; http://dx.doi.org/ 10.1016/j.jconrel.2011.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Y, Shao J, Zaro JL, Shen WC. Proinsulin-transferrin fusion protein as a novel long-acting insulin analog for the inhibition of hepatic glucose production. Diabetes 2014; 63:1779-88; PMID:24353179; http://dx.doi.org/ 10.2337/db13-0973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen X, Lee HF, Zaro JL, Shen WC. Effects of receptor binding on plasma half-life of bifunctional transferrin fusion proteins. Mol Pharm 2011; 8:457-65; PMID:21291258; http://dx.doi.org/ 10.1021/mp1003064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Glendorf T, Sorensen AR, Nishimura E, Pettersson I, Kjeldsen T. Importance of the solvent-exposed residues of the insulin B chain alpha-helix for receptor binding. Biochemistry 2008; 47:4743-51; PMID:18376848; http://dx.doi.org/ 10.1021/bi800054z [DOI] [PubMed] [Google Scholar]