

Abstract
Besides enhancing aqueous solubilities, cocrystals have the ability to fine-tune solubility advantage over drug, supersaturation index, and bioavailability. This review presents important facts about cocrystals that sets them apart from other solid-state forms of drugs, and a quantitative set of rules for the selection of additives and solution/formulation conditions that predict cocrystal solubility, supersaturation index, and transition points. Cocrystal eutectic constants are shown to be the most important cocrystal property that can be measured once a cocrystal is discovered, and simple relationships are presented that allow for prediction of cocrystal behavior as a function of pH and drug solubilizing agents. Cocrystal eutectic constant is a stability or supersatuation index that: (a) reflects how close or far from equilibrium a cocrystal is, (b) establishes transition points, and (c) provides a quantitative scale of cocrystal true solubility changes over drug. The benefit of this strategy is that a single measurement, that requires little material and time, provides a principled basis to tailor cocrystal supersaturation index by the rational selection of cocrystal formulation, dissolution, and processing conditions.
Keywords: cocrystal, co-crystal, enhancing solubility, supersaturation, solubilizing agent, surfactant, micelles, lipids, formulation, pH, biorelevant media, solubilization, transition point, stability, supersaturation index, supersaturation potential, solubility advantage
Graphical Abstract
1. Introduction
Cocrystals have emerged as a means of fine-tuning solubility, dissolution, bioavailability, and other physicochemical properties of drug substances, without changing their molecular structure. Cocrystals are a class of multicomponent solids containing two or more different molecular components in a single homogenous crystalline phase with well-defined stoichiometry [1–5]. They are distinguished from solvates in that the cocrystal components are solids at room temperature.
Hydrogen-bonded assemblies between the neutral molecules of the drug and the cocrystal coformer often guide cocrystal formation [2,4], which is why they are of particular interest due to their ability to modify the solubility properties of nonionizable drugs that cannot otherwise form pharmaceutical salts. Over the last decades cocrystals have received significant attention from the pharmaceutical industry, and numerous pharmaceutical cocrystals have been reported. A pharmaceutical cocrystal is composed of an active pharmaceutical ingredient (API) and a benign molecule or other APIs as coformers. Coformers are commonly selected from substances appearing on the GRAS (Generally Regarded As Safe) status list or those that have been demonstrated to be non-toxic and have regulatory approval [6, 7].
Physicochemical criteria, such as those defined by the Lipinski rule of five, are typically used to predict whether lead molecules, frequently found from high throughput or biological screening, will become drug candidates with adequate permeability, solubility, and bioavailability. Drugs that are highly permeable, are lipophilic and often exhibit poor aqueous solubility [8].
Solid-state modifications and formulation design allow for the improvement of the physicochemical properties of a drug substance while maintaining the same chemical entity and pharmacological interaction. Polymorphs, solvates and salts are the common solid forms employed for product development. However, consideration of cocrystals and cocrystalline salts as viable solid forms for development would significantly expand the number and diversity of solid drug forms available, and improve the likelihood of finding a solid form with the required physicochemical properties [9]. A schematic representation of the different classes of multicomponent solids is shown in Fig. 1.
Fig. 1.

Comparison of multicomponent solid form modifications that can be used to alter the properties of a drug [10]. Reproduced by permission of the American Chemical Society, http://pubs.acs.org/doi/pdf/10.1021/cg900129f.
Unlike salts, cocrystals do not rely on ionic interactions and cocrystals can be made for non-ionizable drugs. Also, for cocrystal formation the number of suitable coformers can exceed the number of suitable counterions for salt formation. In contrast to amorphous pharmaceutical forms, cocrystals can achieve thermodynamic stability in the solid state while providing large solubility advantage over drug. Compared to polymorphs, cocrystals have the ability to increase solubility by orders of magnitude above the drug solubility.
Cocrystals owe their large solubility range to the numerous structures, diverse molecular characteristics of cocrystal components, and speciation of its components in solution [1, 11]. Both cocrystal and solution conditions determine how soluble a cocrystal is compared to the parent drug. Solution pH, the presence and concentration of drug solubilizing agents, and coformer concentration all work in concert to change cocrystal solubility. Fig. 2 shows how each of these solution variations can alter cocrystal solubility, cocrystal solubility relative to the parent drug, and as a result cocrystal thermodynamic stability regions. Unlike amorphous or polymorphic solids, cocrystal solubility advantage over drug can be increased or decreased to the desired value by just changing the solution pH, drug solubilizing agent concentrations, or coformer concentration.
Fig. 2.

Cocrystal solubility can be fine-tuned by (a) pH, (b) drug solubilizing agents, and (c) coformer concentration, where dashed line represents stoichiometric concentrations of (1:1) cocrystal. Solution conditions change the cocrystal solubility relative to drug solubility and so the cocrystal thermodynamic stability. The cocrystal is thermodynamically stable when Scocrystal ≤ Sdrug. The cocrystal solubility advantage over drug (Scocrystal/Sdrug) when Scocrystal > Sdrug is however critical to achieve higher drug concentrations during cocrystal dissolution.
Because of the cocrystal versatility in changing solubility with environmental conditions, it provides unique opportunities to align thermodynamic and kinetic phenomena to manage conversions to less soluble forms. An essential consideration in this regard is to dial-in a cocrystal solubility advantage that is not unnecessarily higher than that desired to meet a pharmacokinetic response or therapeutic effect.
This review will present the mechanisms by which cocrystals modulate solubility and the properties that in combination with simple mathematical relationships can guide cocrystal formation, formulation, and development. A major goal is to establish the link between key concepts and practical implications for fine-tuning cocrystal solubility, supersaturation, and stability.
2. Cocrystal Design and Formation
Prior to carrying out cocrystal screens, which are costly in material and time, potential coformers can be identified based on molecular recognition interactions. Molecular recognition events are responsible for the self-assembly of two or more components through noncovalent interactions with energetically favorable geometries [12]. The Cambridge Structural Database (CSD) can be used to perform supramolecular retrosynthetic analysis, which involves identifying intermolecular units for a target cocrystal structure. Coformers can be selected to cocrystallize with a drug based on knowledge of geometries and preferred orientations of existing intermolecular interactions. Synthons are the common noncovalent intermolecular interactions of specified geometries identified in the literature that make up the structural units within a supramolecular structure; a few examples of synthons are shown in Fig. 3.
Fig. 3.

Common supramolecular synthons formed from carboxylic acids, amides, pyridines, and other aromatic nitrogens [13–15].
Synthons can form between identical functional moieties (homosynthon) or different functional moieties (heterosynthon). Cocrystal structures may contain different combinations of homosynthons and heterosynthons [13, 16]. Additionally, these intermolecular interactions may be homomeric, between the same molecule, or heteromeric, between different molecules [17]. In the case of carbamazepine (CBZ), coformers were selected to form cocrystals based on two design strategies. The first strategy was to maintain the cyclic homomeric carboxamide homosynthon and find coformers that could interact with the exterior hydrogen-bond donors and acceptors. An example of this is the 1:1 cocrystal carbamazepine-saccharin (CBZ-SAC) which is shown in Fig. 4a. The second strategy was to disrupt the carboxamide homosynthon by forming a heteromeric synthon with the carboxamide. This was accomplished by forming a heterosynthon between the carboxamide with a carboxylic acid coformer. An example of the second strategy is the 2:1 carbamazepine-succinic (CBZ-SUC) acid cocrystal, which is shown in Fig. 4b.
Fig. 4.

Examples of two strategies used to form cocrystals of carbamazepine (a) CBZ-SAC which maintain cyclic carboxamide homosynthon (b) CBZ-SUC which disrupts carboxamide homosynthon in favor of a heterosynthon between carboxamide and the dicarboxylic acid [18].
Coformers are often selected based on functional groups capable of complimentary hydrogen bonding with the drug substance. Due to their directional interactions, hydrogen bonds most strongly influence molecular recognition. Etter and Donohue developed general guidelines to predict hydrogen bond interactions that result in crystal formation [12, 15]. These guidelines are based on the analysis of the hydrogen bond interactions and the packing motifs of numerous molecular structures: (1) the hydrogen bonding in the crystal structure will include all acidic hydrogen atoms, (2) all good hydrogen bond acceptors will participate in hydrogen bonding if there is an adequate supply of hydrogen bond donors, (3) hydrogen bonds will preferentially form between the best proton donor and acceptor, and (4) intramolecular hydrogen bonds in a six-membered ring form in preference to intermolecular hydrogen bonds [12, 14, 15].
In addition to these rules, the stereochemistry and competing interactions between molecules may need to be considered for cocrystal design. Other considerations in designing stable crystal structures include minimizing electrostatic energies and the free volume within the crystal [19]. Analysis of cocrystal structures from the CSD suggests that components that cocrystallize often have similar shapes and polarities [20]. While these strategies offer a good basis to select coformers for cocrystal screening and synthesis, they are not able to ab initio determine the cocrystal structure, molecules that will cocrystallize, conditions that promote cocrystallization or the physicochemical properties of the cocrystals based on their supramolecular structure. Several cocrystals have been discovered by using a combination of supramolecular retrosynthetic analysis and cocrystal screening techniques. Some examples of pharmaceutical cocrystals are shown in Table 1.
Table 1.
Examples of pharmaceutical cocrystals reported in the literature.
| Drug | Coformer | Reference |
|---|---|---|
| Carbamazepine | 4-aminobenzoic, saccharin, salicylic, succinic, benzoic, ketoglutaric, maleic, glutaric, malonic, oxalic, adipic, (+)-camphoric, 4- hydroxybenzoic, 1-hydroxy-2-napthoic, DL- tartaric, L-tartaric, fumaric, DL-malic, L-malic, acetic, butyric, 5-nitroisphthalic, formic | [18, 21] |
| Curcumin | resorcinol, pyrogallol | [22, 23] |
| Danazol | vanillin, 4-hydroxybenzoic acid | [24, 25] |
| Indomethacin | saccharin, nicotinamide, D/L mandelic, lactamide, benzamide | [1, 26] |
| Itraconazole | succinic, fumaric, L-malic, L-tartaric, DL-tartaric | [27] |
| Ketoconazole | succinic, fumaric, adipic | [28] |
| Lamotrigine | acetamide, nicotinamide, methylparaben | [29] |
| Meloxicam | aspirin, 1-hydroxy-1-napthoic acid, salicylic, 4- hydroxybenzoic, glutaric, maleic, L-malic, benzoic, DL-malic, hydrocinnamic, fumaric | [11, 30] |
| Nevirapine | maleic acid, saccharin, salicylic acid, tartaric acid, glutaric acid | [31] |
| Paracetamol | oxalic, theophylline, phenazine, naphthalene | [5] |
| Piroxicam | saccharin, L-tartaric, citric, fumaric, adipic acid, succinic, benzoic, 4-hydroxybenzoic, oxalic, ketoglutaric, salicylic, pyroglutamic acid, DL-tartaric, maleic, DL-malic, L-malic | [32, 33] |
| Pterostilbene | piperazine, glutaric acid, caffeine | [34, 35] |
3. Cocrystal Screening and Synthesis
Determining if a cocrystal exists has been an empirical exercise that requires a broad experimental search space. Cocrystal screening has been carried out using a variety of techniques such as slow solvent evaporation [18, 36–39], slurry conversion[40], solid-state grinding [41, 42], solvent drop grinding [43–45], melt and sublimation [46, 47]. One of the major limitations of current screening methods is that they often lead to crystallization of individual components, are not transferable to larger scale cocrystal formation, require large amount of materials, and are time consuming. Basic concepts of crystallization can be applied to understand and control the nucleation and growth of cocrystals. For an in-depth description of cocrystallization mechanisms and methods, readers are referred to other publications [48–52]. Here we summarize the main principles that guide cocrystal formation.
Cocrystal formation requires that at least two components crystallize in a single phase in stoichiometric ratio according to the reaction
| (1) |
This reaction describes cocrystal precipitation (right to left) and dissolution (left to right). Ksp, or solubility product is the thermodynamic equilibrium constant of the reaction and is given by
| (2) |
the product of activity coefficients (γ) of components A and B multiplied by the component concentrations, terms in brackets. Under ideal conditions, where γ=1, Ksp,a the activity based solubility product is replaced by a concentration product, Ksp.
The driving force for nucleation and growth is the supersaturation, which for a cocrystal is expressed in terms of activity or concentration product as
| (3) |
Therefore supersaturation with respect to cocrystal is dependent on solution composition. In other words, supersaturation can be achieved by changing the concentrations of cocrystal components in solution. It is important to realize that it is the dissolved components that determine supersaturation, not the solid phases of the components.
Fig. 5 presents how supersaturation is generated and cocrystals are formed by simply dissolving cocrystal components in solution, A + B → ABsolid. This method is called reaction crystallization method (RCM) [53] and is based on generating supersaturation with respect to cocrystal while the solution is only at saturation or below with respect to cocrystal components, according to equation 3. Thus, there is no need to select solvents that match reactant solubilities for this approach to succeed. Cocrystals can be discovered in solvents in which they are thermodynamically unstable (more soluble than the drug) by reversing the cocrystal to drug thermodynamic stability as coformer concentration increases. Phase diagrams are essential to guide cocrystal discovery and synthesis as they indicate the conditions and solid forms that can potentially crystallize.
Fig. 5.

Schematic phase solubility diagram indicating regions where cocrystal can form or dissolve and a possible cocrystal formation pathway. Lines represent solubilities of drug A, coformer B, and cocrystal AB. Cocrystal solubility decreases with coformer concentration [B]T. Subscript T represent analytical or total concentrations. Arrows represent a path along which cocrystal is the only phase that can crystallize. Region IV: solution is supersaturated with respect to cocrystal, and drug can convert to cocrystal. Region I: solution is supersaturated with respect to drug, and cocrystal can convert to drug. Region II: solution is supersaturated with respect to both drug and cocrystal, and both can crystallize. Region III: solution is below saturation and drug, cocrystal, and coformer dissolve. Crystallization pathway involves: (1) solution saturated with respect to coformer (the most soluble component in this example), (2) dissolution of drug, and (3) cocrystal formation. This method of cocrystal formation is called reaction crystallization method (RCM).
RCM has been carried out in many solvents, however green solvents are favored including water and alcohols. Also, because RCM is based on thermodynamically stable cocrystal conditions it is an attractive process for any reaction scale. In situ monitoring of cocrystal formation by RCM has recently been demonstrated for cocrystals and can be used to effectively control and scale-up cocrystallization processes [21, 54, 55]. We have shown the application of RCM to cocrystal screening in microliter volumes (96 well plates) by in situ Raman microscopy as presented in Fig. 6. First, several solvents are presaturated with the coformers of interest. Second, the solid drug (above its solubility) is added to the presaturated coformer solutions. Solid phase changes are then monitored in situ by Raman Microscopy or other appropriate method. Since crystallization, if it occurs, is a result of drug dissolution into a highly concentrated coformer solution, a new solid phase must be a solvate, salt, or cocrystal, or combinations of these.
Fig. 6.

Rapid in situ cocrystal screening by RCM in microliter (96 well plates) by Raman microscopy, indicating spectral changes between drug crystals (carbamazepine) and its cocrystals [21].
Solution crystallization processes involve several other methods. Supersaturation can be generated by methods that (1) change cocrystal solubility (pH, temperature, drug to coformer ratio, poor solvent), or (2) changing concentrations (evaporation). Evaporation and solvo-thermal methods have the inherent risk of crystallizing individual components instead of cocrystal and screening with a large number of solvents and experimental conditions (rates of evaporation or cooling, initial concentrations, etc.) is generally required. Table 2 summarizes some of the methods used in cocrystal formation.
Table 2.
Common methods used for cocrystal screening and synthesis.
| Method | Mechanism | Characteristic |
|---|---|---|
| Reaction crystallizationa | Solution process based on generating supersaturation with respect to cocrystal by dissolving reactants and or changing pH | Useful for both screening and synthesis; amenable for large and small scales |
| Solvo-thermalb | Solution process based on generating supersaturation with respect to cocrystal through temperature change | Requires screening for solvents with similar reactant solubilities to minimize their crystallization |
| Sonic slurryc | Solution process based on generating supersaturation with respect to cocrystal by subjecting a slurry of reactants to ultrasound pulses | May reduce the supersaturation for nucleation and increase nucelation rate |
| Co-grindingd | Mechanical stresses enhance molecular molibity and lead to transformation of reactants to cocrystal | Solvent free method useful for screening |
| Liquid assisted grindinge | Cocrystal formation through solution and/or solid phase mediated process | Useful for screening but requires larger amounts of materials than RCM |
| Moisture/vapor sorptionf | Solution process involves generating supersaturation by exposing solid reactants to deliquescent materials | Suitable for screening by vapor sorption of solid mixtures |
| Melt crystallizationg | Cocrystal formation occurs through a melted phase | Useful for screening with small quantities of reactants by DSC and microscopy |
| Tween screw extrusion (TSE) and Hot melt extrusion (HME)h | High screw mixing can lead to cocrystal formation with (HME) or without (TSE) melting reactants | Continuous, single-step, solvent free and readily scalable process |
Solid-state cocrystal synthesis methods have been used in which the crystalline or amorphous components are combined. Mechanical activation by grinding cocrystal components together is a common method to form cocrystals [12, 14, 21, 41–45, 56]. Cocrystal formation has been shown to proceed through intermediate amorphous phases in some cases [57]. The glass transition temperature (Tg) and melt temperature of components are important material properties for mechanochemical methods that induce phase transformations and therefore should be considered along with process temperature in the synthetic outcome. Co-grinding reactants with addition of solvent drops referred to as solvent drop grinding or liquid assisted grinding can lead to cocrystal formation through solution and/or solid phase mediated processes.
The effectiveness of adding drops or very small volume of solvent to a solid mixture of reactants in forming cocrystals appears to be due to the dissolution mediated cocrystal formation, whereby the solution becomes saturated with respect to reactants and supersaturated with respect to cocrystals as in RCM. Cocrystal formation by moisture sorption [58] and solvent sorption [59, 60] of solid mixtures containing cocrystal reactants are also explained by a similar mechanism of reactive crystallization as shown for CBZ, nicotinamide (NCT), and sucrose, in Figs. 7 and 8. The carbamazepine-nicotinamide (CBZ-NCT) cocrystal is about 150 times more soluble than CBZ in water and readily converts to CBZ, yet this cocrystal forms in water at high concentrations of NCT relative to CBZ. The highly soluble coformer, dissolves to a greater extent than the drug, generating supersaturation with respect to cocrystal as a result of non-stoichiometric concentration in solution.
Fig. 7.

Illustration of the moisture uptake process leading to deliquescence, reactant dissolution and cocrystal formation. A and B are cocrystal reactants, Ds is solid deliquescent additive and Dl is the solution phase created by deliquescence at RH (relative humidity) greater than DRH (deliquescent relative humidity) Reprinted with permission from [58]. Copyright 2007 American Chemical Society.
Fig. 8.

Optical microscopy images showing moisture sorption, deliquescence, dissolution, and cocrystallization in CBZ/NCT/sucrose system at 25°C and 95% RH. Symbols C, N, and S represent carbamazepine (CBZ), nicotinamide (NCT), and sucrose, respectively. Reprinted with permission from [58]. Copyright 2007 American Chemical Society.
Figure 8 shows photomicrographs of deliquescence-induced cocrystal formation of a ternary mixture of CBZ, NCT, and sucrose crystals when exposed to relative humidities above the deliquescence relative humidity of the mixture. These images reveal that the transformation mechanism to cocrystal involves: moisture uptake, dissolution, cocrystal nucleation and growth. Similar behavior has been shown for CBZ, caffeine (CAF), theophylline (THP), and sulfadimidine with carboxylic acid coformers even when these drugs can form hydrates. Differences in the behavior of deliquescent solids (coformers or excipients) were explained by the composition on the deliquesced solution, cocrystal and component solubilities, according to phase diagrams.
Moisture uptake by amorphous polymers such as polyvinylpyrrolidone (PVP) has also been shown to form cocrystals [61]. Cocrystal formation increased with moisture uptake by PVP and with decreasing the PVP molecular weight. The underlying mechanism for this process is the increased mobility of water and PVP leading to more effective dissolution of components and higher supersaturation with respect to cocrystal. Photomicrographs in Fig. 9 show water uptake by PVP and cocrystal formation for CBZ-NCT cocrystal.
Fig. 9.

Optical microscopy images showing dissolution of CBZ and NCT (A–C) and CBZ-NCT cocrystal formation (D and E) during moisture sorption of PVP K12 (50 wt%) in a mixture of equimolar composition of CBZ and NCT at 75% RH and 25 C. Reproduced from ref.[61] with permission from The Royal Society of Chemistry.
During moisture uptake, as with small amounts of solvent added to solid mixtures, the cocrystal supersaturation is generated by the dissolution of cocrystal components, as illustrated in the phase diagram in Fig. 10. The cocrystal and drug solubilities, and cocrystal and coformer solubilities intersect at eutectic points. Regions of stability for each solid phase or mixtures of solid phases are characterized by eutectic points. In the case of CBZ-NCT, CBZ and NCT, the eutectic point corresponding to drug/cocrystal equilibria changed with the concentration of dissolved PVP such that the ratio of cocrystal to drug solubility or solubility advantage (SA) decreased with increasing PVP. The polymer has two competing effects. It provides the medium for crystallization and it also hinders crystallization by altering the translational diffusion of water and cocrystal components. However, at higher water contents, or lower SA, lower supersaturation levels will lead to slower cocrystal formation. At even higher water content, the cocrystal can transform to drug.
Fig. 10.

Triangular phase diagram illustrating the dissolution paths (arrows) that can lead to cocrystal stability regions (shaded areas). Solution compositions between the two eutectic points, eu1 and eu2, are favorable for cocrystal formation. Moisture content, drug, and coformer solubilities determine the dissolution path and supersaturation levels reached. The highest supersaturation for cocrystal is achieved by saturation with respect to both drug and coformer, conditions associated with water contents below the eutectic points. Reproduced from ref.[61] with permission from The Royal Society of Chemistry.
Twenty-seven carbamazepine cocrystals containing eighteen carboxylic acids were discovered by four different screening methods: high throughput evaporation, solvent drop grinding, sonic slurry, and RCM [21]. While grinding experiments are attractive because of the small quantities of components required and rapid synthesis, some limitations include the difficulty of readily discerning the formation mechanisms or pathways, the chemical stability of components subjected to high mechanical energy processes, purity of products (i.e. extent of transformation), empirical nature, and challenges regarding scalability.
Cocrystal formation in melts has also been used in cocrystal screening [46, 47] and large-scale processes. The application of hot melt extrusion appears to be a promising alternative to formation of cocrystals where chemical instability is not an issue [62].
4. Important Facts about Cocrystals
4.1. Cocrystal solubilities can exceed drug solubilities by orders of magnitude
Cocrystal aqueous solubility has been reported to be as high as 1,000 times the drug solubility (Fig. 11). A trend in cocrystal solubility advantage (SA=Scocrystal/Sdrug) with coformer solubility over drug solubility (Scoformer/Sdrug) has also been observed. The cocrystal solubility advantage, SA, is introduced as a dimensionless solubility number to characterize the ability of a given cocrystal to alter the solubility of a drug at a given pH, solubilizing agent concentration, temperature, etc [4].
Fig. 11.

Cocrystal solubility advantage (Scocrystal/Sdrug) in aqueous media is related to the coformer and drug solubility ratio (Scoformer/Sdrug). It is observed that Scoformer/Sdrug > 10 leads to Scocrystal/Sdrug >1. The solubility values refer to a specific pH value shown by the numbers above the data points, at 25 °C [4, 35, 67, 68].
Pharmaceutical cocrystals are generally made of a hydrophobic drug molecule and a hydrophilic coformer molecule. The mechanism by which cocrystals go into solution involves three main steps: (1) breaking intermolecular bonds in the cocrystal, (2) breaking intermolecular bonds in the solvent, and (3) forming intermolecular bonds between cocrystal molecules and solvent molecules. The limiting step in dissolving cocrystals of hydrophobic drug molecules in aqueous media has been shown to be solvation and not breaking away from the crystal lattice. Coformers appear to decrease the solvation barrier of cocrystals of hydrophobic drugs to an extent proportional to that of the pure coformer. Consequently, coformer aqueous solubility is correlated with cocrystal solubility. On the other hand, melting points are not good indicators of cocrystal aqueous solubilities, since it is drug hydrophobicity and not cocrystal lattice strength that limits solubility [65, 66].
4.2. Cocrystal solubility advantage over drug (Scocrystal/Sdrug) varies with solubilizing agents and pH
Cocrystals will encounter aqueous solutions of varying pH and often with solubilizing agents such as lipids, polymers, synthetic and endogenous surfactants during processing, formulation and dissolution. Does cocrystal SA change under these conditions? Are cocrystal SA measurements in aqueous media transferable to solutions of different pH, or with drug solubilizing agents? The answer to the first question is: yes. Consequently, the answer to the second questions is: no.
Fig. 12 demonstrates how cocrystal and drug solubilities can change in the presence of drug solubilizing agents. Danazol cocrystals, danazol-4-hydroxybenzoic acid (DNZ-HBA) and danazol-vanillin (DNZ-VAN) are much more soluble than danazol (DNZ) in aqueous media with SAaq of 770 and 370 for 4-hydroxybenzoic acid (HBA) and vanillin (VAN) cocrystals, but this SA is decreased to 10 and 6 in solution of Tween 80. Similar behavior is observed for pterostilbene cocrystals with caffeine (PTB-CAF) and with piperazine (PTB-PIP). Cocrystals of PTB exceed the solubility of pterostilbene (PTB) with SAaq of 26 and 6 for the CAF and PIP cocrystals, but this SA is overturned (SA < 1) in the presence of lipids to values of 0.3 and 0.2. This means that SA is not an inherent property of the cocrystal and that it is greatly influenced by drug solubilizing agents.
Fig. 12.

Effect of drug solubilizing agents on cocrystal and drug solubilities for DNZ and its cocrystals with HBA, and VAN, and for PTB and its cocrystals with CAF and with PIP. Tween 80 aqueous solution (150 mM, pH 5.0) used for danazol and lipid formulation for PTB (Captex 355/Capmul MCM (1/3): Cremophor EL (3:7)). Numbers represent cocrystal solubility advantage (SA= Scocrystal/Sdrug) in buffer with and without solubilizing agents. Both drugs are highly solubilized by the additives but their cocrystals are solubilized to a much lesser extent than drugs. Drug solubilizing agents decrease SA and can overturn it as in the case of PTB, where cocrystals become less soluble than PTB in the lipid formulation studied. Adapted with permission from M.P. Lipert and N. Rodríguez-Hornedo from ref. [70]. Copyright 2015 American Chemical Society.
Fig. 13 shows how cocrystal and drug solubilities change with solubilizing agent concentration. CBZ-SAC cocrystal is more soluble than the drug CBZ in water, but the cocrystal solubility advantage decreases as the solubilization by surfactant (sodium lauryl sulfate, SLS) increases. Cocrystal SA values decrease from 4.4 in water to <1 as surfactant concentration increases above 44 mM. The reason for this behavior is that the cocrystal solubility dependence on surfactant concentration is weaker than that of the drug solubility. This phenomena affords the unique ability to fine-tune cocrystal SA by a predictable value.
Fig. 13.

Cocrystal solubility values approach drug solubility values with increasing solubilizing agent concentration, as shown for the CBZ-SAC cocrystal and carbamazepine dihydrate (CBZD) in SLS solutions. Cocrystal solubility advantage over drug decreases with increasing drug solubilization, and can reach a value above which cocrystal is less soluble than drug. The intersection of solubility curves represents a transition point. This transition point is characterized by a solubility value (S*) where Scocrystal=Sdrug and a solubilizing agent concentration referred to as critical stabilization concentration (CSC). Cocrystal or drug solubilities above S* indicate that the cocrystal is above the transition point and drug is more soluble than cocrystal. Curves represent simulations according to the solubility equations for cocrystal and drug [69]. Adapted with permission from M. P. Lipert and N. Rodríguez-Hornedo from ref. [70]. Copyright 2015 American Chemical Society.
How large of an SA do you want or do you need? For some drugs the SA in aqueous media can be as high 100 or 1,000, but these high SA values may present unnecessary risks associated with cocrystal instability and potential for conversion to the less soluble drug. Why not then lower the SA to 10 or to a value that still maintains an increased cocrystal dissolution or solubility relative to the drug, while avoiding unnecessary stability risks? SA values can be quantitatively changed with the extent of drug solubilization. In other words, a cocrystal solubility relative to drug can be dialed up or down based on the power and selectivity of drug solubilizing agents.
Cocrystal solubility advantage is a “supersaturation index” (supersaturation with respect to drug). SA is an indicator of the potential of cocrystals to convert to the constituent drug (drug precipitation) when cocrystal is in contact with solutions, such as during dissolution or pharmaceutical processes. In practical terms, if a cocrystal has a supersaturation index that leads to fast conversions, a lower supersaturation index can be dialed and to a predictable level, by a combination of solubilizing agents and/or pH. Current approaches to harness the cocrystal superior solubility over drug have however neglected this important cocrystal property.
Another important parameter that determines cocrystal solubility is solution pH. When cocrystal constituents ionize in solution, the cocrystal solubility will be modulated by the extent of ionization. Cocrystals can impart solubility-pH dependence to non-ionizable drugs, (CBZ cocrystals with acidic coformers such as saccharin (SAC), succinic (SUC), and salicylic (SLC) acids, [21, 69, 71] and to alter the solubility-pH dependence of ionizable drugs such as gabapentin (GBP) [72], indomethacin (IND) [65], itraconazole (ITZ) [27], nevirapine (NVP) [73] and ketoconazole (KTZ) [74].
Fig. 14 shows the solubility-pH dependence of NVP and its cocrystals with acidic coformers: SAC, maleic (MLE) and SLC acids. Cocrystals do not only enhance solubility over drug, but do so on a pH dependent fashion. In this case, NVP is highly soluble at pH 1 but its solubility decreases by about 2 orders of magnitude to a very low and constant solubility value at pH > 4. Cocrystals, in contrast, lead to a “U shaped” solubility-pH curve, with exponentially decreasing and increasing solubility as pH is increased. Furthermore, cocrystal and drug solubility curves may intersect at a pH referred to as pHmax.
Fig. 14.

Solubility of the basic drug NVP and its cocrystals with acidic coformers: (1:1) cocrystal NVP-MLE, and (2:1) NVP-SAC and NVP-SLC as a function of pH. Symbols represent solubilities determined from solutions saturated with NVP and/or cocrystal at 25°C. pH values correspond to equilibrium pH. As pH in increased the cocrystal and drug solubility curves approach each other and intersect at pHmax. The pH value at the intersection of the drug and cocrystal (NVP-SAC and NVP-SLC) solubility curves corresponds to pHmax or transition point above which a less soluble cocrystal becomes more soluble than drug. Curves were calculated from cocrystal and drug solubility-pH dependence according to equations and and parameter values presented in the text and in Table 3. Symbols represent: NVP solubility (NVP hydrate-open circles, NVP anhydrous-filled circles) and cocrystal solubilities from eutectic points (squares). Reproduced from ref. [73] with permission from The Royal Society of Chemistry.
It is important to note that cocrystal solubilities and their solubility relative to drug are highly dependent on pH, but pH is not always measured when studying cocrystals. Table 3 presents the NVP cocrystal SA values measured from cocrystal dissolution in water without considering pH, and those obtained from solubilities in Fig. 14 that consider pH. The reported SA values in water (unknown pH) show SA values close to 1 for NVP-SAC and NVP-SLC cocrystals, and SA about 5 for NVP-MLE cocrystal. These cocrystals have coformers that are much more soluble than the drug, consequently their modest SA values (around 1) were surprising, since their melting points did not justify such behavior. These results are explained when one considers the influence of pH on cocrystal solubility, drug solubility, and SA (Fig. 14). SA values close to 1 occur at pH values close to pHmax, where a cocrystal SA is equal to 1. These observations confirm the importance of measuring pH since SA can experience large changes with small variations of pH. Therefore, cocrystal dissolution and solubility measurements are not meaningful unless solution pH is measured.
Table 3.
NVP cocrystals: Ksp, pHmax, and Scocrystal/Sdrug.
| Cocrystal | Kspa (M2 or M3) b | pHmaxc | Scocrystal/Sdrugd pH 1 to 5 |
Scocrystal/Sdruge pH ? |
|---|---|---|---|---|
| NVP-MLE (1:1) | (2.0±0.5) × 10−5 | none | 3.4–906 | 5.3 |
| NVP-SAC (2:1) | (1.0±0.6) × 10−10 | 1.1 | 0.9–47 | 1.4 |
| NVP-SLC (2:1) | (4.0±0.9) × 10−11 | 1.7 | 0.6–11 | 1.1 |
Calculated from equilibrium solubility measurements at cocrystal/drug eutectic points at 25°C.
Units of M2 for 1:1 and M3 for 2:1 cocrystals.
Obtained from the intercept of drug and cocrystal solubility curves in Fig. 14.
Obtained from equilibrium solubility calculation, S vs pH curves in Fig. 14.
From Caira et al., [31] obtained from cocrystal dissolution in water, pH unknown, and NVP solubility in water (0.36mM) at 37°C. The influence of temperature on Scocrystal/Sdrug is expected to be small compared to the influence of pH. Sdrug hydrate increases by about 2 fold between 25 and 37°C [75] and the change in Scocrystal/Sdrug may be even smaller if at all.
4.3. Cocrystal have transition points
The same cocrystal can display higher, equal, or lower solubility than the constituent drug, depending on solution conditions such as pH, drug solubilizing agents, and coformer concentration (Figs. 2, 13, and 14) [4, 69, 71, 76, 77]. As a result of this phenomena, cocrystals exhibit transition points at which the cocrystal and drug solubilities are equal, and above or below which the cocrystal solubility advantage over drug is eliminated (SA≤1). These transition points are characterized by:
pHmax: caused by ionization of cocrystal components, and or
S* and critical stabilization concentration (CSC): caused by drug solubilizing agents, preferential solubilization of drug over coformer.
4.3.1. Transition points induced by pH: pHmax
Cocrystals of NVP, ITZ, GBP, piroxicam (PXC), and lamotrigine (LGT) among others exhibit a pHmax in aqueous solutions [71–73, 78, 79]. pHmax, is also an important parameter that identifies stability regions of pharmaceutical salts [80–82]. Cocrystals with basic drugs and acidic coformers as demonstrated for NVP cocrystals in Fig. 14, are more soluble than the drug at pH > pHmax.
4.3.2. Transition points induced by solubilizing agents: S* and CSC
Cocrystals of CBZ, DNZ, IND, and PTB exhibit transition points in solutions with drug solubilizing agents [67, 69, 70, 76, 77, 83]. As indicated in Fig. 13 for CBZ-SAC cocrystal, a transition point occurs at the intersection of the cocrystals and drug solubility curves. This cocrystal transition point is characterized by a solubilizing agent concentration CSC and a solubility (S*) at which both drug and cocrystal have equal solubilities [84]. We recently reported that CSC is dependent on the effectiveness of the drug solubilizing agents and SAaq, whereas S* is not dependent on solubilizing agents as long as coformer is not solubilized by agent. S* is determined by SAaq, and is an indicator of the highest solubility value below which a cocrystal will be more soluble than drug (SA>1), at a given pH and temperature.
Transition points can be experimentally determined by several approaches that rely on cocrystal solubility measurements in media with and without the additives of interest. As indicated in Fig. 12 for PTB cocrystals in lipids, both cocrystals are above the transition point (and below drug solubility) at the lipid concentrations studied. Transition points are also determined from solubility vs solubilizing agent plots similar to Fig. 13, as shown for CBZ-SAC cocrystal. Mathematical relationships can also be used to predict transition points, from knowledge of SA (in the absence or presence of solubilizing agents) and drug solubilization (SRdrug) and in this way reduce the number of experiments required. These relationships are further described below. Comparison of transition point values determined by different approaches is discussed in the literature [69, 71, 85].
4.4. Cocrystal solubilization in solubilizing media is lower than drug solubilization
In an earlier section we focused on how cocrystal SA is influenced by the presence of drug solubilizing agents. Here we will describe how cocrystal solubilization (SRcocrystal), where SR is defined as
| (4) |
can be predictably changed with drug SR. SR is the total solubility in drug solubilizing media (ST) divided by the aqueous solubility (Saq). ST represents the sum of the concentrations of all species dissolved (ST = Saqueous + Ssolubilizing agent). Saq represents the cocrystal aqueous solubility at a given pH in the absence of solubilizing agent (Saq = Snonionized,aq + Sionized,aq) and is the sum of the nonionized and ionized contributions to the aqueous solubility.
Cocrystals and drugs are not solubilized to the same extent by drug solubilizing agents. This is demonstrated by the DNZ and PTB cocrystals in synthetic solubilizing agents (Fig. 12), and by cocrystals of DNZ, IND, PXC, and CBZ in FeSSIF, (Fig. 15). Drugs are solubilized to a greater extent than cocrystals in FeSSIF even though cocrystals are more soluble than drugs in both FeSSIF and buffer. Cocrystals of the more hydrophobic drug DNZ show a large SA in buffer that is reduced in FeSSIF (770 vs 25). Similar trends are observed for cocrystals of the less hydrophobic drugs but the magnitude of the differences is much smaller. These findings highlight the risks associated with the expectation of constant cocrystal SA and SR values across solutions with different pH, additives, solubilizing agents, including endogenous surfactants.
Fig. 15.

Solubilization ratios in FeSSIF for cocrystals and their constituent drugs at 25°C. The initial pH was 5.00 in both buffer and FeSSIF. Greater solubilization of drug leads to lower solubilization of cocrystal. The final pH of solubility measurements in FeSSIF and buffer are as follows: DNZ (5.01±0.05 and 4.96±0.01), DNZ-VAN (5.00±0.01 and 4.96±0.01), and DNZ-HBA (4.46±0.06 and 4.47±0.01). IND (4.98±0.06 and 4.96±0.03), IND-SAC (3.65±0.05 and 3.66±0.02). PXC (H) (5.03±0.02 and 4.98±0.01), and PXC-SAC (3.79±0.02 and 3.64±0.02)CBZ (H) (4.86±0.05 and 4.95±0.01), CBZ-4ABA-HYD (4.94±0.02 and 4.84±0.03), CBZ-SLC (4.29±0.02 and 4.37±0.02), CBZ-SAC (3.11±0.02 and 3.08±0.03). Reprinted with permission from Elsevier [25].
4.5. Cocrystals modulate microenvironment pH during dissolution
It is well known that the pH at the dissolving surface of ionizable drugs can be different from the bulk solution and this microenvironment pH is very important in determining the rate of dissolution [82, 86–92]. Cocrystals usually contain ionizable components, which means that they also have the ability to change the microenvironment pH. Depending on the ionization properties of the coformers, the microenvironment pH of the cocrystals can be modulated to different extents from that of the parent drug. For example, CBZ is nonionizable, so it has no ability to alter the pH at the dissolving surface and this means that the microenvironment pH is the same as the bulk pH. However, when CBZ cocrystallizes with acidic coformers, SAC and SLC, the microenvironment pH behavior is significantly different as shown in Fig. 16. For both cocrystals, the microenvironment pH decreases as the coformers ionize and both reach constant values at bulk pH 4 to 8 due to their self-buffering ability [93]. In contrast, the microenvironment pH of CBZ just remains the same as the bulk pH. Due to the alteration in microenvironment pH, the cocrystal dissolution rate may not follow their pH dependent solubility. Instead of increasing with bulk pH like solubility, the dissolution rates of both CBZ-SAC and CBZ-SLC would reach constant values at the buffering region [93] as shown in Fig. 16.
Fig. 16.

Microenvironment pH (a) and flux (b) of CBZ (red) and its two cocrystals, CBZ-SAC (blue) and CBZ-SLC (orange) predicted using developed mass transport models as a function of bulk pH. The dotted lines in (b) represents the flux prediction with the assumption that microenvironment pH is the same as bulk pH. The solubility product of CBZ–SAC is 1.00 mM2 and CBZ–SLC is 0.4 mM2 at 25°C. The pKa values of SAC and SLC are 1.6 and 3.0, respectively [93].
5. Basic Principles and Relationships
Behind the ability of cocrystals to modulate solubility and dissolution, and solve problems of low and erratic drug bioavailability, lies a series of simple molecular processes that are not difficult to quantify. During cocrystal development, one therefore tries to find correlations between cocrystal properties (solubility, dissolution, stability, etc) and experimental conditions (for instance those encountered in formulations, processing, storage and dissolution). Relevant to these correlations are the molecular mechanisms by which cocrystals dissolve.
The solubility of a cocrystal is not a unique value determined by its solid-state chemisty, but a series of values determined by what happens to cocrystal molecules when they dissolve. In our earlier work, we examined cocrystal dissolution in terms of reaction equilibria and associated equilibrium constants that led to solubility concepts that explain the interplay between solution conditions and cocrystal properties. The interested reader can find a full treatment of cocrystal solubility mechanisms in several publications [25, 65, 69–72, 77]. More recently, we have developed simple relationships that allow for estimation of cocrystal solubilities, solubility advantage over drug, supersaturation, and transition points, without having to measure equilibrium constants and do not require use of the more rigorous equations. This approach involves knowledge of commonly used drug solubility descriptors and readily accessible cocrystal property measurements while still allowing for quantitative conclusions.
5.1. Mechanistic basis of cocrystal solubility
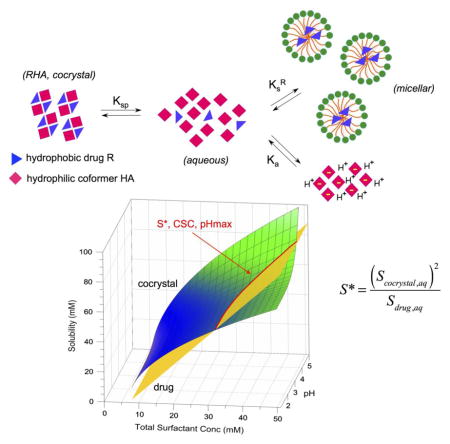
Unlike simple molecular solids that dissolve to give the aqueous molecular constituents, cocrystal solubility is complicated by a delicate interplay of solution composition, solute interactions, and pH. To address this problem we have considered the reaction mechanisms by which cocrystals dissolve, such as dissociation, ionization, and solubilization, summarized in Fig. 17.
Fig. 17.

Cocrystal solubility is determined by the fate of its molecular constituents in solutions. This diagram shows cocrystal-solution phase interactions for a cocrystal RHA of a non-ionizable drug (R) and an ionizable coformer (HA) and associated equilibria commonly encountered by pharmaceutical dosage forms, such as dissociation, complexation, ionization and micellar solubilization. Ksp represents cocrystal solubility product, Ka is the ionization constant, Kc is the complexation constant and are the micellar solubilization constants for HA, A− and R, respectively. Adapted from ref. [66] with permission from The Royal Society of Chemistry.
We have derived mathematical relationships that describe cocrystal solubility in terms of the equilibrium constants associated with cocrystal dissociation (Ksp), coformer ionization (Ka), and micellar solubilization (Ks). For the case of (1:1) cocrystals of a non-ionizable drug with an ionizable coformer, the cocrystal solubility as a function of pH is given by
| (5) |
The presence of drug solubilizing agents introduces another term to the equation above, so that cocrystal solubility is related to pH and micellar concentration, for example, according to
| (6) |
where [M] represents the micellar surfactant concentration. Equations of this type allow for the quantitative prediction of cocrystal solubility as a function of ionization and micellar solubilization, from independently measured cocrystal and component properties. Studies of several cocrystals by these methods showed excellent agreement between the observed and predicted cocrystal solubility values. Equations that consider other cocrystals stoichiometries, ionization, and solubilization are presented in other references [69, 71, 77, 94].
These solubility equations are the basis for the more practical relationships between cocrystal solubility advantage and cocrystal solubilization in the presence of drug solubilizing agents.
5.2. Cocrystal solubilization can be estimated from only drug solubilization
The influence of solubilizing agents on cocrystal solubility is determined by the drug solubilization according to [84]
| (7) |
for 1:1 cocrystals. For a 2:1 cocrystals (drug:coformer) the relationship is
| (8) |
Plots of log (SRcocrystal) vs log (SRdrug) produce straight lines with slopes of 1/2 for 1:1 cocrystals and 2/3 for 2:1 cocrystals as shown in Fig. 18. There is excellent agreement between predicted and observed values across different cocrystals and drug solubilizing agents. These findings imply that SRcocrystal can be predicted without any cocrystal analysis or even discovery of a cocrystal, just from knowledge of drug solubilization.
Fig. 18.

SRcocrystal dependence on SRdrug for (a) 1:1 cocrystals and (b) 2:1 cocrystals. Lines represent theoretical relationships between SRcocrystal and SRdrug according to equations 15 and 18 in log form for 1:1 and 2:1 cocrystals, respectively. The slope of the line is predicted to be 1/2 for 1:1 cocrystals and 2/3 for 2:1 cocrystals. Symbols represent experimentally determined SR values from cocrystal and drug solubilities measured under equilibrium conditions in solubilizing agents as indicated in the legend. Reprinted with permission from M. P. Lipert and N. Rodríguez-Hornedo from ref. [70]. Copyright 2015 American Chemical Society.
A central assumption of these simple relationships is that coformer is not solubilized by additives. This assumption is justified for most cocrystals with hydrophobic drugs and hydrophilic coformers, as the drugs are preferentially solubilized by the additives. Positive deviations observed for several 1:1 cocrystals at high values of SRdrug are a result of coformer solubilization and can be quantified by a factor ε as described in a subsequent section.
5.3. Cocrystal supersaturation index and transition point can be estimaded from SA and SRdrug
Cocrystal supersaturation index, described by SA, varies with drug solubilization according to
| (9) |
or
| (10) |
where SA is the total cocrystal solubility at drug solubilization SR, and SA is the aqueous cocrystal solubility advantage in the absence of drug solubilization. When SRdrug=1, SA=SAaq in the absence of drug solubilization. The above expression clearly suggests a way of fine-tuning cocrystal supersaturation by changing drug solubilization, through addition of polymers, surfactants, lipids or additives that preferentially solubilize drug over coformer.
Fig. 19 shows the cocrystal SA as a function of drug solubilization according to equation 11 in logarithmic form:
| (11) |
Fig. 19.

Cocrystal solubility advantage over drug or supersaturation index (SA) decreases in a predictable way with increasing (SRdrug). Full lines represent (1:1) cocrystals with SAaq = 2, 10, and 100. The dashed line indicates SA=1. The intersection of the cocrystal SA and SA=1 lines represents the SRdrug at which Scocrystal=Sdrug, and identifies transition points, which in these example are at SRdrug = 4, 100, and 10,000 for the corresponding cocrystals. Below the SRdrug limit, cocrystal is more soluble than drug but becomes less soluble than drug above this SRdrug value.
The plot of log (SA) vs log (SRdrug) is characterized by: (1) lines with slope of −1/2 where the position of each line is determined by the cocrystal SA value, (2) the drug solubilization associated with a given cocrystal SA, and (3) the regions of drug solubilization over which the cocrystal is more soluble, equally soluble or less soluble than drug, SA > = or < 1. The intersection of a cocrystal SA line with the SA=1 line establishes the SRdrug limit below which cocrystal can generate supersaturation with respect to drug or transition point. Consequently, the level of supersaturation with respect to drug in this SRdrug range can be selected from knowledge of the additive influence on SRdrug. It is the interplay between supersaturation and absolute solubilities that determines the nucleation rate and therefore, SA is one of the central parameters to be considered. It is well recognized that higher solubilities at the same supersaturation or SA results in faster nucleation rates.
Another important conclusion that can be drawn from examining Fig. 19 is that the lower the cocrystal SAaq is, the lower is the SRdrug at the transition point. This means that cocrystals with modest solubility advantage over drug are more susceptible to be overturned by low extents of drug solubilization. Therefore, it is important to know what is the transition SRdrug, before formulating cocrystals, so that inadvertent reversals in cocrystal SA do not occur.
Deviations from Equations (9–11) will occur when coformer is solubilized by additive or when there are other interactions between drug, coformer, and additives. As a first approximation, this analysis provides very useful information to anticipate the influence of drug solubilization on cocrystal SA, supersaturation with respect to drug, as well as prediction of cocrystal transition points, where SA=1.
Fig. 20 compares the predicted and experimental measurements of cocrystal SA for CBZ, DNZ and PTB as a function of SRdrug in different surfactant systems as indicated in the plot. Both cocrystal SA and SRdrug are well predicted from the simple equations using only the SAaq experimental value. In fact, the results also anticipate the observed lower solubility of PTB cocrystal compared to drug in a lipid formulation as presented earlier. SRdrug for PTB in this formulation was measured to be 12,200. This value is above the SRdrug at the transition point, and therefore Scocrystal is lower than Sdrug. A similar analysis for the DNZ cocrystals indicated that for these cocrystals the SA values were lower, but the cocrystal solubility still exceeded drug solubility. As shown in Fig. 20, cocrystals are more soluble than drug over the range of SRdrug where SA >1, and less soluble than drug over the range of SRdrug where SA <1.
Fig. 20.

Cocrystal solubility advantage as a function of drug solubilization for CBZ-SAC, PTB-CAF and DNZ-HBA cocrystals, decreases with increasing SR in a predictable way that identifies the SR limit above which cocrystal SA is overturned, the transition point. Full lines represent cocrystal SA predicted from equation (9) using only the experimentally determined cocrystal SAaq. The dotted line indicates the line of equal cocrystal and drug solubilities. Solubilizing agents correspond to those in Figs. 12 and 13.
5.4. Cocrystal solubility at transition points (S*)
S* establishes the drug solubility above which the cocrystals is no longer more soluble than drug. The value of S* is determined by the cocrystal and drug aqueous solubilities according to
| (12) |
where m=2 and n=1 for 1:1 cocrystals; and m=3 and n=2 for 2:1 cocrystals. S* identifies the solubility value of drug or cocrystal above which the cocrystal SA is overturned. S* values for several cocrystals of CBZ, DNZ and PTB are shown in Table 4. Not only is there excellent agreement between observed and predicted values, but S* is shown to be useful guide for additive selection, so that the desired SA is attained and more importantly that it is not overturned. Considering the case of PTB cocrystals, one can clearly see that the S* values of PTB cocrystals (58.6 and 17.8 mM for PTB-CAF and PTB-PIP respectively) are below the solubility of PTB in the lipid formulation (1M), indicating that the cocrystals will be less soluble than drug under these conditions as observed in the reported studies (Fig. 12). Therefore, from knowledge of S* one can calculate the drug solubilities below which cocrystals maintain a solubility advantage over drug.
Table 4.
S* values for CBZ, DNZ, and PTB cocrystals in solutions with drug solubilizing agents, SLS, Tween, and lipid formulation.a [70].
| Cocrystal | pH | S*predb (mM) | S* obsc (mM) |
|---|---|---|---|
| CBZ-SLC (1:1) | 3.0 | 3.3 | 4.6 |
| CBZ-SAC (1:1) | 2.2 | 10.5 | 12.0 |
| CBZ-4ABA-HYD (2:1) | 4.0 | 21.8 | 22.0 |
| CBZ-SUC (2:1) | 3.1 | 48.0 | 47.6 |
| DNZ-HBA (1:1) | 5.0 | 90.0 | >32 |
| DNZ-VAN (1:1) | 5.0 | 20.3 | >17 |
| PTB-CAF (1:1) | nr | 58.6 | <222 |
| PTB-PIP (2:1) | nr | 17.8 | <246 |
Solubilizing agents are: aqueous solution of SLS for CBZ cocrystals, Tween 80 (150 mM, pH 5.0) for DNZ, and lipid formulation (Captex 355/Capmul MCM (1/3): Cremophor EL (3:7)) for PTB as described in the text.
S* predicted from equation 12 for 1:1 and 2:1 cocrystals.
Determined from the intersection of Scocrystal,T and Sdrug,T curves or measurement of cocrystal and drug solubilities in solubilizing agents.
After ref. [70].
5.5. Implications of coformer solubilization on cocrystal SRcocrystal and S*
One of the main sources of deviation in these calculations is the coformer solubilization by additives. This deviation can be quantified by a factor ε so that for a 1:1 cocrystal the expressions of cocrystal solubilization becomes
| (13) |
where
| (14) |
where SRcocrystal is predicted from equation 7 that assumes coformer solubilization is zero (Kscoformer = 0).
The relationship for S* is given by
| (15) |
where
| (16) |
where S* is predicted using equation 12 that assumes coformer solubilization is zero. The values of ε for CBZ cocrystals in SLS were modest ranging from 1.0 to 1.4. The highest value corresponds to the highest observed coformer solubilization, salicylic acid in the CBZ-SLC cocrystal.
The factor ε is determined by the equilibrium constants for solubilization and ionization of coformer as given by
| (17) |
for a monoprotic weakly acidic coformer such as saccharin and salicylic acid. The ε values predicted from this equation were in excellent agreement with those calculated from the deviations of SR and S*. As a first approximation, the simple equations provide qualitative guidance so that cocrystals might be formulated in the right environment, and more accurate predictions are achieved by using the ε factor.
5.6. Cocrystal dissolution mechanism
Dissolution rate is one of the important factors that governs the bioavailability of oral drugs, especially for those with dissolution rate limited absorption [95]. The dissolution process is driven by the concentration gradient that establishes across the diffusion layer adjacent to the dissolving surface [96, 97]. At the dissolving surface, the drug concentration is at its equilibrium solubility, while its concentration is usually assumed to be zero in the bulk solution under sink conditions [96, 97]. The rate at which the drug diffuses across the diffusion layer can be described by the Noyes-Whitney and Nernst-Brunner equation [98, 99]:
| (18) |
where D is the diffusion coefficient, S is the equilibrium solubility of the drug, A is the surface area of the solid drug and h is the thickness of the diffusion layer.
The dissolution mechanism for single components has been well studied and it has been shown that one of the determining factors for the dissolution rate of ionizable drug is the microenvironment pH. The dissolution rates of three carboxylic acids, benzoic acid, 2-naphthoic acid and IND under unbuffered conditions have been shown to have poor correlation with the pH in the bulk solution as demonstrated in Figure 21 [86]. There is a region in which the flux of each acid remains constant regardless of bulk pH [86]. These findings suggest that the pH at the dissolving surface is not necessary the same as the bulk solution pH [86]. These carboxylic acids can liberate hydrogen ions that can lower the pH at the dissolving solid surfaces compared to the bulk solution [86]. They also have the ability to self-buffer the pH microenvironment at the interface for a range of bulk pH as shown in Figure 21 [86]. The minimal changes in microenvironment pH at the buffering region result in constant flux in that region [86]. The main influencing factor for microenvironment pH is the degree of ionization at the interface and this is determined by the concentration and pKa of the dissolving substance [86]. IND is the least soluble and weakest acid among the three acids, so its ability in lowering and self-buffering the microenvironment pH is the lowest. This study has demonstrated how microenvironment pH can alter the solubility at the dissolving solid surface and ultimately change the rate at which the compound dissolves.
Fig. 21.

a) Microenvironment pH as a function of bulk pH for 1: indomethacin, 2: 2-naphthoic acid and 3: benzoic acid. b) Flux ratios for indomethacin (▲), 2-naphthoic acid (■) and benzoic acid (●). Reprinted with permission from Elsevier [86].
Knowing that the cocrystal components can be ionizable, it is important to investigate the dissolution mechanism to determine how the microenvironment pH can be modulated through cocrystallization. The concentration of the dissolving substance at the interface is the key determinant for both microenvironment pH and dissolution rate [86–88]. For single component dissolution, this surface concentration is dictated by the solubility of that component [86–88]. We have recently discovered that the concentrations of the components at the dissolving solid surface are dependent on both the solubility and the diffusion coefficients of the components for multi-component dissolution like cocrystals [93].
For most cocrystals, the drug has larger molecular weight than the coformer and as a result, the drug diffuses slower than the coformer. CBZ has a molecular weight about 1.7x higher than SLC and this leads to 1.4x slower diffusion rate compared to SLC. KTZ diffuses at a rate that is 2.4x slower than that of its coformer, FUM because the molecular weight of KTZ is about 4.6x higher than FUM. The difference in diffusion coefficients could result in unequal surface concentrations between the cocrystal components [93].
A schematic representation of the dissolution process for a 1:1 cocrystal, RHA with R as the non-ionizable drug and HA as the monoprotic acidic coformer is shown in Figure 22 [93]. As the cocrystal dissolves into solution, it dissociates into its components R and HA. Because HA is acidic, so it undergoes ionization in basic solution to form A− and the total concentration of the coformer is defined as [A]T, which is the sum of [HA] and [A−]. Before the cocrystal components diffuse away from the solid surface, the saturated layer adjacent to the dissolving surface consists of equal concentrations of R and Atot at the stoichiometric solubility of the cocrystal. As diffusion begins, both components diffuse away from the dissolving surface into the bulk solution according to their own diffusion coefficients and in this case, R is assumed to have a smaller diffusion coefficient compared to HA. Being the slower diffusing component, the drug is able to maintain the same concentration as the solubility of the cocrystal, however, the coformer concentration at the surface is depleted because it has a faster diffusion. The surface concentrations of the cocrystal components are important parameters required in the mass transport analysis for determining the microenvironment pH and rate of the cocrystal dissolution [93].
Fig. 22.

Dissolution process of 1:1 cocrystal with non-ionizable drug R and acidic coformer HA. Subscript 0 denotes the interface and h is the bulk solution. [R] is the total drug concentration and [A]T is the sum of [HA] and [A−]. Sink conditions are assumed in the bulk solution. Reproduced with permission of the American Chemical Society, http://pubs.acs.org/doi/abs/10.1021/acs.molpharmaceut.5b00862 [93].
The dissolution pH dependence of CBZ-SAC and CBZ-SLC has been experimentally demonstrated to follow the microenvironment pH predictions shown in Fig. 23 [93]. The flux of both cocrystals increase as bulk pH increases because of the acidity of the coformers, however, both reach plateau values at the buffering region, where microenvironment pH has minimal changes [93]. The pH effect on the dissolution rate of CBZ-SAC is more profound than that of CBZ-SLC because SAC is more acidic than SLC. There is an excellent agreement between the experimental and theoretical values as shown in Fig. 23. By modeling microenvironment pH, the pH effect on the dissolution rate of cocrystal can be adequately described by the mass transport models [93].
Fig. 23.

Flux of CBZ-SAC at 400 mM SLS (a) and CBZ-SLC at 150 mM SLS (b) as a function of bulk pH. CBZ is highly soluble among low solubility drugs and its critical supersaturation is about 3. For the purpose of studying cocrystal dissolution mechanisms without any conversion to CBZ, SLS concentrations slightly above transition points were used. Reproduced with permission of the American Chemical Society, http://pubs.acs.org/doi/abs/10.1021/acs.molpharmaceut.5b00862 [93].
6. Meaningful Characterization
An important question to answer once a cocrystal is discovered is whether the cocrystal is more soluble than the drug and if so by how much? A second question one will need to address is: will its solubility advantage over drug survive with the addition of other ingredients or solubilizing agents? However, one does not need to measure the full phase solubility diagram for cocrystals (ternary composition: solution, drug and coformer) in order to establish the stability regions and its solubility. One only needs to measure the point of mutual stability of two solid phases of interest, for instance drug and cocrystal phases. This point is an easily measurable stability index, referred to as the eutectic point.
6.1. Key thermodynamic stability indicators
Table 5 summarizes key stability indicators commonly used in the characterization of pharmaceutical solids. Stability indicators are thermodynamic parameters as the term suggests, define regions of thermodynamic stability and instability, which are essential for the development of such materials. For instance, in the case of hydrate/anhydrous forms of a drug, the critical relative humidity is a key indicator of the regions of stability of each form. Similarly other indicators such as pHmax for salts, Tg for amorphous solids, transition temperature for enantiotropic polymorphs are used. Cocrystals, however, are commonly characterized by studying their dissolution behavior without a reliable stability indicator.
Table 5.
Key thermodynamic stability indicators for pharmaceutical solid-state forms
| Solid-state form | Thermodynamic parameter |
|---|---|
| Polymorphs | Transition temperature |
| Hydrates/anhydrous | Critical water activity or critical RH |
| Salts | pHmax |
| Amorphous | Tg, glass transition temperature |
| Cocrystals | Keu, or [coformer]eu and [drug]eu, pHmax, S* and CSC |
6.2. Dissolution measurement and cocrystal supersaturation index
Dissolution is the most common method used in evaluating cocrystal solubility [100–103]. However, this method has limited utility by itself for two main reasons. First, the drug concentration-time profile may be a result of the cocrystal conversion kinetics (dissolution and crystallization rates) and a maximum drug concentration (Cmax) does not correlate with cocrystal solubility (an equilibrium value) [66] (Fig. 24). Second, the findings are not transferable to other conditions (pH or solubilizing agents for example) without time consuming studies on a case-by-case basis for each and every condition. However, cocrystal dissolution assessment can be streamlined from knowledge of the cocrystal SA, supersaturation index, or solubility determined from eutectic measurements [4]. In this way one can select nucleation inhibitors and conditions on the basis of a supersaturation index that will provide a meaningful assessment of cocrystal dissolution kinetics. Without this information, it becomes a trial and error exercise in search of additives and conditions that will provide acceptable dissolved drug levels. It is well recognized that the efficacy of nucleation inhibitors is highly dependent and inversely proportional to supersaturation.
Fig. 24.

Dissolution methods may provide Cmax for moderately soluble cocrystals and may not detect highly soluble cocrystals. As cocrystals dissolve and drug precipitates, drug concentrations can reach a maximum in the case of moderately soluble cocrystals, whereas highly soluble cocrystals may undergo such rapid conversion that eludes detection and drug concentration is maintained close to or at the drug solubility. Adapted from ref.[66] with permission from The Royal Society of Chemistry.
The rate at which a cocrystal converts to the solid drug is dependent of various factors including cocrystal and drug solubility, supersaturation with respect to drug, cocrystal dissolution rate and drug crystallization rate [66]. The cocrystal conversion rate can be reduced by decreasing the cocrystal SA, and this can be achieved by using drug solubilizing agents. In fact, physiologically relevant surfactants can affect the cocrystal supersaturation index and consequently the dissolution of cocrystals as demonstrated in Figs. 25 and 26 for the dissolution of IND-SAC and piroxicam saccharin (PXC-SAC) cocrystals in FeSSIF [68]. IND-SAC achieved a peak concentration of 0.36 mM at 10 minutes during the dissolution in pH 5 buffer and then it rapidly decreased to a constant concentration close to the solubility of the parent drug, which was an indication of the rapid conversion back to the drug form during dissolution [68]. In contrast, the solution mediated transformation of IND-SAC was prevented during dissolution in FeSSIF [68]. IND-SAC was able to achieve and maintain a peak concentration of 4.1 mM for the duration of the experiment as shown in Fig. 25 [68]. Although IND-SAC generated a supersaturation of 15.5 during the dissolution in buffer, it rapidly decreased to 1.5 as it converted back to IND due to the high driving force for crystallization [68]. On the other hand, IND-SAC generated a lower supersaturation level (5.5) during the dissolution in FeSSIF and it was maintained for the duration of the experiment as shown in Fig. 25 [68].
Fig. 25.

IND-SAC dissolution in FeSSIF (red square) and buffer (blue diamond) at 25 °C. (a) [IND]T vs time profile for dissolution and (b) supersaturation generated by IND-SAC during dissolution ([IND]T/STIND [68].
Fig. 26.

PXC-SAC dissolution in FeSSIF (red square) and buffer (blue diamond) at 25 °C. (a) [PXC]T vs time profile for dissolution and (b) supersaturation generated by PXC-SAC during dissolution [PXC]T/SPXC,T [68].
Similar behavior was also observed for PXC-SAC as shown in Fig. 26 [68]. In pH 5 buffer, PXC-SAC rapidly converts to the stable drug form, while the cocrystal was stable throughout the dissolution in FeSSIF [68]. The peak supersaturation for the dissolution of PXC-SAC in buffer was 3 [68]. However, in FeSSIF, PXC-SAC achieved a supersaturation of 14 and maintained for the duration of the experiment [68]. The different cocrystal SA in these dissolution media explain the different dissolution behavior observed for both cocrystals. In pH 5 buffer solution, the SA of IND-SAC was 132 (eutectic pH: 3.66) and this advantage was reduced to 24 in FeSSIF (eutectic pH: 3.65) [68]. For PXC-SAC, a SA of 52 in pH 5 buffer (eutectic pH: 3.64) was reduced to 37 in FeSSIF (eutectic pH: 3.79) [68]. The reduction in SA in FeSSIF is due to the preferential solubilization of the drug by the mixed micelles formed by sodium taurocholate and lecithin in FeSSIF [68]. There was a 5.5 fold reduction in the SA of IND-SAC in FeSSIF compared to buffer, while PXC-SAC only had a 1.4 fold reduction [68]. The larger reduction in solubility advantage of IND-SAC is due to the greater extent of solubilization of IND by FeSSIF [68]. The reduction in cocrystal SA lowers the driving force for phase transformation, and consequently decreases the crystallization kinetics of the drug and prolongs the supersaturation during dissolution.
6.3. Eutectic points as indicators of Scocrystal, SA, and transition points
The eutectic point is characterized by the solution concentrations of drug and coformer ([drug]eu and [coformer]eu) at the point where the solution is doubly saturated with respect to drug and cocrystal. The nature of the eutectic point dictates that when a cocrystal and drug are equally soluble then [coformer]eu = [drug]eu, for a 1:1 cocrystal. The eutectic point is independent of the mass of each phase at equilibrium, and is dependent on temperature, pH, solvent, and additives. Departure of the solution coformer and drug stoichiometric ratio from that of the cocrystal, indicates that the cocrystal is more soluble, then the drug when [drug]eu < [coformer]eu, or that a cocrystal is less soluble, then the drug when [drug]eu > [coformer]eu.
At least two eutectic points exist for a cocrystal, which are differentiated by the phases at equilibrium such as drug and cocrystal, or coformer and cocrystal. Eutectic points offer an experimentally accessible method to assess cocrystal solubility and stability regardless of the solubility relationship between cocrystal and drug [4, 71, 94]. The eutectic points referred here are those between cocrystal and solid drug, unless otherwise stated. Eutectic points as critical indicators of cocrystal solubility have been discussed thoroughly elsewhere [4, 71]. The eutectic point solution compositions have several important features: (1) indicates the thermodynamic stability of cocrystal relative to drug crystal, (2) provides the cocrystal solubility under non-stoichiometric conditions and enables estimation of thermodynamic cocrystal solubility in solution compositions where cocrystal is unstable, and (3) provides the drug solubility under the conditions of the eutectic point measurement (coformer concentration, pH, etc.).
The experimental methods to measure eutectic points have been thoroughly described in the literature [4, 76, 85] and they only require (1) small amounts of cocrystal and drug solid phases slurried in a solution of interest (at desired pH, temperature, additives, etc.), (2) that the suspension reaches saturation or equilibrium with respect to the two solid phases and the solution liquid phase, and (3) measurement of the concentrations of both coformer and drug at this equilibrium [drug]eu and [coformer]eu. It is essential to record the pH and temperature at the eutectic as well as confirm the solid phases at equilibrium. A flowchart of the processes used to determine cocrystal eutectic concentrations is presented in Fig. 27.
Fig. 27.

Flowchart of representative methods used to determine equilibrium solution concentrations of cocrystal components at the eutectic point. In this case the solid phases at equilibrium are cocrystal and solid drug [4].
Eutectic point measurements can be used to access the equilibrium solubility of cocrystals under different solution conditions, such as pH and the presence of solubilizing agents. Besides equilibrium solubility, eutectic concentrations of cocrystal components can also be used to evaluate the solubility relationships between the cocrystal and the parent drug, and the existence of transition points. Eutectic measurements provide meaningful cocrystal characterization as indicated by the relationships in Fig. 28.
Fig. 28.

How Scocrystal, Scocrystal/Sdrug and transition points can be obtained from eutectic point measurements. Eutectic points here refer to 1:1 cocrystal and drug solid phases in equilibrium with solution at a given pH, additive concentrations, and temperature.
6.3.1. Eutectic constant, Keu
The following examples illustrate how solution conditions can influence the eutectic concentrations and the stability of the cocrystal. Fig. 29 shows the drug and coformer concentrations at the eutectic point for carbamazepine-salicylic acid cocrystal (CBZ-SLC) in pure water and in a 1% sodium lauryl sulfate (SLS) solution. It can be seen that in water [drug]eu is lower than [coformer]eu and that addition of surfactant reverses this relationship. [CBZ]eu and [SLC]eu denote the total analytical concentrations of drug and coformer at the eutectic point. In the absence of surfactant, [SLC]eu is higher than [CBZ]eu, indicating that the cocrystal requires excess coformer concentration (4.8x [drug]eu]) to be at equilibrium with pure drug. This situation is reversed in the 1% SLS solution, where [CBZ]eu is higher than [SLC]eu. This simple experiment reveals two very important findings with regards to the cocrystal and drug solubilities and thermodynamic stabilities: 1) in water the cocrystal is more soluble than the drug and in 1% SLS the cocrystal is less soluble than the drug; and 2) this cocrystal shows a transition point in the presence of solubilizing agent, S* and CSC [77].
Fig. 29.

Concentrations of drug (carbamazepine, CBZ) and coformer (salicylic acid, SLC) at the eutectic point for the CBZ-SLC cocrystal and CBZ dihydrate system in water and in 1% SLS. In the absence of surfactant, [SLC]eu > [CBZ]eu, indicating that the cocrystal is more soluble than the drug. This situation is reversed in 1% SLS, where [CBZ]eu > [SLC]eu indicating that the cocrystal is less soluble than the drug. Solid phases at the eutectic point are the cocrystal and CBZ dihydrate, which is the drug solid form in equilibrium with cocrystal in aqueous media. Evaluation of Keu and Scc are described in the text.
Fig. 30 shows the influence of pH on eutectic points of NVP cocrystals [73]. It is observed that for the MLE cocrystal the [coformer]eu is higher than [drug]eu at all pH values. However, for the SAC and SLC cocrystals the [coformer]eu is lower than [drug]eu at pH 1.2 and there is a reversal in this trend as pH increases. This means that the MLE cocrystal is more soluble than drug at these pH values and its solubility increases with pH. In contrast, SAC and SLC cocrystals exhibit lower, equal or higher solubility than the drug depending on pH and as a consequence, these cocrystals exhibit a pHmax [73].
Fig. 30.

Drug and coformer eutectic concentrations at different pH values for a) NVP-MLE, b) NVP-SAC and c) NVP-SLC. MLE cocrystal has higher coformer concentration at eutectic than the drug at all pH values. SAC and SLC cocrystal have a reversal in this trend as pH increase that indicate a pHmax. Reproduced from ref.[73] with permission from The Royal Society of Chemistry.
The eutectic constant, Keu, serves as a key cocrystal stability indicator [85]. Keu is defined as the ratio of coformer to drug activities (a) at the eutectic point and under the assumption of ideal solutions it can be approximated by the concentration ratio as
| (19) |
Keu > 1 for a 1:1 cocrystal indicates that cocrystal is thermodynamically unstable (more soluble) with respect to drug under stoichiometric solution conditions. Keu values below the cocrystal stoichiometric ratio, e.g., < 1 for a 1:1 cocrystal, or < 0.5 for a 2:1 cocrystal indicates that cocrystal is more stable (less soluble) than drug [85]. The decrease in Keu values for CBZ-SLC cocrystal in water compared to 1% SLS (from 4.8 to 0.6) as shown in Fig. 29 is evidence of an unstable cocrystal in water becoming stable by addition of SLS.
The influence of pH on the Keu of nevirapine cocrystals, as shown in Fig. 31, demonstrate that Keu increases with increasing pH for 1:1 cocrystal and this increase is even more pronounced for 2:1 cocrystals. Cocrystal solubilities increase relative to drug solubility in aqueous solution as the [coformer]eu also increases relative to [drug]eu. For the NVP-MLE, in all pH values Keu > 1 meaning that the cocrystal is the more soluble, less stable phase. The greater the coformer ionization, the higher the Keu and the cocrystal SA are. For NVP-SAC and NVP-SLC, at pH 1.2, Keu < 0.5 meaning that the cocrystal is the less soluble, thermodynamically stable phase. This situation is reversed for NVP-SAC at pH values of 2.4 and 2.7, where Keu > 0.5. The same occurs to NVP-SLC at pH values of 3.2 and 4, demonstrating that there is a maximum pH (pHmax) for these cocrystals where the cocrystal is the stable phase relative to the drug phase [73].
Fig. 31.

Predicted and experimental values of Keu and cocrystal solubility advantage (Scocrystal/Sdrug) for 1:1 NVP-MLE and 2:1 NVP-SAC and NVP-SLC cocrystals. Keu is a key indicator of Scocrystal/Sdrug. Keu dependence on pH reveals the cocrystal pHmax as well as the cocrystal increase in solubility over drug as pH increases. At pHmax, Keu = 1 for 1:1 cocrystals and Keu = 0.5 for 2:1 cocrystals. Log axes are used due to the large range of values. Symbols represent experimental values. Numbers next to data points indicate pH at eutectic point or equilibrium pH. Lines were generated according to equations 20 and 21. Solid lines represent 1:1 cocrystals and dashed lines 2:1 cocrystals. Reproduced from ref.[73] with permission from The Royal Society of Chemistry.
The cocrystal SA (Scocrystal/Sdrug) can also be obtained from Keu according to the following equations. For 1:1 cocrystals the relationship is
| (20) |
and for 2:1 cocrystals
| (21) |
These relationships have been shown to be excellent predictors of cocrystal SA. Fig. 31 shows the experimental and predicted Keu dependence on SA and pH for NVP cocrystals. [73] These results demonstrate the remarkably different Keu values for each cocrystal at different pH values and its relationship to cocrystal solubility advantage.
6.3.2. Cocrystal solubility and Ksp
An important property of cocrystals is that their solubility is dependent on the solution concentrations of cocrystal components. During dissolution, cocrystals dissociate into their molecular components and the equilibrium between the solid cocrystal and its components in solution is described by an equilibrium constant referred to as a solubility product, Ksp. It is important to recognize that term “dissociation” refers to the equilibrium equation above and does not mean precipitation of components.
Cocrystal stoichiometric solubility can be obtained from the eutectic solution concentrations of drug and coformer [76] for 1:1 cocrystal according to
| (22) |
For a 2:1 cocrystal the cocrystal solubility is given by
| (23) |
Subscript T refers to total concentration (or analytical concentration) at equilibrium, and is given by the sum of all the drug and coformer species in solution. This may include ionized and nonionized, as well as aqueous and solubilized species. A detailed discussion of micellar solubilization and ionization effects on cocrystal stoichiometric solubilities is presented elsewhere [69, 71].
Cocrystal solubility is then used to calculate Ksp from the appropriate solubility equations as summarized in Fig. 28. It is important to note that Ksp refers to concentration product of free and non-ionized aqueous drug and coformer concentrations. We have developed relationships that allow for determination of Ksp from measurement of Scocrystal,T under ionizing and solubilizing conditions [69, 71, 77].
An example calculation for Ksp from eutectic point measurement follows. For NVP-MLE (1:1 cocrystal) the solid phases in equilibrium at the eutectic are cocrystal and drug. The measured eutectic concentrations at pH 1.3 and 25° C were [NVP]eu,T = 0.0036 M and [MLE]eu,T = 0.1806 M. According to equation 22, the cocrystal solubility under stoichiometric conditions is . The drug solubility at the eutectic point is [NVP]eu,T under the eutectic solution conditions (pH, temperature, coformer concentration). A comparison of drug solubility at the eutectic point with that measured in the absence of coformer provides information of how coformer concentration influences drug solubility.
The cocrystal Ksp can be calculated by solving for Ksp from the equation that describes cocrystal solubility as a function of ionization of the cocrystal constituents, weakly basic drug (NVP) and diprotic acidic coformer (MLE), according to
| (24) |
using Scocrystal evaluated from eutectic point measurements (equation 22) at pH 1.3 and ionization constant values for NVP and MLE reported in the literature: pKa,drug = 2.8 [104] and pKa1,coformer =1.9 and pKa2,coformer = 6.6 [105], gives Ksp = 1.7×10−5 M2. The eutectic concentrations presented here were taken from an average of measurements at one pH value. Small deviations can be seen in Ksp obtained from average or linear regression of measurements at more than one pH value.
6.4. Cocrystal Solubility and Phase Solubility Diagram
The meaning of eutectic points is best appreciated by considering the phase solubility diagrams (PSD) that include cocrystal and drug (or coformer) solid phases in equilibrium with solution phases. This type of phase diagram is characterized by two eutectic points: (1) cocrystal and drug solid phases, and (2) cocrystal and coformer solid phases. The pharmaceutically relevant eutectic point commonly involves the equilibrium of drug and cocrystal solid phases because drugs are often the least water soluble cocrystal component (e.g. class II BCS drugs). Thus conversions between cocrystal and solid drug are more relevant than conversions between cocrystal and coformer. The eutectic point is determined by the intersection of solubility curves as shown in Fig. 32.
Fig. 32.

Schematic phase solubility diagram indicating the eutectic points (*) where cocrystal and drug solid are in equilibrium with solution [4]. Ceu represents the eutectic concentrations of drug and coformer. Two different cocrystals are considered based on their stability with respect to drug under stoichiometric conditions: a stable cocrystal (cocrystal 1) and metastable (cocrystal 2) where cocrystal generates supersaturation with respect to drug. Drug solubility is indicated and is much lower than the solubility of the coformer, which is not shown. Circles represent the solubility of cocrystals in pure solvent. Dashed line illustrates stoichiometric concentrations of cocrystal components which dissolution could follow. This line represents a drug to coformer ratio equal to the cocrystal stoichiometric ratio of the components. Adapted with permission from D. J. Good and N. Rodríguez-Hornedo from ref. [4]. Copyright 2009 American Chemical Society.
The PSD in Fig. 32 represents two different cocrystals, which are either stable (cocrystal 1: low solubility and Ksp) or metastable (cocrystal 2: high solubility and Ksp) with respect to the pure drug form in a given solvent. These curves represent cocrystal solubility product (Ksp) behavior with the drug concentration as a function of coformer concentrations given by Ksp = [drug][coformer] [4]. The drug solubility (horizontal line) is assumed to be much lower than the coformer solubility, which is not shown. A dashed line represents stoichiometric solution concentrations or stoichiometric dissolution of cocrystal in pure solvent and its intersection with the cocrystal solubility curves (marked by circles) indicates the maximum drug concentration associated with the cocrystal solubilities. Unless otherwise specified the term cocrystal solubility refers to stoichiometric solubility. For a metastable cocrystal (cocrystal 2) the drug concentration associated with the cocrystal solubility is greater than the solubility of the stable drug form (horizontal line). The solubility of a metastable cocrystal is not typically a measurable equilibrium and these cocrystals are referred to as incongruently saturating. As a metastable cocrystal dissolves the drug released into solution can crystallize due to supersaturation. This supersaturation is a necessary, but not sufficient condition for crystallization. In certain instances slow nucleation or other kinetic factors might delay crystallization of the favored thermodynamic form and enable measurement of the true equilibrium solubility. In the other case, a congruently saturating cocrystal (cocrystal 1) has a lower drug concentration than the pure drug form at their respective solubility values. Therefore, the solubility of congruently saturating cocrystals can be readily measured from solid cocrystal dissolved and equilibrated with solution.
The eutectic points are the points of intersection of the drug and cocrystal solubility curves in Fig. 32. The eutectic points identify regions of stability of cocrystal and drug. Other eutectic points and associated concentrations exist for the equilibria of cocrystals with different stoichiometry, cocrystal solvates, as well as between cocrystal and solid coformer.
7. Cocrystal Biopharmaceutical Properties
7.1. Challenges in comparing in vitro and in vivo behavior
Inadequate solubility of drug candidates is an ongoing issue in drug development and methodologies to improve solubility are commonly pursued. Cocrystals can generate supersaturation with respect to the less soluble parent drug, and solve bioavailability problems in particular for Biopharmaceutical Classification System (BCS) class II drugs (low aqueous solubility, and high permeability). Pharmacokinetic studies of about 60 cocrystals have been reported and while there is clear improvement on bioavailability for many cocrystals, our limited understanding of their solution and permeability behavior prevents more pharmaceutical cocrystals from being used in drug products [106]. Cocrystals have the potential to supersaturate with respect to less soluble drug and will require a combination of thermodynamic and kinetic approaches to harness their solubility advantage. Several examples of improved solubility, dissolution, and pharmacokinetic behavior are presented.
The IND-SAC cocrystal was found to achieve higher solution concentrations than the parent drug during dissolution [1, 107], and is 13–65 times more soluble than the parent drug in a range of pH 1–3, as determined by equilibrium solubility measurements [65]. The cocrystal was found to improve bioavailability relative to the unformulated parent drug when dosed in dogs [107]. Cocrystals of ITZ increased drug concentration relative to the free drug (4 to 20-fold), and showed similar dissolution to the marketed amorphous formulation (Sporanox®) [27].
There are several examples of cocrystals that exhibit enhanced bioavailability relative to the parent drug, as measured by an increase in the area under the curve (AUC) of the time course of the drug in the plasma. Smith et al. showed that four cocrystals of quercetin had superior bioavailability relative to the parent drug; the highest increase in AUC was achieved by the quercetin-theobromine dihydrate cocrystal and was 10 times higher than that of the parent drug [108]. McNamara et al. showed that a glutaric acid cocrystal of a poorly soluble development compound, 2-[4-(4-chloro-2-fluorophenoxy)phenyl]pyrimidine-4-carboxamide, enhanced the intrinsic dissolution rate by 18-fold which translated to a 3-fold higher AUC relative to drug when dosed in dogs [46]. Other cocrystals reported to increase AUC relative to the parent drug include meloxicam-aspirin (4.4-fold increase in AUC) [11], meloxicam-1-hydroxy-2napthoic acid (1.5-fold increase in AUC) [103], IND-SAC (1.9-fold increase in AUC) [107] as long as drug and cocrystal were compared using the same formulation.
There are cases in which cocrystals generate higher solution concentrations during dissolution relative to the parent drug, but do not show an improvement in bioavailability. For example, lamotrigine-nicotinamide monohydrate exhibited a lower AUC and Cmax when dosed in rats despite demonstrating improved in vitro dissolution in water and acidic media (water 0.1 M HCl, pH =1) [29]. The cocrystal and drug were dosed in a suspension (5% PEG and 95% Methyl cellulose aqueous solution) without consideration of the influence of pH and counterions or coformer on cocrystal solubility and thermodynamic stability under these conditions. The CBZ-SAC cocrystal has a higher solubility than carbamazepine dihydrate (CBZD) [4], however, the pharmacokinetic parameters determined from a bioavailability study of CBZ-SAC in dogs were not statistically different compared to those of the marketed formulation of CBZ (Tegretol®) [109]. Most reported studies do not take into account the huge changes in cocrystal solubility and dissolution as a result of pH, additives or excipients. Cocrystal dissolution studies are often carried out under conditions different than those encountered during dissolution of formulated cocrystals and in vivo dissolution, thus meaningful correlations between cocrystal properties with in vitro and in vivo conditions are scarce.
The importance of formulation on cocrystal dissolution and bioavailability was demonstrated for DNZ-VAN cocrystals [24]. Bioavailability was enhanced 10-fold compared to drug when cocrystal was formulated with 1% vitamin E-TPGS and 2% Klucel LF Pharm hydroxypropylcellulose. The unformulated cocrystal, however, had a modest in vivo improvement of 1.7-fold higher bioavailability compare to the drug. These results also reflected the in vitro dissolution behavior of the cocrystal.
Cocrystals are supersaturating drug delivery systems that like amorphous forms and salts are predisposed to conversion to less soluble drug forms. Consequently, they will require formulation additives to mitigate such conversions while still maintaining high levels of drug during dissolution.
8. Conclusions
This article has focused on establishing the key cocrystal properties that must be measured for the purpose of measuring and fine-tuning cocrystal solubility, supersaturation index, transition points, and thermodynamic stability. Cocrystal solubility is a multi-mechanism process (not just a number) that is highly dependent on environmental conditions, such as pH and the presence of dug solubilizing agents. Even the cocrystal solubility advantage over drug (Scocrystal/Sdrug) can change and be reversed as cocrystal components ionize and interact with endogenous or formulation additives to different extents. Currently used strategies are inefficient since they only measure the kinetic response of cocrystals to solution conditions, which by themselves may not reflect the cocrystal true potential for increasing solubility and dissolution. Thus there is generally an empirical search for cocrystal formulation excipients, dissolution additives, and even crystallization inhibitors that require a large number of experiments with, little if any, translation to new situations. Unlike the commonly used descriptor of cocrystal kinetic solubility, cocrystal eutectic constants provide a value whereby cocrystals can be ranked in terms of their solubility advantage over drug. A strong case is made for eutectic constants to be measured as the most important cocrystal property, since they provide a stability index (reflects how close or far from equilibrium a cocrystal is), whether there are transition points, and provide a quantitative scale of cocrystal true solubility changes with formulation, dissolution, and processing conditions.
Acknowledgments
We gratefully acknowledge partial support from CAPES (PVE Project A065_2013) and from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award number R01GM107146. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations and terms
- RCM
reaction crystallization method
- PSD
phase solubility diagraml
- FeSSIF
fed state simulated intestinal fluid
- [drug]eu
drug concentration at the eutectic point
- [coformer]eu
coformer concentration at the eutectic point
- Keu
eutectic constant, Keu ≡ [coformer]eu/[drug]eu
- Scocrystal
cocrystal solubility
- Sdrug
drug solubility
- Scoformer
solubility of coformer
- ST
total solubility in media with solubilizing agents, ST = Saq + Ss
- Saq
aqueous solubility at a given pH, Saq = Snonionized,aq + Sionized,aq
- Scocrystal,aq
cocrystal solubility in aqueous media
- Scocrystal,T
total cocrystal solubility in solubilizing agent media
- Sdrug,aq
drug solubility in aqueous media
- Sdrug,T
total drug solubility in solubilizing agent media
- pHmax
pH at which both drug and cocrystal have equal solubilities
- CSC
critical stabilization concentration
- S*
solubility at which both drug and cocrystal have equal solubilities
- SR
solubilization ratio, SR=ST/Saq
- SRcocrystal
cocrystal solubilization ratio, (ST/Saq)cocrystal
- SRdrug
drug solubilization ratio, (ST/Saq)drug
- SA
cocrystal solubility advantage, SA = Scocrystal/Sdrug
- CMC
critical micellar concentration
- Ksp
solubility product
- Ks
solubilization constant
- Ka
dissociation constant
- [M]
micellar surfactant concentration
dissolution rate
- J
flux (mass/surface area/time)
- D
diffusion coefficient
- S
equilibrium solubility
- A
surface area of solid drug
- h
thickness of diffusion layer
- pH0
microenvironment pH
- pHbulk
bulk solution pH
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basavoju S, Boström D, Velaga S. Indomethacin–Saccharin Cocrystal: Design, Synthesis and Preliminary Pharmaceutical Characterization. Pharm Res. 2008;25:530–541. doi: 10.1007/s11095-007-9394-1. [DOI] [PubMed] [Google Scholar]
- 2.Aitipamula S, Banerjee R, Bansal AK, Biradha K, Cheney ML, Choudhury AR, Desiraju GR, Dikundwar AG, Dubey R, Duggirala N, Ghogale PP, Ghosh S, Goswami PK, Goud NR, Jetti R, Karpinski P, Kaushik P, Kumar D, Kumar V, Moulton B, Mukherjee A, Mukherjee G, Myerson AS, Puri V, Ramanan A, Rajamannar T, Reddy CM, Rodríguez-Hornedo N, Rogers RD, Row TNG, Sanphui P, Shan N, Shete G, Singh A, Sun CQC, Swift JA, Thaimattam R, Thakur TS, Thaper RK, Thomas SP, Tothadi S, Vangala VR, Variankaval N, Vishweshwar P, Weyna DR, Zaworotko MJ. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst Growth Des. 2012;12:2147–2152. [Google Scholar]
- 3.Gao Y, Zu H, Zhang J. Enhanced dissolution and stability of adefovir dipivoxil by cocrystal formation. J Pharm Pharmacol. 2011;63:483–490. doi: 10.1111/j.2042-7158.2010.01246.x. [DOI] [PubMed] [Google Scholar]
- 4.Good DJ, Rodríguez-Hornedo N. Solubility Advantage of pHarmaceutical Cocrystals. Crystal Growth & Design. 2009;9:2252–2264. [Google Scholar]
- 5.Karki S, Friščić T, Fábián L, Laity PR, Day GM, Jones W. Improving Mechanical Properties of Crystalline Solids by Cocrystal Formation: New Compressible Forms of Paracetamol. Adv Mater. 2009;21:3905–3909. [Google Scholar]
- 6.Wouters J, Rome S, Quere L. Monographs of most Frequent Co-Crystal Formers. Rsc Drug Discov. 2011:338–382. [Google Scholar]
- 7.Blagden N, Colesb SJ, Berry DJ. Pharmaceutical co-crystals - are we there yet? Crystengcomm. 2014;16:5753–5761. [Google Scholar]
- 8.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 9.Aakeroy CB, Fasulo ME, Desper J. Cocrystal or salt: Does it really matter? Mol Pharmaceut. 2007;4:317–322. doi: 10.1021/mp060126o. [DOI] [PubMed] [Google Scholar]
- 10.Schultheiss N, Newman A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst Growth Des. 2009;9:2950–2967. doi: 10.1021/cg900129f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheney ML, Weyna DR, Shan N, Hanna M, Wojtas L, Zaworotko MJ. Coformer selection in pharmaceutical cocrystal development: A case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J Pharm Sci. 2011;100:2172–2181. doi: 10.1002/jps.22434. [DOI] [PubMed] [Google Scholar]
- 12.Etter MC. Hydrogen bonds as design elements in organic chemistry. The Journal of Physical Chemistry. 1991;95:4601–4610. [Google Scholar]
- 13.Desiraju GR. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angewandte Chemie International Edition in English. 1995;34:2311–2327. [Google Scholar]
- 14.Etter MC. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc Chem Res. 1990;23:120–126. [Google Scholar]
- 15.Donohue J. The Hydrogen Bond in Organic Crystals. The Journal of Physical Chemistry. 1952;56:502–510. [Google Scholar]
- 16.Desiraju GR. Designer crystals: intermolecular interactions, network structures and supramolecular synthons. Chem Commun. 1997:1475–1482. [Google Scholar]
- 17.Almarsson O, Zaworotko MJ. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines? Chem Commun. 2004:1889–1896. doi: 10.1039/b402150a. [DOI] [PubMed] [Google Scholar]
- 18.Fleischman SG, Kuduva SS, McMahon JA, Moulton B, Bailey Walsh RD, Rodríguez-Hornedo N, Zaworotko MJ. Crystal Engineering of the Composition of Pharmaceutical Phases: Multiple-Component Crystalline Solids Involving Carbamazepine. Cryst Growth Des. 2003;3:909–919. [Google Scholar]
- 19.Kitaigorodskii A. The principle of close packing and the condition of thermodynamic stability of organic crystals. Acta Crystallographica. 1965;18:585–590. [Google Scholar]
- 20.Fábián Ls. Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals. Cryst Growth Des. 2009;9:1436–1443. [Google Scholar]
- 21.Childs SL, Rodríguez-Hornedo N, Reddy LS, Jayasankar A, Maheshwari C, McCausland L, Shipplett R, Stahly BC. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. Crystengcomm. 2008;10:856–864. [Google Scholar]
- 22.Sanphui P, Goud NR, Khandavilli UBR, Nangia A. Fast Dissolving Curcumin Cocrystals. Cryst Growth Des. 2011;11:4135–4145. [Google Scholar]
- 23.Chadha R, Saini A, Arora P, Jain DS, Dasgupta A, Guru Row TN. Multicomponent solids of lamotrigine with some selected coformers and their characterization by thermoanalytical, spectroscopic and X-ray diffraction methods. Crystengcomm. 2011;13:6271–6284. [Google Scholar]
- 24.Childs SL, Kandi P, Lingireddy SR. Formulation of a Danazol Cocrystal with Controlled Supersaturation Plays an Essential Role in Improving Bioavailability. Mol Pharmaceut. 2013;10:3112–3127. doi: 10.1021/mp400176y. [DOI] [PubMed] [Google Scholar]
- 25.Lipert MP, Roy L, Childs SL, Rodríguez-Hornedo N. Cocrystal Solubilization in Biorelevant Media and its Prediction from Drug Solubilization. J Pharm Sci. 2015 doi: 10.1002/jps.24640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima T, Tsutsumi S, Yamamoto K, Ikeda Y, Moriwaki T. High-throughput cocrystal slurry screening by use of in situ Raman microscopy and multi-well plate. Int J Pharm. 2010;399:52–59. doi: 10.1016/j.ijpharm.2010.07.055. [DOI] [PubMed] [Google Scholar]
- 27.Remenar JF, Morissette SL, Peterson ML, Moulton B, MacPhee JM, Guzmán HR, Almarsson Ö. Crystal Engineering of Novel Cocrystals of a Triazole Drug with 1,4-Dicarboxylic Acids. J Am Chem Soc. 2003;125:8456–8457. doi: 10.1021/ja035776p. [DOI] [PubMed] [Google Scholar]
- 28.Otte A, Boerrigter SX, Pinal R. Cocrystallization of Ketoconazole with Dicarboxylic Acids. AAPS; Chicago, IL: 2012. [Google Scholar]
- 29.Cheney ML, Shan N, Healey ER, Hanna M, Wojtas L, Zaworotko MJ, Sava V, Song S, Sanchez-Ramos JR. Effects of Crystal Form on Solubility and Pharmacokinetics: A Crystal Engineering Case Study of Lamotrigine. Cryst Growth Des. 2009;10:394–405. [Google Scholar]
- 30.Cheney ML, Weyna DR, Shan N, Hanna M, Wojtas L, Zaworotko MJ. Supramolecular Architectures of Meloxicam Carboxylic Acid Cocrystals, a Crystal Engineering Case Study. Cryst Growth Des. 2010;10:4401–4413. [Google Scholar]
- 31.Caira MR, Bourne SA, Samsodien H, Engel E, Liebenberg W, Stieger N, Aucamp M. Co-crystals of the antiretroviral nevirapine: crystal structures, thermal analysis and dissolution behaviour. Crystengcomm. 2012;14:2541–2551. [Google Scholar]
- 32.Childs SL, Hardcastle KI. Cocrystals of Piroxicam with Carboxylic Acids. Crystal Growth & Design. 2007;7:1291–1304. [Google Scholar]
- 33.Bhatt PM, Ravindra NV, Banerjee R, Desiraju GR. Saccharin as a salt former. Enhanced solubilities of saccharinates of active pharmaceutical ingredients. Chem Commun. 2005:1073–1075. doi: 10.1039/b416137h. [DOI] [PubMed] [Google Scholar]
- 34.Bethune SJ, Schultheiss N, Henck JO. Improving the Poor Aqueous Solubility of Nutraceutical Compound Pterostilbene through Cocrystal Formation. Cryst Growth Des. 2011;11:2817–2823. [Google Scholar]
- 35.Schultheiss N, Bethune S, Henck JO. Nutraceutical cocrystals: utilizing pterostilbene as a cocrystal former. Crystengcomm. 2010;12:2436–2442. [Google Scholar]
- 36.Weyna David R, Shattock Tanise, Vishweshwar Peddy, Zaworotko MJ. Synthesis and Structural Characterization of Cocrystals and Pharmaceutical Cocrystals: Mechanochemistry vs Slow Evaporation from Solution. Crystal Growth & Design. 2009;9:1106–1123. [Google Scholar]
- 37.Yu ZQ, Chow PS, Tan RBH. Operating Regions in Cooling Cocrystallization of Caffeine and Glutaric Acid in Acetonitrile. Crystal Growth & Design. 2010;10:2382–2387. [Google Scholar]
- 38.Yu ZQ, Chow PS, Tan RBH, Ang WH. Supersaturation Control in Cooling Polymorphic Co-Crystallization of Caffeine and Glutaric Acid. Crystal Growth & Design. 2011;11:4525–4532. [Google Scholar]
- 39.Childs SL, Chyall LJ, Dunlap JT, Smolenskaya VN, Stahly BC, Stahly GP. Crystal Engineering Approach to Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Aicds. Journal of American Chemical Society. 2004;126:13335–13342. doi: 10.1021/ja048114o. [DOI] [PubMed] [Google Scholar]
- 40.Zhang GGZ, Henry RF, Borchardt TB, Lou X. Efficient Co-crystal Screening Using Solution-Mediated Phase Transformation. Journal of Pharmaceutical Sciences. 2006;96:990–995. doi: 10.1002/jps.20949. [DOI] [PubMed] [Google Scholar]
- 41.Karki S, Friscic T, Jones W. Control and interconversion of cocrystal stoichiometry in grinding: stepwise mechanism for the formation of a hydrogen-bonded cocrystal. Crystengcomm. 2009;11:470–481. [Google Scholar]
- 42.Friscic T, Jones W. Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding. Cryst Growth Des. 2009;9:1621–1637. [Google Scholar]
- 43.Trask AV, Jones W. Crystal engineering of organic cocrystals by the solid-state grinding approach. Top Curr Chem. 2005;254:41–70. [Google Scholar]
- 44.Shan N, Toda F, Jones W. Mechanochemistry and co-crystal formation: effect of solvent on reaction kinetics. Chem Commun. 2002:2372–2373. doi: 10.1039/b207369m. [DOI] [PubMed] [Google Scholar]
- 45.Trask AV, Motherwell WDS, Jones W. Solvent-drop grinding: green polymorph control of cocrystallisation. Chem Commun. 2004:890–891. doi: 10.1039/b400978a. [DOI] [PubMed] [Google Scholar]
- 46.McNamara DP, Childs SL, Giordano J, Iarriccio A, Cassidy J, Shet MS, Mannion R, O’Donnell E, Park A. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm Res. 2006;23:1888–1897. doi: 10.1007/s11095-006-9032-3. [DOI] [PubMed] [Google Scholar]
- 47.Lu E, Rodríguez-Hornedo N, Suryanarayanan R. A rapid thermal method for cocrystal screening. Crystengcomm. 2008;10:665–668. [Google Scholar]
- 48.Rager T, Hilfiker R. Cocrystal Formation from Solvent Mixtures. Crystal Growth & Design. 2010;10:3237–3241. [Google Scholar]
- 49.Gagniere E, Mangin D, Puel F, Bebon C, Klein J-P, Monnier O, Garcia E. Cocrystal Formation in Solution: In Situ Solute Concentration Monitoring of the Two Components and Kinetic Pathways. Crystal Growth & Design. 2009;9:3376–3383. [Google Scholar]
- 50.Sheikh AY, Rahim SA, Hammond RB, Roberts KJ. Scalable Solution Cocrystallization: Case of Carbamazepine-Nicotinamide I. CrystEngComm. 2008;11:501–509. [Google Scholar]
- 51.Rager T, Hilfiker R. Application of Phase Diagrams in Co-crystal Search and Preparation. In: Johan Wouters LQ, editor. Rsc Drug Discov. Royal Society of Chemistry; 2011. pp. 280–299. [Google Scholar]
- 52.Coquerel G. Limits of the Co-crystal Concept and Beyond. In: Quere JWaL., editor. Pharmaceutical Salts and Co-crystal. Royal Society of Chemistry; 2011. pp. 300–317. [Google Scholar]
- 53.Rodríguez-Hornedo N, Nehm SJ, Seefeldt KF, Pagán-Torres Y, Falkiewicz CJ. Reaction Crystallization of Pharmaceutical Molecular Complexes. Mol Pharmaceut. 2006;3:362–367. doi: 10.1021/mp050099m. [DOI] [PubMed] [Google Scholar]
- 54.Gagniere E, Mangin D, Puel F, Bebon C, Klein JP, Monnier O, Garcia E. Cocrystal Formation in Solution: In Situ Solute Concentration Monitoring of the Two Components and Kinetic Pathways. Cryst Growth Des. 2009;9:3376–3383. [Google Scholar]
- 55.Gagniere E, Mangin D, Puel F, Rivoire A, Monnier O, Garcia E, Klein JR. Formation of co-crystals: Kinetic and thermodynamic aspects. J Cryst Growth. 2009;311:2689–2695. [Google Scholar]
- 56.Friscic T, Childs SL, Rizvi SAA, Jones W. The role of solvent in mechanochemical and sonochemical cocrystal formation: a solubility-based approach for predicting cocrystallisation outcome. Crystengcomm. 2009;11:418–426. [Google Scholar]
- 57.Jayasankar A, Somwangthanaroj A, Shao ZJ, Rodríguez-Hornedo N. Cocrystal formation during cogrinding and storage is mediated by amorphous phase. Pharm Res. 2006;23:2381–2392. doi: 10.1007/s11095-006-9110-6. [DOI] [PubMed] [Google Scholar]
- 58.Jayasankar A, Good DJ, Rodríguez-Hornedo N. Mechanisms by which moisture generates cocrystals. Mol Pharmaceut. 2007;4:360–372. doi: 10.1021/mp0700099. [DOI] [PubMed] [Google Scholar]
- 59.Braga D, Giaffreda SL, Rubini K, Grepioni F, Chierotti MR, Gobetto R. Making crystals from crystals: three solvent-free routes to the hydrogen bonded co-crystal between 1,1′-di-pyridyl-ferrocene and anthranilic acid. Crystengcomm. 2007;9:39–45. [Google Scholar]
- 60.Braga D, Giaffreda SL, Grepioni F, Chierotti MR, Gobetto R, Palladino G, Polito M. Solvent effect in a “solvent free” reaction. Crystengcomm. 2007;9:879–881. [Google Scholar]
- 61.Good D, Miranda C, Rodríguez-Hornedo N. Dependence of cocrystal formation and thermodynamic stability on moisture sorption by amorphous polymer. Crystengcomm. 2011;13:1181–1189. [Google Scholar]
- 62.Seefeldt K, Miller J, Alvarez-Nunez F, Rodríguez-Hornedo N. Crystallization pathways and kinetics of carbamazepine-nicotinamide cocrystals from the amorphous state by in situ thermomicroscopy, spectroscopy and calorimetry studies. J Pharm Sci. 2007;96:1147–1158. doi: 10.1002/jps.20945. [DOI] [PubMed] [Google Scholar]
- 63.Childs Scott L, Mougin Patricia, Stahly BC. Screening for Solid Forms by Ultrsound Crystallization and Cocrystallization using. Ultrasound. 2005 [Google Scholar]
- 64.Dhumal RS, Kelly AL, York P, Coates PD, Paradkar A. Cocrystalization and Simultaneous Agglomeration Using Hot Melt Extrusion. Pharmaceutical Research. 2010;27:2725–2733. doi: 10.1007/s11095-010-0273-9. [DOI] [PubMed] [Google Scholar]
- 65.Alhalaweh A, Roy L, Rodríguez-Hornedo N, Velaga SP. pH-Dependent Solubility of Indomethacin-Saccharin and Carbamazepine-Saccharin Cocrystals in Aqueous Media. Mol Pharmaceut. 2012;9:2605–2612. doi: 10.1021/mp300189b. [DOI] [PubMed] [Google Scholar]
- 66.Roy L, Lipert MP, Rodríguez-Hornedo N. Rsc Drug Discov. Royal Society of Chemistry; 2012. Co-crystal Solubility and Thermodynamic Stability; pp. 247–279. [Google Scholar]
- 67.Roy L. Doctoral Dissertation. University of Michigan; 2013. Engineering Cocrystal and Cocrystalline Salt Solubility by Modulation of Solution Phase Chemistry. Retrieved from Deep Blue. ( http://hdl.handle.net/2027.42/98067) [Google Scholar]
- 68.Lipert MP. Doctoral Dissertation. University of Michigan; 2015. Predicting the Influence of Drug Solubilizing Agents on Cocrystal Solubility, Stability, and Transition Points. [Google Scholar]
- 69.Huang N, Rodríguez-Hornedo N. Engineering Cocrystal Solubility, Stability, and pH(max) by Micellar Solubilization. J Pharm Sci. 2011;100:5219–5234. doi: 10.1002/jps.22725. [DOI] [PubMed] [Google Scholar]
- 70.Lipert MP, Rodríguez-Hornedo N. Cocrystal Transition Points: Role of Cocrystal Solubility, Drug Solubility, and Solubilizing Agents. Mol Pharmaceut. 2015;12:3535–3546. doi: 10.1021/acs.molpharmaceut.5b00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bethune SJ, Huang N, Jayasankar A, Rodríguez-Hornedo N. Understanding and Predicting the Effect of Cocrystal Components and pH on Cocrystal Solubility. Cryst Growth Des. 2009;9:3976–3988. [Google Scholar]
- 72.Reddy LS, Bethune SJ, Kampf JW, Rodríguez-Hornedo N. Cocrystals and Salts of Gabapentin: pH Dependent Cocrystal Stability and Solubility. Cryst Growth Des. 2009;9:378–385. [Google Scholar]
- 73.Kuminek G, Rodríguez-Hornedo N, Siedler S, Rocha HVA, Cuffini SL, Cardoso SG. How cocrystals of weakly basic drugs and acidic coformers might modulate solubility and stability. Chem Commun. 2016 doi: 10.1039/c6cc00898d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Y, Rodríguez-Hornedo N. Solubility, pHmax, and Dissolution of Ketoconazole Cocrystals in Aqueous Media. Poster presentation at the 2015 AAPS Annual Meeting and Exposition; Orlando, FLA. 2015. Poster M1208. [Google Scholar]
- 75.Pereira BG, Fonte-Boa FD, Resende JALC, Pinheiro CB, Fernandes NG, Yoshida MI, Vianna-Soares CD. Pseudopolymorphs and intrinsic dissolution of nevirapine. Cryst Growth Des. 2007;7:2016–2023. [Google Scholar]
- 76.Huang N, Rodríguez-Hornedo N. Engineering cocrystal thermodynamic stability and eutectic points by micellar solubilization and ionization. Crystengcomm. 2011;13:5409–5422. [Google Scholar]
- 77.Huang N, Rodríguez-Hornedo N. Effect of Micellar Solubilization on Cocrystal Solubility and Stability. Cryst Growth Des. 2010;10:2050–2053. [Google Scholar]
- 78.Maheshwari C, Andre V, Reddy S, Roy L, Duarte T, Rodrigez-Hornedo N. Tailoring aqueous solubility of a highly soluble compound via cocrystallization: effect of coformer ionization, pH(max) and solute-solvent interactions. Crystengcomm. 2012;14:4801–4811. [Google Scholar]
- 79.Maheshwari C University of Michigan. Pharmaceutical Sciences. Understanding the solution phase chemistry and solid state thermodynamic behavior of pharmaceutical cocrystals. 2012:221. [Google Scholar]
- 80.Stahl PH, Wermuth CG. Handbook of pharmaceutical salts : properties, selection, and use. 2. Wiley-VCH; Weinheim: 2011. International Union of Pure and Applied Chemistry. rev. ed. [Google Scholar]
- 81.Avdeef A, Voloboy D, Foreman A. 5.17 - Dissolution and Solubility. In: Triggle JBTJ, editor. Comprehensive Medicinal Chemistry II. Elsevier; Oxford: 2007. pp. 399–423. [Google Scholar]
- 82.Serajuddin AT. Salt formation to improve drug solubility. Adv Drug Deliv Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 83.Roy L, Rodríguez-Hornedo N. A Rational Approach for Surfactant Selection to Modulate Cocrystal Solubility and Stability. Poster presentation at the 2010 AAPS Annual Meeting and Exposition; New Orleans, LA. 2010. Poster R6072. [Google Scholar]
- 84.Lipert MP, Rodrigez-Hornedo N. Cocrystal transition points: Role of cocrystal solubility, drug solubility, and solubilizing agents. Molecular Pharmaceutics. 2015 doi: 10.1021/acs.molpharmaceut.5b00111. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Good DJ, Rodríguez-Hornedo N. Cocrystal Eutectic Constants and Prediction of Solubility Behavior. Cryst Growth Des. 2010;10:1028–1032. [Google Scholar]
- 86.Mooney KG, Mintun MA, Himmelstein KJ, Stella VJ. Dissolution Kinetics of Carboxylic Acids I: Effect of pH under Unbuffered Conditions. American Pharmaceutical Association. 1981;70:13–22. doi: 10.1002/jps.2600700103. [DOI] [PubMed] [Google Scholar]
- 87.Mooney KG, Mintun MA, Himmelstein KJ, Stella VJ. Dissolution Kinetics of Carboxylic Acids II: Effect of Buffers. American Pharmaceutical Association. 1981;70:22–32. doi: 10.1002/jps.2600700104. [DOI] [PubMed] [Google Scholar]
- 88.Aunins JG, Southard MZ, Myers RA, Himmelstein kJ, Stella VJ. Dissolution of Carboxylic Acids III: The effect of Polyionizable Buffers. Journal of Pharmaceutical Sciences. 1985;74:1305–1316. doi: 10.1002/jps.2600741212. [DOI] [PubMed] [Google Scholar]
- 89.Serajuddin ATM, Jarowski CI. Effect of Diffusion Layer pH and Solubility on the Dissoluiotn Rate of Pharmaceutical Acids and Their Sodium Salts II: Salicylic Acid, Theophylline, and Benzoic Acid. Journal of Pharmaceutical Sciences. 1985;74:148–154. doi: 10.1002/jps.2600740209. [DOI] [PubMed] [Google Scholar]
- 90.Serajuddin ATM, Jarowski CI. Effect of Diffusion Layer pH and Solubility on the Dissolution Rate of Pharmaceutical Bases and Their Hydrochloride Salts I: Phenazopyridine. journal of Pharmaceutical Sciences. 1985;74:142–147. doi: 10.1002/jps.2600740208. [DOI] [PubMed] [Google Scholar]
- 91.Serajuddin ATM, Mufson D. pH-Solubility Profiles of Organic Bases and Their Hydrochloride Salts. Pharmaceutical Research. 1985;2:65–68. doi: 10.1023/A:1016382426347. [DOI] [PubMed] [Google Scholar]
- 92.Ozturk SS, Palsson BO, Dressman JB. Dissolution of Ionizable Drugs in Buffered and Unbuffered Solutions. Pharmaceutical Research. 1988;5:272–282. doi: 10.1023/a:1015970502993. [DOI] [PubMed] [Google Scholar]
- 93.Cao FJ, Amidon GL, Rodríguez-Hornedo N, Amidon GE. Mechanistic Analysis of Cocrystal Dissolution as a Function of pH and Micellar Solubilization. Mol Pharmaceut. 2016;13:1030–1046. doi: 10.1021/acs.molpharmaceut.5b00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jayasankar A, Reddy LS, Bethune SJ, Rodríguez-Hornedo N. Role of Cocrystal and Solution Chemistry on the Formation and Stability of Cocrystals with Different Stoichiometry. Cryst Growth Des. 2009;9:889–897. [Google Scholar]
- 95.Amidon GL, Lennernas H, Shah VP, Crison JR. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 96.Siepmann J, Siepmann F. Mathematical Modeling of Drug Dissolution. Int J Pharm. 2013 doi: 10.1016/j.ijpharm.2013.04.044. [DOI] [PubMed] [Google Scholar]
- 97.Sugano K, Okazaki A, Sugimoto S, Tavornvipas S, Omura A, Mano T. Solubility and Dissolution Profile Assessment in Drug Discovery. Drug Metab Pharmacokinet. 2007;22:225–254. doi: 10.2133/dmpk.22.225. [DOI] [PubMed] [Google Scholar]
- 98.Nernst W. Theorie der Reaktionsgeschwindigkeit in heterogenen systemen. Zeitschrift für Physikalische Chemie. 1904;47:52–55. [Google Scholar]
- 99.Noyes AA, Whitney WR. The Rate of Solution of Solid Substances in Their Own Soluitons. Journal of American Chemical Society. (19):930–934. [Google Scholar]
- 100.Cheney ML, Shan N, Healey ER, Hanna M, Wojtas L, Zaworotko MJ, Sava V, Song S, Sanchez-Ramos JR. Effects of Crystal Form on Solubility and Pharmacokinetics: A Crystal Enginneering Case Study of Lamotrigine. Cryst Growth Des. 2010;10:394–405. [Google Scholar]
- 101.Thakuria R, Delori A, Jones W, Lipert MP, Roy L, Rodríguez-Hornedo N. Pharmaceutical Cocrystals and Poorly Soluble Drugs. International Journal of Pharmaceutics. 2013;453:101–125. doi: 10.1016/j.ijpharm.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 102.Martin FA, Pop MM, Borodi G, Filip X, Kacso I. Ketoconazole Salt and Co-crystals with Enhanced Aqueous Solubility. Cryst Growth Des. 2013;13:4295–4304. [Google Scholar]
- 103.Weyna DR, Cheney ML, Shan N, Hanna M, Zaworotko MJ, Sava V, Song S, Sanchez-Ramos JR. Improving Solubility and Pharmacokinetics of Meloxicam via Multiple-Component Crystal Formation. Mol Pharmaceut. 2012;9:2094–2102. doi: 10.1021/mp300169c. [DOI] [PubMed] [Google Scholar]
- 104.The Merck index. 13. Merck Research Laboratories; Whitehouse Station, NJ: 2001. [Google Scholar]
- 105.Dawson RMC. Data for biochemical research. Clarendon Press; Oxford: 1959. [Google Scholar]
- 106.Shan N, Perry ML, Weyna DR, Zaworotko MJ. Impact of pharmaceutical cocrystals: the effects on drug pharmacokinetics. Expert Opin Drug Met. 2014;10:1255–1271. doi: 10.1517/17425255.2014.942281. [DOI] [PubMed] [Google Scholar]
- 107.Jung M-S, Kim J-S, Kim M-S, Alhalaweh A, Cho W, Hwang S-J, Velaga SP. Bioavailability of indomethacin-saccharin cocrystals. J Pharm Pharmacol. 2010;62:1560–1568. doi: 10.1111/j.2042-7158.2010.01189.x. [DOI] [PubMed] [Google Scholar]
- 108.Smith AJ, Kavuru P, Wojtas L, Zaworotko MJ, Shytle RD. Cocrystals of Quercetin with Improved Solubility and Oral Bioavailability. Mol Pharmaceut. 2011;8:1867–1876. doi: 10.1021/mp200209j. [DOI] [PubMed] [Google Scholar]
- 109.Hickey MB, Peterson ML, Scoppettuolo LA, Morrisette SL, Vetter A, Guzmán H, Remenar JF, Zhang Z, Tawa MD, Haley S, Zaworotko MJ, Almarsson Ö. Performance comparison of a co-crystal of carbamazepine with marketed product. Eur J Pharm Biopharm. 2007;67:112–119. doi: 10.1016/j.ejpb.2006.12.016. [DOI] [PubMed] [Google Scholar]