

ABSTRACT
Tumor necrosis factor related apoptosis-inducing ligand (TRAIL) has tremendous promise in treating various forms of cancers. However, many cancer cells exhibit or develop resistance to TRAIL. Interestingly, many studies have identified several secondary agents that can overcome TRAIL resistance. To expand on these studies, we conducted an extensive drug-re-profiling screen to identify FDA-approved compounds that can be used clinically as TRAIL-sensitizing agents in a very malignant type of brain cancer, Glioblastoma Multiforme (GBM). Using selected isogenic GBM cell pairs with differential levels of TRAIL sensitivity, we revealed 26 TRAIL-sensitizing compounds, 13 of which were effective as single agents. Cardiac glycosides constituted a large group of TRAIL-sensitizing compounds, and they were also effective on GBM cells as single agents. We then explored a second class of TRAIL-sensitizing drugs, which were enhancers of TRAIL response without any effect on their own. One such drug, Mitoxantrone, a DNA-damaging agent, did not cause toxicity to non-malignant cells at the doses that synergized with TRAIL on tumor cells. We investigated the downstream changes in apoptosis pathway components upon Mitoxantrone treatment, and observed that Death Receptors (DR4 and DR5) expression was upregulated, and pro-apoptotic and anti-apoptotic gene expression patterns were altered in favor of apoptosis. Together, our results suggest that combination of Mitoxantrone and TRAIL can be a promising therapeutic approach for GBM patients.
KEYWORDS: Apoptosis, GBM, Mitoxantrone, resistance, TRAIL
Abbreviations
- TRAIL
tumor necrosis factor related apoptosis-inducing ligand
- GBM
glioblastoma multiforme
- TMZ
temozolomide
- IAP
inhibitors of apoptosis protein
- SD
standard deviation
- BBB
Blood-brain barrier
- ATCC
American Tissue Type Culture Collection
Introduction
Glioblastoma multiforme (GBM) is the most common of all malignant brain and CNS tumors and relative survival estimates are quite low; 5.1% of patients survive 5 year post diagnosis.1 The aggressive nature and heterogeneity of GBMs make treatment very difficult. Current therapies include surgery, radiation therapy and administration of Temozolomide (TMZ), an alkylating agent and the most widely used drug for GBM patients. However TMZ can confer a modest increase in patient survival due to inherent or acquired resistance of tumor cells.2 Similarly, most therapeutic approaches applied as a single chemotherapeutic agent fail to significantly increase patient survival, making the use of multiple agents that target multiple hallmarks of cancers a necessity.
In tumors, reactivating dormant apoptotic programs with pro-apoptotic ligands or small molecules is a promising approach to direct tumor cells to self-destruct. As such, the tumor-selective killing capacity of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)3 makes it a potential treatment option in GBM. Binding of TRAIL to death receptors, DR4 and/or DR5, triggers the caspase-dependent extrinsic apoptosis pathway, which also cooperates with the intrinsic apoptosis pathway mediated by mitochondria.4 To exploit the tumor killing ability of TRAIL in therapy, different recombinant forms of human TRAIL or DR4/DR5 agonist antibodies have been developed and are being tested. 4,5 Although TRAIL has the ability to selectively kill cancer cells,3 many cancer cells are intrinsically resistant or acquire resistance to TRAIL. While the mechanisms behind TRAIL resistance are still ill-defined, they include aberrant expression or dysregulation of apoptosis pathway components at the transcriptional level, such as the suppression of death receptors6 or at the protein level, such as the degradation of FLIP.7 Apoptosis resistance can be caused by reduced caspase expression,8 increased expression of anti-apoptotic molecules such as the inhibitors of apoptosis proteins (IAPs), overexpression of Bcl-2 family members and other inhibitors of intrinsic apoptosis pathway.6,9
Accumulating evidence suggest that sensitization of tumor cells to TRAIL is possible10,11 through secondary agents. A widely studied approach for TRAIL-sensitization involves the use of secondary therapeutics in addition to TRAIL, where the timing and sequence of application of each agent might be different. For example, it has been reported that combination of Bortezomib, a proteasome inhibitor, and TRAIL synergize and lead tumor cells to undergo apoptosis, as shown for GBM cell lines and primary tumors.10 However, most studies have traditionally focused on a small number of compounds rather than large-scale libraries. While there is growing interest in the identification of single therapeutic agents through chemical library screens for all cancer types, there has been relatively much lower attention to identification of novel combination therapies in GBM cells.
In this study, we aimed to identify molecules with a capacity to augment TRAIL efficacy and overcome resistance in an isogenic GBM cell line model system, where TRAIL-mid-sensitive and TRAIL-resistant cells were screened in parallel. We utilized drugs that are already FDA-approved with a potential to be translated to clinical repurposing. Our screen among 1200 FDA-approved drugs revealed Mitoxantrone as a potential and powerful candidate as a TRAIL-sensitizing agent in GBM cells.
Results
Screen among 1200 FDA-approved drugs reveals 26 drugs as novel TRAIL-sensitizing agents
To identify drugs with novel TRAIL-sensitizing potential, we first examined the basal level of TRAIL responses of our GBM cells lines. According to the degree of cell death observed with a range of TRAIL concentrations (0–500 ng/ml), we categorized the cell lines as sensitive, mid-sensitive and resistant to TRAIL (Fig. S1). While the A172 cells die completely with TRAIL, the LN229 or U373 cells are resistant and do not die. On the other hand, U87MG cells die up to 40% at the indicated concentrations. We chose U87MG cells for the screen as these cells displayed medium level of sensitivity and had the potential to display higher level of apoptosis when combined with secondary agents.
With the aim of finding new drugs that could cooperate with TRAIL in GBM cells, we utilized a chemical library of 1200 FDA approved drugs. These drugs, all with established therapeutic effects and chemical structures, belonged to 15 different therapeutic classes by the supplier, namely, endocrinology, cardiovascular, immunology, diagnostic, metabolism, allergology, dermatology, gastroenterology, hematology, ophthalmology, neuromuscular, infectiology, respiratory, central nervous system, and oncology. To obtain new TRAIL-sensitizing agents among these drugs, we conducted an ATP-based cell viability screen on U87MG cells with a fixed concentration of TRAIL treatment (25 ng/ml), which was previously defined by dose-response experiments and caused 10–30% cell death as a single agent (Fig. S1). In parallel, we tested the efficacy of each drug on GBM cells as single agents. Since the drug library was supplied in DMSO, DMSO-only treated cells were included in each plate as negative controls, in addition to untreated controls. We included Bortezomib as positive control, since it is a known TRAIL sensitizer in GBM cells.10
First, we assessed the effect of drugs applied at a concentration of 5 µM as single agents on U87MG cell viability (Fig. 1A). Previously, this FDA-approved chemical library was used in a variety of cancer screens in concentrations of 2 – 10 µM 12-15 and 5 µM was the chosen dosage for our TRAIL-sensitizing screen. The average cell viability of the screen with single agents was 88 ± 7.5% compared to controls. Taking 3 standard deviations (SDs) below and above the mean, we defined the lower threshold as 64%. Accordingly, 13 of 1200 agents caused reduction in U87MG cell viability below the threshold after 24 hours of treatment (Fig. 1C). These drugs were Doxorubicin hydrochloride (39 ± 8.1%), Alexidine dihydrochloride (62 ± 5.1%), Monensin sodium salt (62 ± 8.8%), Camptothecine (S,+) (61 ± 6.1%), Lanatoside C (31 ± 7.3%), Digitoxigenin (39 ± 6.3%) Proscillaridin A (42 ± 4.8%), Digoxigenin (52 ± 2.6%), Pyrivinium pamoate (52 ± 1.6%), Niclosamide (41 ± 0.7%), Quinacrine dihydrochloride (61 ± 2.2%), Terfenadine (62 ± 6.7%), and Astemizole (64 ± 3.4%).
Figure 1.
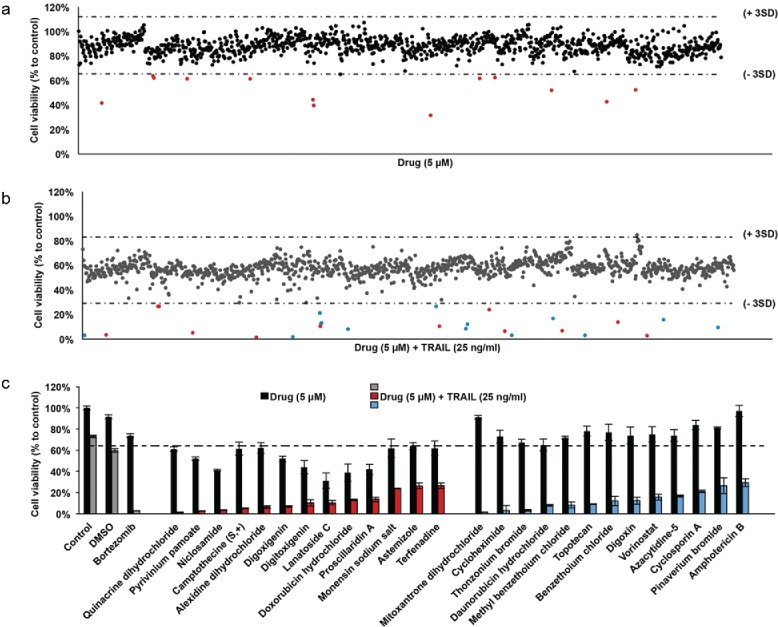
Screening of FDA-approved drugs for TRAIL-sensitizers in GBM cells (A) Scatter plot of U87MG cell viability upon 24 hours of treatment with drug library composed of 1200 compounds. Each dot represents the percent cell viability compared to untreated control samples. Horizontal dashed lines depict the range of viability within 3 SD of the mean viability (64 % and 112%). Red dots are drugs that have significant effect on cell viability as single agents. (B) Scatter plot of U87MG cell viability upon 24 hours of treatment with drug library composed of 1200 compounds and TRAIL together. Each dot represents the combinatorial effect of the individual drug (5 µM) and TRAIL (25 ng/ml) on cell viability compared to untreated control samples. Horizontal dashed lines depict the range of viability within 3 SD of the mean viability (29% and 83%). Red and blue dots indicate the combination effect below the threshold of 3 SD from the mean cell viability. (C) Top 26 hits were determined via cut off value of 29% cell viability after drug and TRAIL addition. DMSO and Bortezomib, negative and positive control respectively, are indicated in gray bars. The effect of drug and TRAIL are grouped among hits, as drugs with significant effect on cell viability as single agents (red bars) and the others (blue bars). Error bars represent “mean ± NSEM.” SD: standard deviation from mean cell viability.
Validation of the effects of TRAIL sensitizing agents on GBM cell viability
To test the combination effect of drugs with TRAIL, each drug (5 µM) was co-applied with TRAIL (25 ng/ml). The mean effect of DMSO+TRAIL treatment on cell viability was 60±3.9%; and the mean effect of drug+TRAIL was 57±9.4% compared to controls. Using 3 SDs, the new threshold was defined as 29%. Accordingly, 26 drugs reduced cell viability below this threshold in combination with TRAIL (Fig. 1B, Table 1). While 13 of these 26 drugs were also effective as single agents (as shown in Fig. 1A), remaining 13 out of 26 drugs were effective only in combination with TRAIL. These drugs, which reduced the cell viability below 29% when combined with TRAIL, were Mitoxantrone dihydrochloride (1.7 ± 0.16%), Cycloheximide (3.2 ± 4.7%), Thonzonium bromide (3.2 ± 0.81%), Daunorubicin hydrochloride (8.0 ± 0.8%), Methyl benzethoium chloride (8.3 ± 2.7%), Topotecan (9.3 ± 0.42%), Benzethoium chloride (12 ± 4.4%), Digoxin (13 ± 3.5%), Vorinostat (16 ± 2.7%), Azacytidine-5 (17 ± 0.91%), Cyclosporin A (10 ± 1.2%), Pinaverium bromide (27 ± 7.3%) and Amphotericin B (30 ± 3.7%) (Fig. 1C, Table 1).
Table 1.
Top hits of U87MG and U87MG-R50 drug screens. Drugs that have significant effect on GBM cells as single agents are indicated in bold.
| U87MG Screen |
U87MG-R50 Screen |
||||
|---|---|---|---|---|---|
| Therapeutic class | Drug Name | Drug alone % cell viability ± SEM (% of control ) | In combination with TRAIL % cell viability ± SEM (% of control) | Drug alone % cell viability ± SEM (% of control ) | In combination with TRAIL % cell viability ± SEM (% of control) |
| Antibacterial | Doxorubicin hydrochloride | 39 ± 8.1 | 13 ± 0.76 | 64 ± 4.1 | 12 ± 0.77 |
| Alexidine dihydrochloride | 62 ± 5.1 | 6.4 ± 1.2 | |||
| Monensin sodium salt | 62 ± 8.8 | 24 ± 0.44 | 69 ± 2.3 | 37 ± 5.1 | |
| Daunorubicin hydrochloride | 65 ± 5.5 | 8.0 ± 0.76 | 58 ± 6.2 | 11 ± 1.3 | |
| Methyl benzethonium chloride | 72 ± 1.4 | 8.3 ± 2.7 | 76 ± 3.7 | 38 ± 22 | |
| Cycloheximide | 73 ± 5.9 | 3.2 ± 4.7 | 73 ± 10 | 12 ± 2.8 | |
| Benzethonium chloride | 77 ± 7.8 | 12 ± 4.4 | 89 ± 18 | 30 ± 12 | |
| Amphotericin B | 84 ± 5.5 | 21 ± 3.7 | |||
| |
Thiostrepton |
|
|
84 ± 1.2 |
35 ± 4.4 |
| Antineoplastic | Camptothecine (S,+) | 61 ± 6.1 | 5.1 ± 0.40 | 66 ± 4.9 | 8.1 ± 0.37 |
| Azacytidine-5 | 74 ± 5.5 | 17 ± 0.91 | |||
| Vorinostat | 75 ± 7.3 | 16 ± 2.7 | 81 ± 14 | 26 ± 0.10 | |
| Topotecan | 78 ± 4.5 | 9.3 ± 0.42 | 66 ± 7.3 | 14 ± 2.9 | |
| |
Mitoxantrone dihydrochloride |
91 ± 1.8 |
1.7 ± 0.16 |
76 ± 1.8 |
3.1 ± 0.68 |
| Cardiac glycosides | Lanatoside C | 31 ± 7.3 | 10 ± 2.1 | 37 ± 2.1 | 15 ± 0.37 |
| Digitoxigenin | 44 ± 6.3 | 10 ± 3.2 | 41 ± 2.5 | 12 ± 2.3 | |
| Proscillaridin A | 42 ± 4.8 | 14 ± 2.2 | 38 ± 1.9 | 19 ± 1.4 | |
| Digoxigenin | 52 ± 2.6 | 6.9 ± 0.70 | 52 ± 4.7 | 11 ± 1.1 | |
| |
Digoxin |
74 ± 7.9 |
13 ± 3.5 |
35 ± 1.7 |
13 ± 2.6 |
| Immuno-suppressant |
Cyclosporin A |
84 ± 4.3 |
21 ± 1.2 |
|
|
| Metabolism |
Pyrvinium pamoate |
52 ± 1.6 |
2.8 ± 0.28 |
56 ± 10 |
7.6 ± 1.7 |
| Antihelmintic | Niclosamide | 41 ± 0.74 | 3.4 ± 0.21 | 70 ± 2.6 | 11 ± 3.6 |
| |
Quinacrine dihydrochloride |
61 ± 2.2 |
1.5 ± 0.37 |
60 ± 7.8 |
3.0 ± 0.77 |
| Antihistaminic | Terfenadine | 62 ± 6.7 | 27 ± 2.8 | ||
| |
Astemizole |
64 ± 3.4 |
26 ± 2.7 |
|
|
| Antiseptic |
Thonzonium bromide |
67 ± 3.1 |
3.1 ± 0.81 |
62 ± 7.0 |
8.1 ± 4.2 |
| Antispastic | Pinaverium bromide | 81 ± 0.97 | 27 ± 7.3 | ||
| Threshold for % cell viability | 64% | 29% | 67% | 40% | |
To further characterize the drugs that were classified as TRAIL-sensitizers, we first tested the response of U87MG cells to varying doses of drugs that included as low as 10 nM and as high as 10 µM (Fig. S2). Accordingly, the drugs that were singly effective at 5 µM also reduced cell viability at higher doses as expected. To then assess the TRAIL-cooperative action of the drugs, we tested the combined effect of 3 different doses of TRAIL and 3 different doses of the selected drugs on U87MG cell viability (Fig 2A, Fig 3B, Fig. S3, Fig S5). Accordingly, the combinatorial effects of drug+TRAIL were validated for each drug.
Figure 2.
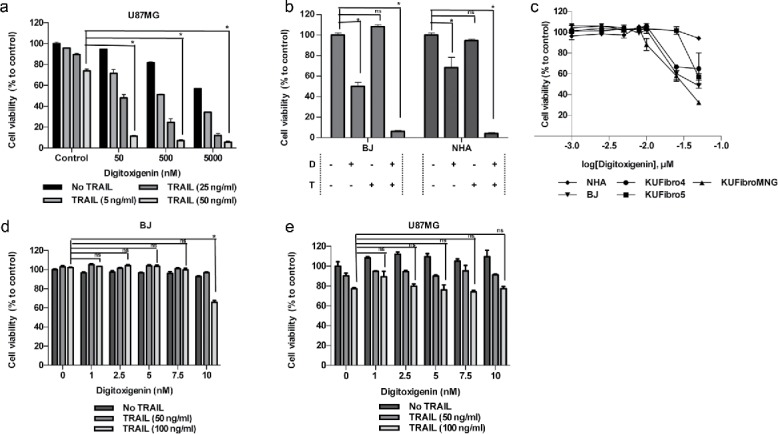
Effect of a cardiac glycoside, Digitoxigenin, on GBM and non-malignant cell viability. Cell viability detection after 24 hours of Digitoxigenin and/or TRAIL addition. (A) The effect of different doses of Digitoxigenin and TRAIL combinations on U87MG. (B) The chosen dose combination of 500 nM Digitoxigenin and 50 ng/ml TRAIL on BJ and NHA. (C) Low doses of Digitoxigenin (1–50 nM) tested on a panel of human fibroblasts: KUFibro4, KUFibro5, KUFibroMNG, BJ and Human Astrocytes (NHA). (D) Doses up to 10 nM were combined with 50 and 100 ng/ml TRAIL on BJ cells. (E) Low dose combination of Digitoxigenin on U87MG cells. * indicates p <0.05 and ns indicates nonsignificant (p>0.05).
As cardiac glycosides constituted one of the largest therapeutic classes among our hits, we focused on characterizing their efficacy in GBM cells. To this end, 6 different doses of 5 cardiac glycosides (0.3, 0.6, 1.25, 2.5 and 5 µM) were tested on 3 different GBM cell lines, U87MG, U373 and LN229. Accordingly, all cardiac glycosides reduced the viability of all GBM cells in a dose-dependent manner (Fig. S4). To further validate the cooperation of the cardiac glycosides with TRAIL, combined effect of different drug and TRAIL doses were tested and shown to markedly reduce GBM cell viability (Fig. 2A, Fig. 5A-D). Doses for the combination treatment were chosen (Fig S5E) such that drug alone did not induce cell death more than 10% as a single agent, whereas the combination with TRAIL reduced cell viability below 29%. Since our eventual aim with the combinatorial treatment is to target tumor cells specifically, we included non-malignant cells in our further experiments. To this end, the chosen doses of cardiac glycosides and their combination with TRAIL were tested on BJ fibroblasts and Normal Human Astrocytes (NHAs) (Fig. 2B, Fig. S6A). After observing that they reduced BJ cell viability with TRAIL, we sought to determine the overall effect of these drugs on an expanded panel of non malignant cells, including primary patient-derived cells and NHAs (Fig. 2C,Fig. S6C). A selected cardiac glycoside, Digitoxigenin, reduced the viability of all non-malignant cells even at concentrations (7.5–50 nM) that were far below 5 µM used previously. The doses that did not reduce cell viability significantly in non-malignant cells (Fig. 2D) were not effective in reducing GBM cell viability (Fig. 2E). Similar effects were observed with other cardiac glycosides Digoxigenin and Proscillaridin A (Fig. S6). Given the effects of cardiac glycosides alone on non-malignant and GBM cells, we decided to explore other candidates as TRAIL-sensitizing agents.
Figure 5.
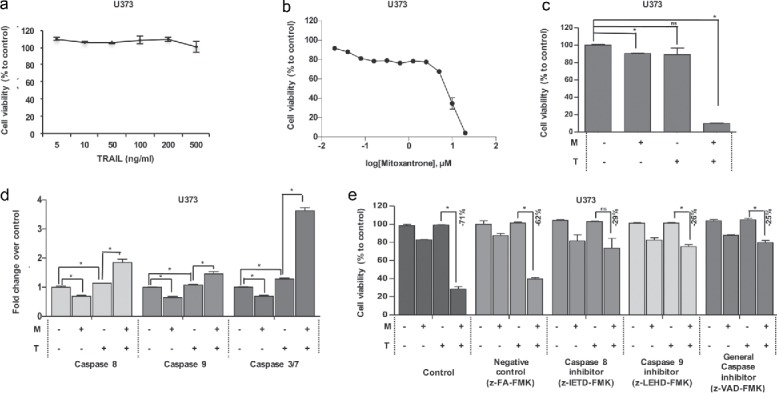
The effect of Mitoxantrone on TRAIL resistant cell line, U373. (A) The effect of TRAIL (5–500 ng/ml) on intrinsically TRAIL resistant cells U373. (B) Effects of 0.02–20 μM of Mitoxantrone on U373 viability. (C) The effect as single agents and of the combination of the chosen dose of Mitoxantrone and TRAIL on U373 cells. (D) Caspase-8, −9 and −3/7 level detections of Mitoxantrone and/or TRAIL treated U373 cells. (E) The effect of caspase-8 or −9 inhibitors and general caspase inhibitors on U373 cells treated with Mitoxantrone, TRAIL and the combination. * indicates p <0.05 and ns indicates nonsignificant (p>0.05).
Mitoxantrone and TRAIL combination reduces GBM cell viability and induces apoptosis
With an interest in drugs that do not have significant effect on cell viability as single agents, but can cooperate with TRAIL specifically on tumor cells, we shifted our focus to the DNA-damaging agent Mitoxantrone, which belonged to the antineoplastic oncology drugs. Mitoxantrone was not effective as a single agent in the screen, but it reduced the viability of U87MG cells to 2% when combined with TRAIL (Fig. 1C, Table 1). It was previously identified as a TRAIL-sensitizing agent for pancreatic cancer, prostate cancer and breast cancer.16,17 Up to 5 μM, Mitoxantrone did not reduce U87MG cell viability as a single agent (Fig. 3A). To investigate the ideal dosage combination, 3 different doses of Mitoxantrone (50, 500 and 5000 nM) were combined with 5, 25 and 50 ng/ml TRAIL (Fig. 3B); and 500 nM Mitoxantrone and 50 ng/ml TRAIL was chosen for further studies. In addition, combinatorial effect of Mitoxantrone and TRAIL on cell viability was validated by a secondary non-ATP based colorimetric assay (Fig. S8A). To obtain a TRAIL-sensitizing agent that does not affect viability of non-malignant cells, the effect of Mitoxantrone was tested on our panel of fibroblasts and NHAs. Accordingly, Mitoxantrone did not affect viability of fibroblasts and NHAs significantly up to 1 µM; however, higher doses reduced viability of 3 of the 5 lines tested (Fig. 3C). Besides, the combination of Mitoxantrone and TRAIL did not have a major effect on BJ fibroblasts and NHAs at concentrations that synergized to reduce the viability of GBM cells (Fig. 3D). To then study the time-dependent effects of Mitoxantrone and/or TRAIL, U87MG cells were treated with Mitoxantrone and/or TRAIL and observed with live cell imaging for 24 hours (Fig. 3E). Accordingly, the number of viable cells decreased with TRAIL treatment at 9 hours and continued decreasing during the rest of the treatment. However, combination of Mitoxantrone and TRAIL exerted their effects at 6 hours, which reached maximum killing efficiency at 12 hours (Supp. Videos 1–4).
Figure 3.
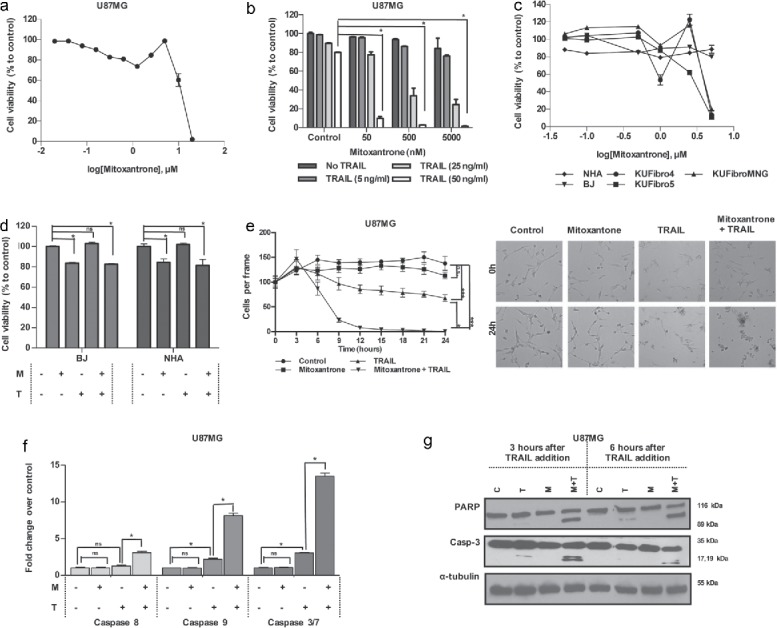
Effect of a DNA damaging agent, Mitoxantrone, on GBM and non-malignant cell viability (A) Viability of U87MG cells upon 0.02–20 μM of Mitoxantrone treatment. (B) The effect of different dose combinations of Mitoxantrone and TRAIL on U87MG cell viability. (C) The effects of Mitoxantrone doses between 50–5000 nM on primary human fibroblasts: KUFibro5, KUFibroMNG and established cell lines, BJ and human astrocytes (NHA). (D) The effect of the combination Mitoxantrone (500 nM) and TRAIL 50ng/ml) on BJ and NHA. (E) Live cell imaging of U87MG cells treated with Mitoxantrone, TRAIL and the combination for 24 hours. Left: graph depicting viable cell numbers, Right: representative snapshot images at t = 0 and t = 24 hours. Statistics were performed by 2-way ANOVA with Tukey's post hoc testing. (F) Caspase-8, −9 and −3/7 level activity of Mitoxantrone and/or TRAIL-treated U87MG cells. (G) Detection of apoptosis markers, cleaved PARP and cleaved caspase-3 protein levels, by Western Blot. *Indicates p <0.05, *** indicates p<0.0001 and ns stands for nonsignificant.
To assess the features of Mitoxantrone and TRAIL-induced apoptosis in GBM cells, we performed assays to measure the activation of caspases. While Caspase-8 levels showed approximately 3-fold increase in U87MG cells in the combinatorial treatments, Caspase-9 activity increased 8-fold, and Caspase-3/7 activity increased 13-fold compared to controls suggesting the involvement of caspase activation in Mitoxantrone+TRAIL mediated cell death (Fig. 3F). The inhibition of these caspases with specific inhibitors partially rescued the combinatorial effects on viability (Fig. S8B). The activation of apoptosis was further evidenced with increased levels of cleaved PARP and cleaved caspase-3 protein only in the combination of Mitoxantrone and TRAIL compared to individual treatments or controls (Fig. 3G).
Screen on a resistant subpopulation of GBM cells leads to the identification of same drugs as TRAIL-sensitizers
Resistance to treatment is a major problem for the treatment of cancer. To test whether a TRAIL-resistant cell line can be re-sensitized to TRAIL with secondary agents, we performed a similar screen on a TRAIL-resistant subpopulation of U87MG cells, U87MG-R50. This TRAIL-resistant population, which was selected through chronic TRAIL treatment (manuscript in preparation), is less affected by TRAIL even at high doses (Fig. 4A). The drug screen performed on U87MG-R50 cells revealed 20 hits, which were all common with the hits from screen performed on U87MG cells except Thiostrepton (Fig. 4B, Fig. S7, Table 1). The overall effect of drug treatment alone was 91 ± 8 % compared to controls (Fig. S7A). Taking 3SDs, the lower threshold of single agent effect was determined as 67%. 12 out of 20 hits were below this threshold as single agents (5 µM). When combined with TRAIL, the overall effect of all drugs+TRAIL was 77 ± 12 %, and the lower threshold was determined as 40% (Fig. 4B,Fig. S7). Table 1 contains all the hits and their cell viability percentages after 24 hours of treatment. Accordingly, Mitoxantrone was the most effective agent, where it reduced the viability to 2 ± 0 % when combined with TRAIL.
Figure 4.
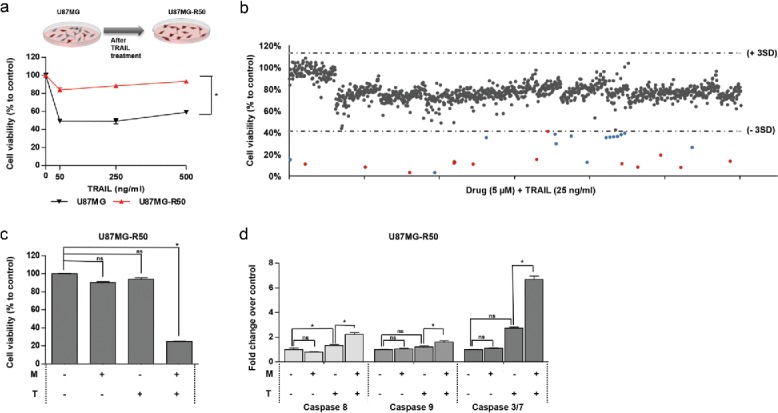
The effect of Mitoxantrone on TRAIL-resistant subpopulation, U87MG-R50. (A) The difference of U87MG cells and U87MG-R50 cells response to different doses (50, 250 and 500 ng/ml) of TRAIL. (B) Scatter plot of U87MG cell viability upon 24 hours of treatment with drug library composed of 1200 compounds and TRAIL together. Each dot represents the combinatorial effect of the individual drug (5 µM) and TRAIL (25 ng/ml) on cell viability compared to untreated control samples. Horizontal dashed lines depict the range of viability within 3 SD of the mean viability (41% and 113%). Red and blue dots indicate the combination effect below the threshold of 3 SD from the mean cell viability. (C) The effect of Mitoxantrone (500 nM) and/or TRAIL (50 ng/ml) on U87MG-R50 cells. (D) Detection of Caspase-8, −9 and −3/7 activity levels of Mitoxantrone and/or TRAIL treated U87MG-R50 cells. * indicates p <0.05 and ns indicates nonsignificant (p>0.05).
Mitoxantrone and TRAIL combination induces cell death in TRAIL-resistant GBM cell lines
To characterize the effects of Mitoxantrone in resistant cells further, U87MG-R50 cells were sequentially treated with Mitoxantrone followed by TRAIL and cell viability was detected. Cell death up to 80% was observed in the combination treatment (Fig. 4C). Both Caspase-8 and −9 showed approximately 3-fold activation in U87MG-R50 cells in the combination treatment compared to controls. Approximately 7-fold increase was observed in Caspase-3/7 levels (Fig. 4D).
Next, we sought to investigate the effect of our combination on an innately TRAIL-resistant cell line, U373. TRAIL concentration up to 500 ng/ml in this cell line indicated no significant cell death (Fig. 5A). To test the effect of Mitoxantrone alone, we tested dosages between 0.02–20 μM and validated that up to 5 μM, no significant increase in U373 cell death is observed (Fig. 5B). With the same treatment regimen of U87MG and U87MG-R50, U373 cells showed a notable decrease of cell viability when treated with Mitoxantrone and TRAIL together (Fig. 5C). We also observed that Caspase-8 showed approximately 3-fold increase in the combination, whereas Caspase-9 activity levels were not markedly altered in U373 cells. 16-fold increase was observed in Caspase-3/7 levels (Fig. 5D). Inhibition of Caspase-8, −9 or all caspases using specific caspase inhibitors significantly rescued the Mitoxantrone and TRAIL induced cell death in U373 cells (Fig. 5E).
Mitoxantrone causes DNA damage and alters the expression of apoptosis mediators and death receptors in GBM cells
To validate the DNA-damaging effects of Mitoxantrone on GBM cells, U87MG and U373 cells were treated with 500 and 5000 nM Mitoxantrone for 24 hours and stained for γH2AX as a well-established marker for the presence of double-stranded DNA breaks. As expected, Mitoxantrone caused the accumulation of γH2AX-positive puncta in both cell types (Fig. 6A, 6B). TRAIL sensitization has been shown to be linked to TRAIL receptor levels,6,18 thus we evaluated the receptor expression levels in all 3-cell lines' upon Mitoxantrone treatment. U87MG cells have shown >10-fold increase in DR4, without any effect of Mitoxantrone on DR5 gene expression levels. Among decoy receptors, DcR1 and DcR2 levels were elevated, whereas OPG levels were reduced. These patterns were similar for U87MG-R50 cells, where DR4 was the most affected among the receptors (Fig. 6C). These changes were validated at the protein level by Western blotting, where Mitoxantrone treatment caused marked increase in DR4 and a modest increase in DR5 levels in both the parental and TRAIL-resistant subpopulation of U87MG cells (Fig. 6D). In U373 cells, we were not able to detect DR4 levels and no significant shift in DR5 levels were observed. However, DcR2 and OPG levels were slightly increased with Mitoxantrone treatment (Fig. 6C). To test the functional link between death receptors and Mitoxantrone-mediated TRAIL sensitization, we employed shRNA to knockdown DR4 or DR5 expression in U87MG and U373 cells. While we did not observe a significant change with DR4 knockdown, we observed a significant decrease in cell death in response to Mitoxantrone and TRAIL combination with DR5 knockdown in U87MG cells (Fig. S9A, S9B). DR5 knockdown of U373 cells resulted in a 10% decrease in cell death in cells treated with Mitoxantrone and TRAIL combination (Fig. S10A). Next, in order to get a better insight into the mode-of-action of Mitoxantrone, we investigated the expression changes of a panel of apoptosis-related genes in U87MG and U373 cells upon treatment. These genes included the pro- and anti-apoptotic Bcl-2 family members such as Bik, Bak, Bid, Bim, Bag1, Bag3, Puma, Noxa, Hrk, Bcl-2, Bcl-XL, Bclaf1, Bcl10, Bcl2L10 and Mcl1. In addition, apoptosis regulators such as XIAP, Birc2, Birc3, Birc5, Birc8, FADD, Bnip2 and Bnip3 were included; as well as NfKB and its associated p100 and p65. Accordingly, in U87MG cells, Puma, Noxa and Hrk were among the pro-apoptotic genes that were most upregulated with Mitoxantrone treatment, and Bcl2L10, Birc5, Birc3 and Bcl2 were among the downregulated anti-apoptotic genes (Fig. 6E). In U373 cells, Bim, Noxa, Bik and Hrk were among markedly upregulated pro-apoptotic genes (Fig. 6F). Together, Mitoxantrone treatment altered the gene expression in favor of apoptosis in both U87MG and U373 cells. To test the function of HRK or Puma in Mitoxantrone-mediated TRAIL sensitization, we employed loss-of-function experiments using shHRK or shPuma. HRK knockdown of U87MG cells partially rescued the cells from TRAIL induced cell death, but the response to the combination of Mitoxantrone and TRAIL treated cells was similar to the controls (Fig. S9C). On the other hand, neither Puma nor HRK knockdown changed the overall response to Mitoxantrone and TRAIL combination in U373 cells (Fig. S10B, S10C). Taken together, these results suggest that Mitoxantrone treatment has the ability to prime GBM cells for TRAIL induced apoptosis by changing the apoptosis-related gene expression in favor of enhanced apoptosis, and that DRs may serve as a major functional target of Mitoxantrone in this process.
Figure 6.
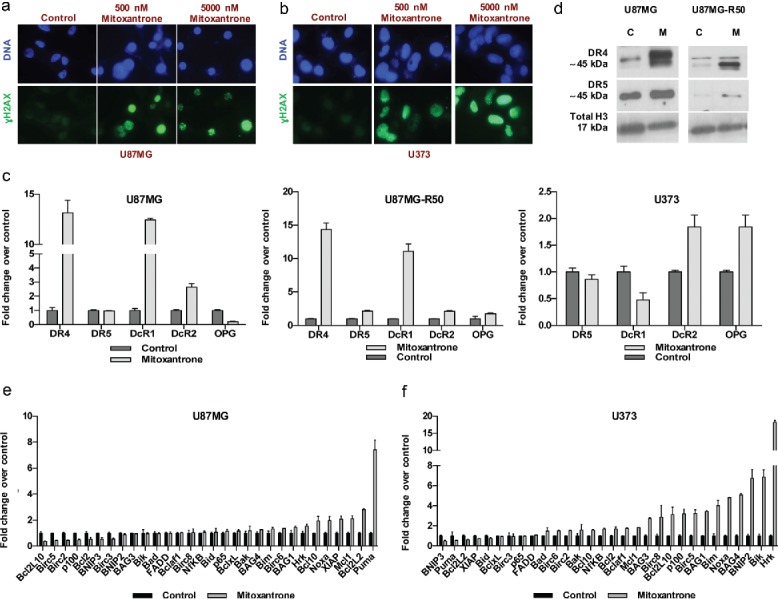
The effect of Mitoxantrone on apoptosis pathway. (A-B) γH2AX staining of U87MG cells (A) and U373 cells (B) treated with 0, 500 and 5000 nM Mitoxantrone. (C) Gene expression levels of death receptors of U87MG, U87MG-R50 and U373 cells treated with Mitoxantrone for 24 hours. (D) DR4 and DR5 protein levels of U87MG and U87MG-R50 cells treated with Mitoxantrone for 24 hours. (E-F) The expression of pro-apoptotic and anti-apoptotic genes of U87MG cells (E) and U373 cells (F) treated with Mitoxantrone for 24 hours.
Discussion
In this study, we searched for clinically approved drugs that had the potential to cooperate with TRAIL as anti-cancer therapies. Using three different GBM cell line models; one that exhibits medium level sensitivity to TRAIL (U87MG), one that is selected as a TRAIL-resistant subpopulation from U87MG cells (U87MG-R50), and one that is innately resistant (U373), we demonstrated that a DNA-damaging agent, Mitoxantrone, has the ability to prime all GBM cells to TRAIL-induced apoptosis in a cooperative manner.
Our screen was performed with 1200 FDA-approved compounds, which was with the motivation of repurposing clinically approved drugs for GBM therapy. While screening with naïve compounds without prior characterization, very laborious and costly biological evaluations would be needed such as in vitro and in vivo efficacy and safety. However, a repurposed drug already comes with the systemic toxicity knowledge, reducing cytotoxicity problems.19 Indeed, repurposed drugs contain 30% of FDA approved drugs in the last years.20 Therefore, we selected an already-approved drug library as our starting point for identifying TRAIL-sensitizing agents.
Specifically, our screen included the test of agents as single agents or in combination with TRAIL. Accordingly, we identified 13 drugs that were effective as single agents. Some of them were previously suggested as therapeutic candidates for GBM as single agents such as Doxorubicin hydrochloride,21 Camptothecine (S,+),22 Proscillaridin A,15 Pyrivinium pamoate,23 and Niclosamide.24 We report Alexidine dihydrochloride, Monensin sodium salt, Lanatoside C, Digitoxigenin, Digoxigenin, Digoxin, Quinacrine dihydrochloride, Terfenadine and Astemizole as novel drugs that can be therapeutic candidates for GBM. Notably, we did not identify Temozolomide (TMZ), the most common drug used as anti-GBM therapy,2,25 as an effective agent in our screen although it was included, attesting to the unmet need for identifying novel drugs.
While TRAIL is a prime therapeutic candidate, its translation to clinics suffers from the problem of innate or acquired TRAIL resistance. Therefore, identifying the mechanisms of TRAIL resistance and finding secondary agents that can overcome this resistance is promising. To this end, there have been reports on the use of several drugs as TRAIL-sensitizers in various cancer types.16,26 However, an important consideration to make while performing combinatorial strategies is the effect on non-malignant cells. Thus, while validating our candidate TRAIL sensitizing drugs from the screen, we sought to identify drugs with minimal toxicity on normal cells as single agents or in combination with TRAIL.
The 26 hits from our screen belonged to distinct pharmacological classes including antibacterials, antineoplastics, antihelmintics and cardiotonics. Nine of these hit drugs (Doxorubicin, Daunorubicin, Camptothecine (S,+), Azacytidine-5, Vorinostat, Topotecan, Mitoxantrone, Cycloheximide and Quinacrine dihydrochloride) were previously indicated as TRAIL-sensitizing agents in several cancers;17,27-36 and 6 of them (Cycloheximide, Monensin sodium salt, Doxorubicin, Topotecan, Digoxin and Lanatoside C) were also studied as TRAIL-sensitizers in GBM.37-42 The novel TRAIL-sensitizers for GBM were Alexidine dihydrochloride, Daunorubicin, Methyl benzethonium chloride, Benzethonium chloride, Amphotericin B, Camptothecine (S,+), Azacytidine-5, Vorinostat, Mitoxantrone, Digitoxigenin, Proscillaridin A, Digoxigenin, Cyclosporin A, Pyrivinium pamoate, Niclosamide, Quinacrine dihydrochloride, Terfenadine, Astemizole, Thonzonium bromide, and Pinaverium bromide in our screen.
There have been many studies toward combining TRAIL with secondary agents, and one major class of drugs that is known to cooperate with TRAIL is histone deacetylase (HDAC) inhibitors.43 Indeed, MS-275, an HDAC inhibitor, was previously shown to have a significant effect on GBM cell viability when combined with TRAIL.44 In our library of 1200 drugs, there was only one HDAC inhibitor, Vorinostat, which was previously indicated as an anti-cancer agents and also studied as a TRAIL sensitizing agent in various cancers including leukemia and lung cancer.43,45,46 Consistent with these observations, Vorinostat cooperated with TRAIL and scored as a hit in our screen, highlighting the validity of our screen findings. Given the growing interest in the field of epigenetic enzyme inhibitors as therapies, Vorinostat and TRAIL combination can also offer promise in the future for GBMs.
Cardiac glycosides constituted a major therapeutic class of hit compounds in our screen; therefore we first studied their effects on GBM cells. Indeed, this family of drugs were previously suggested as potential cancer therapeutics,47 and also shown to be effective in GBMs.15 However, the need for the use of high doses blocked their introduction to clinic. In addition, it has been shown by different groups that the combination of low dose cardiac glycosides with TRAIL can trigger cell death in various cancers, including GBM.37,42,48 Our studies are in line with these previous findings, validating the utility and consistency of our screen. However, when we applied the low dosages of the cardiac glycoside Digitoxigenin, Proscillaridin A and Digoxin in combination with TRAIL on non-malignant cells such as human fibroblasts and normal human astrocytes, we observed equal amount of cell death in these cells, highlighting a possible toxicity issue. However, given the wide use of cardiac glycosides for the treatment of heart disorders in patients, these clinically approved drugs might offer promise as single agents in GBMs. The effect of cardiac glycoside and TRAIL combination prompts further studies in in vivo models and in clinical settings to thoroughly investigate their efficacy and possible toxicity.
The second functional class of drugs that scored as hits included DNA damaging agents such as Daunorubicin, Doxorubicin, and Topotecan. Doxorubicin was already identified as a TRAIL sensitizing agent for GBM, however cardiac toxicity of Doxorubicin and poor drug delivery across blood-brain barrier (BBB) is a challenge.40 Of the DNA-damaging agents, Mitoxantrone was the only one that reduced cell viability only in combination with TRAIL, but not on its own. Mitoxantrone-induced DNA damage is known to induce expression of the p53 protein and result in cellular apoptosis.17 Here we showed that Mitoxantrone could cooperate with TRAIL and lead to GBM cell death in 3 GBM cell lines, U87MG, U87MG-R50, and U373. While we do not see a direct correlation between cell lines' p53 status and response to Mitoxantrone and/or TRAIL combination, it will be of major interest to investigate the role of p53 in TRAIL sensitization in an expanded panel of established and patient-derived GBM cell lines in the future. While enhanced caspase activation, and therefore apoptosis, was observed in all cell lines with combination, inhibition of caspases was sufficient to fully rescue death in U373 cells, but not in U87MG cells. Therefore, additional forms of cell death, such as necroptosis and autophagy, or caspase-independent apoptotic pathways might be at play during Mitoxantrone-induced TRAIL sensitization in different cell lines.49 At the low doses that Mitoxantrone was used as a TRAIL sensitizing agent, the toxicity to fibroblasts was also very minimal compared to cardiac glycosides. Therefore, Mitoxantrone and TRAIL combination might be very promising therapy approach for GBMs.
There are several reports on the mechanism of TRAIL sensitization by secondary agents. These include the up-regulation of the death receptors DR4 and DR5, downregulation of cell survival proteins such as IAPs, rescued caspase-8 activity, upregulation of pro-apoptotic elements like Bax, Bid, etc. While some of these changes were reported to be through transcriptional regulation of apoptosis pathway components; translational and posttranslational regulation of the expression and stability of apoptosis-related proteins was often reported.7 In this study, we validated the DNA-damaging action of Mitoxantrone and showed the formation of double strand breaks in our GBM cell lines by γH2AX staining. However, the ability of Mitoxantrone's TRAIL sensitization and its relation to p53 activation prompts further detailed studies. We found that treatment of U87MG and U87MG-R50 cells with Mitoxantrone results in elevated expression of a significant DR4 receptor upregulation both at the transcriptional level and the translational level. In addition, DR5 receptor levels were slightly upregulated, suggesting that receptor activation could be one possible mechanism of TRAIL sensitization by Mitoxantrone. Further knockdown experiments of DR4 and DR5 in U87MG cells revealed that DR5 knockdown resulted in decrease in cell death in Mitoxantrone and TRAIL combination treatment, whereas DR4 knockdown did not cause a significant change. In innately-resistant U373 cells, receptor levels were not dramatically altered, suggesting another modulatory mechanism of Mitoxantrone. Yet, DR5 knockdown in U373 cells slightly decreased cell death of the combination treatment compared to controls, suggesting the role of death receptors in these cell line also. While a single molecular component of apoptosis pathway can be altered and be responsible for different responses to TRAIL, ideally, a combination of changes at the gene and protein levels and function would be more effective in changing the apoptotic tendency of GBM cells. To this end, we observed that marked changes in gene expression levels of pro-apoptotic and anti-apoptotic genes occurred with Mitoxantrone treatment. Puma and Hrk were most affected genes in U87MG and U373 cells, respectively. Although the individual silencing of Puma or Hrk did not change the response of GBM cells to Mitoxantrone and TRAIL, our future efforts will be directed to understand to functional role of the apoptosis-related molecules in further detail.
In GBMs, although the in vitro biological activity of many drugs offers promise, their application to clinics is a challenge due to the existence of BBB.50 Because Mitoxantrone is a very small molecule, it is able to cross the blood–brain barrier and interact with cells in the CNS.51 Therefore, it will be of interest to investigate the combination of Mitoxantrone and TRAIL in in vivo models of GBM to further characterize the utility of this therapeutic approach. To this end, combining Mitoxantrone with local delivery of TRAIL utilizing neural stem cells can be a candidate therapy approach.11 Overall, our results suggest that Mitoxantrone and TRAIL combination might offer a promising alternative therapy in the future.
Materials and methods
Cell culture
Human glioblastoma cells U87MG (p53 wild-type, PTEN mutated), LN229 (p53 mutated, PTEN wild-type), A172 (p53 wild-type, PTEN mutated) and U373 (p53 mutated52, PTEN mutated 53) were supplied and authenticated by American Tissue Type Culture Collection (ATCC). Normal Human Astrocytes (NHA) were purchased from Lonza. TRAIL-resistant subpopulation of U87MG cells (U87MG-R50) was generated by selection of U87MG cells under TRAIL treatment (50 ng/ml) for one month (manuscript in preparation). BJ human fibroblast line was kindly gifted by Dr. Tamer Onder (Koc University, TURKEY). Primary GBM patient-derived fibroblasts (KUFibroMNG, KUFibro4 and KuFibro5) were generated in collaboration with Dr. Ihsan Solaroglu (Koc University, TURKEY) according to guidelines and protocols approved by the Institutional Review Board (IRB2013.198.IRB2.61). Accordingly, 1-mm2 skin biopsies around incision site were obtained during surgery, and cultured for 2 weeks for fibroblasts to grow out of explants. All cells except NHA were grown in DMEM medium (Gibco) with 10% fetal bovine serum (Invitrogen) and 1% Penicillin-Streptomycin (Gibco). NHA was grown in AGM BulletKit (Lonza) according to the manufacturer's instructions. All cells were cultured in were cultured in 37°C humidified incubator with 5% CO2.
Reagents
The drug library composed of 1,200 FDA (Food and Drug Administration of US) and EMA (European Medicines Agency)-approved drugs. The selected individual drugs (Mitoxantrone dihydrochloride, Digitoxigenin, Digoxin, Lanatoside C, Digoxigenin, Proscillaridin A) and Bortezomib were purchased from Prestwick Chemicals (France), and Selleck Chemicals (US), respectively. TRAIL was supplied from Enzo Life Sciences or produced from 293T cells as described.54
Drug screen
2,000 cells/well were seeded to 384-well black plates via Multidrop™ Combi Reagent Dispenser (Thermo-Scientific). Next day, a single concentration of TRAIL (25 ng/ml) was applied to cells alone or with individual drugs at a final concentration of 5 µM. Untreated cells and DMSO-treated cells were used as negative controls. Bortezomib (50 nM) was used as a positive control. For drug treatment, final concentration of 5 μM was used in 384-well plates. Each treatment was performed in triplicates. Cell viability was determined after 24 hours.
Cell viability assays
Cells were treated with the respective drug and TRAIL simultaneously for 24 hours. Cell viability was detected by an ATP based assay; Cell Titer-Glo® (CTG) Luminescent Cell Viability Assay (Promega) according to manufacturer's instructions using Luminometric measurements at a plate reader (Synergy H1 Reader, BioTek). For validation experiments that assessed combination of Mitoxantrone and TRAIL, cells were treated with Mitoxantrone for 24 hours, followed by its removal and addition of TRAIL (50 ng/ml). Cell viability was detected after 24 hours with CTG. In addition, cell viability was measured with Cell Proliferation Reagent WST-1 (Roche), a non-ATP-based assay. Biological triplicate cells were treated with Mitoxantrone and/or TRAIL for 24 hours and cell viability was quantified.
Live cell imaging
200,000 U87MG cells were plated to 6-well plates. All live-cell imaging experiments were carried out by Xcellence Pro inverted microscope (Olympus) with a 10X air objective in a chamber at 37°C, supplied with 5% CO2. Five random positions in each well were recorded per sample. After addition of reagents, live cell images were captured every 4 minutes for 24 hours. Time-lapse images were acquired and converted using Xcellence RT 1.2 software. Cell viability was statistically analyzed by counting the number of live cells using ImageJ (NIH Image). Statistics were performed by 2-way ANOVA with Tukey's post hoc testing calculated by GraphPad Prism. *Indicates p<0.05 and *** indicates p< 0.0001
Caspase activity assays
10,000 cells/well were seeded to 96-well black plates. Cells were treated with Mitoxantrone (500 nM) for 24 hours followed by TRAIL (50 ng/ml) for 3 hours. Caspase Glo 8, 9 and 3/7 (Promega) were used to detect caspase activity levels according to manufacturer's instructions. All conditions were performed in triplicate. Results were presented as fold changes normalized to untreated control cells.
Caspase inhibition assays
10,000 cells/well were seeded to 96-well black plates. Respective wells were treated with Mitoxantrone (500 nM) and the indicated caspase inhibitors. After 24 hours, Mitoxantrone was removed and TRAIL (50 ng/ml) was added to respective wells along with freshly added caspase inhibitors: Caspase-8 inhibitor; z-IETD-FMK (550380, BD PharMingen), Caspase-9 inhibitor; z-LEHD-FMK (550381, BD PharMingen), General Caspase inhibitor; z-VAD-FMK (550377, BD PharMingen), Negative Control for Caspase Inhibitors; z-FA-FMK (550411, BD PharMingen). After 24 hours, cell viability was detected as described above.
Real-time PCR
RNA was collected via Qiagen RNeasy Mini Kit following manufacturer's instructions (Qiagen). 500 ng RNA was used to synthesize cDNA with M-MLV Reverse Transcriptase (Invitrogen). Relative gene expression levels of TRAIL receptors were detected by using LightCycler® 480 SYBR Green I Master (Roche). Gene expression levels of anti-and pro-apoptotic genes were assessed by EvaGreen® (Biotium). The primers used in these experiments are listed in Table S1.
Western blot analysis
Cells were seeded to 6-well plates at a density of 300,000 cells/well. The corresponding wells were treated with Mitoxantrone for 24 hours. Next day, after the removal of Mitoxantrone, TRAIL was added. Following 3 or 6 hours, cells were trypsinized, collected with centrifugation at 1500 rpm for 5 minutes, and pellets were lysed in an appropriate volume of cell lysis buffer [1% NP-40, 150 mM NaCl, 1mM EDTA, 50 mM Tris-HCl (pH 7.8), 1 mM NaF, 0.5 mM PMSF and 1X protease inhibitor cocktail (cOmplete Protease Inhibitor Cocktail Tablets, Roche]. Following 30 minutes of incubation on ice and centrifugation at 14,000 rpm for 15 min at 4°C, protein-containing supernatants were quantified with BCA Protein Assay Kit (Thermo-Scientific). 25 µg of proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred onto a PVDF membrane by Trans-Blot® Turbo™ RTA Mini PVDF Transfer Kit (#170–4272, Biorad). The membranes were blocked with 5% nonfat dry milk in TBS-T (20 mM TrisHCl, pH 7.8, 150 mM NaCl, 0.1%, v/v Tween-20) at RT for 1 hour, primary antibodies were added and incubated overnight at 4°C. PARP (#9664, Cell Signaling) and caspase-3 (#9665, Cell Signaling) were used in 1:1000. DR4 (1139, ProSci) and DR5 (2019, ProSci) were used in 1:500. Histone H3 (#9715, Cell Signaling) and α-tubulin (T9026, Sigma-Aldrich) were used in 1:1000 as loading controls. Secondary antibodies conjugated to HRP, anti-rabbit and anti-mouse were used in 1:3000 (#7074 and #7076 respectively, Cell Signaling). Membranes were developed using Clarity™ Western ECL Substrate (#170–5061, Biorad) and CL-XPosure Film (Thermo-Scientific).
γH2AX staining
6 × 104 cells were seeded on 12 mm round cover slips and treated with 0, 500 or 5000 nM Mitoxantrone next day. Following 24 hour-treatment, cells were stained for γH2AX as described previously.55 γ-H2AX antibodies (1:400 diluted in 0.2% gelatin/PBS, Cell Signaling, #9718) at +4°C, overnight and FITC-conjugated secondary antibodies (1:100 diluted in PBS, Abcam) for 2 hours at RT were used for immunofluorescence. The nuclei were counter stained with 5 µg/mL DAPI and visualized using a Leica DMI 6000 microscope. All images were taken using the same camera settings.
Statistics
All normalizations were performed to untreated control cells, denoted as 100% using GraphPad Prism version 6 and Microsoft Excel. Significance was detected by student's t-test, ANOVA and at p value <0.05.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. İhsan Solaroğlu and Zeynep Kahya for generating primary fibroblasts, Dr. Hakan Örer and Dr. Tamer Önder for helpful discussions, Zeynep Kaya and Fidan Şeker for technical help with live-cell imaging experiments. This work was supported by TUBITAK-112S555 (TBO), Marie Curie CIG-618673 (TBO) and Istanbul Kalkınma Ajansı (NAL).
References
- 1.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol 2015; 17 Suppl 4:iv1-62; PMID:26511214; http://dx.doi.org/ 10.1093/neuonc/nov189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today 2015; 7(20):889-905; PMID:25744176; http://dx.doi.org/8663110 10.1016/j.drudis.2015.02.011 [DOI] [PubMed] [Google Scholar]
- 3.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 1996; 271:12687-90; PMID:8663110; http://dx.doi.org/ 10.1074/jbc.271.22.12687 [DOI] [PubMed] [Google Scholar]
- 4.Lim B, Allen JE, Prabhu VV, Talekar MK, Finnberg NK, El-Deiry WS. Targeting TRAIL in the treatment of cancer: new developments. Expert Opin Ther Targets 2015; 1-15; 9(19):1171-1185; PMID:26004811; http://dx.doi.org/22531313 10.1517/14728222.2015.1049838 [DOI] [PubMed] [Google Scholar]
- 5.den Hollander MW, Gietema JA, de Jong S, Walenkamp AME, Reyners AKL, Oldenhuis CNAM, de Vries EGE. Translating TRAIL-receptor targeting agents to the clinic. Cancer Lett 2013; 332:194-201; PMID:22531313; http://dx.doi.org/ 10.1016/j.canlet.2012.04.007 [DOI] [PubMed] [Google Scholar]
- 6.Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013; 32:1341-50; PMID:22580613; http://dx.doi.org/ 10.1038/onc.2012.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schröter M, et al.. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997; 386:517-21; PMID:9087414; http://dx.doi.org/ 10.1038/386517a0 [DOI] [PubMed] [Google Scholar]
- 8.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 2013; 5:a008656; PMID:23545416; http://dx.doi.org/ 10.1101/cshperspect.a008656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev 1999; 13:239-52; PMID:9990849; http://dx.doi.org/ 10.1101/gad.13.3.239 [DOI] [PubMed] [Google Scholar]
- 10.Unterkircher T, Cristofanon S, Vellanki SHK, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin K-M, Fulda S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res 2011; 17:4019-30; PMID:21525171; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0075 [DOI] [PubMed] [Google Scholar]
- 11.Bagci-Onder T, Du W, Figueiredo J-L, Martinez-Quintanilla J, Shah K. Targeting breast to brain metastatic tumours with death receptor ligand expressing therapeutic stem cells. Brain 2015; 6:138:1710-1721; PMID:25564487; http://dx.doi.org/ 10.1093/brain/awv094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finlay D, Richardson RD, Landberg LK, Howes AL, Vuori K. Novel HTS strategy identifies TRAIL-sensitizing compounds acting specifically through the caspase-8 apoptotic axis. PLoS One 2010; 5:e13375; PMID:20967281; http://dx.doi.org/ 10.1371/journal.pone.0013375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yip KW, Mao X, Au PYB, Hedley DW, Chow S, Dalili S, Mocanu JD, Bastianutto C, Schimmer A, Liu F-F. Benzethonium chloride: a novel anticancer agent identified by using a cell-based small-molecule screen. Clin Cancer Res 2006; 12:5557-69; PMID:17000693; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0536 [DOI] [PubMed] [Google Scholar]
- 14.Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, Halfmann R, Naidoo J, Wang L, Li L, et al.. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol 2013; 9:84-9; PMID:23292651; http://dx.doi.org/ 10.1038/nchembio.1153 [DOI] [PubMed] [Google Scholar]
- 15.Denicolaï E, Baeza-Kallee N, Tchoghandjian A, Carré M, Colin C, Jiglaire CJ, Mercurio S, Beclin C, Figarella-Branger D. Proscillaridin A is cytotoxic for glioblastoma cell lines and controls tumor xenograft growth in vivo. Oncotarget 2014; 5:10934-48; PMID:2540011; http://dx.doi.org/22044796 10.18632/oncotarget.2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor DJ, Parsons CE, Han H, Jayaraman A, Rege K. Parallel screening of FDA-approved antineoplastic drugs for identifying sensitizers of TRAIL-induced apoptosis in cancer cells. BMC Cancer 2011; 11:470; PMID:22044796; http://dx.doi.org/ 10.1186/1471-2407-11-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grandhi TSP, Potta T, Taylor DJ, Tian Y, Johnson RH, Meldrum DR, Rege K. Sensitizing cancer cells to TRAIL-induced death by micellar delivery of mitoxantrone. Nanomedicine (Lond) 2014; 9:1775-88; PMID:24195660; http://dx.doi.org/ 10.2217/nnm.13.125 [DOI] [PubMed] [Google Scholar]
- 18.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ 2014; 21:1350-64; PMID:24948009; http://dx.doi.org/ 10.1038/cdd.2014.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong CR, Sullivan DJ. New uses for old drugs. Nature 2007; 448:645-6; PMID:17687303; http://dx.doi.org/ 10.1038/448645a [DOI] [PubMed] [Google Scholar]
- 20.Jin G, Wong STC. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today 2014; 19:637-44; PMID:24239728; http://dx.doi.org/ 10.1016/j.drudis.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang P, Mukthavaram R, Mukthavavam R, Chao Y, Bharati IS, Fogal V, Pastorino S, Cong X, Nomura N, Gallagher M, et al.. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J Transl Med 2014; 12:13; PMID:24433351; http://dx.doi.org/ 10.1186/1479-5876-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee B-S, Amano T, Wang HQ, Pantoja JL, Yoon CW, Hanson CJ, Amatya R, Yen A, Black KL, Yu JS. Reactive oxygen species responsive nanoprodrug to treat intracranial glioblastoma. ACS Nano 2013; 7:3061-77; PMID:23557138; http://dx.doi.org/ 10.1021/nn400347j [DOI] [PubMed] [Google Scholar]
- 23.Hothi P, Martins TJ, Chen L, Deleyrolle L, Reynolds B, Foltz G. High-Throughput Chemical Screens Identify Disulfiram as an Inhibitor of Human Glioblastoma Stem Cells Abstract: 2012; 3:1124-36; PMID:23165409; http://dx.doi.org/ 10.18632/oncotarget.707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wieland A, Trageser D, Gogolok S, Reinartz R, Höfer H, Keller M, Leinhaas A, Schelle R, Normann S, Klaas L, et al.. Anticancer effects of niclosamide in human glioblastoma. Clin Cancer Res 2013; 19:4124-36; PMID:23908450; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2895 [DOI] [PubMed] [Google Scholar]
- 25.Iacob G, Dinca EB. Current data and strategy in glioblastoma multiforme. 2009; Nov 15; 2(4): 386–393; PMID:20108752 [PMC free article] [PubMed] [Google Scholar]
- 26.Booth NL, Sayers TJ, Brooks AD, Thomas CL, Jacobsen K, Goncharova EI, McMahon JB, Henrich CJ. A cell-based high-throughput screen to identify synergistic TRAIL sensitizers. Cancer Immunol Immunother 2009; 58:1229-44; PMID:19089423; http://dx.doi.org/ 10.1007/s00262-008-0637-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon N, Park MS, Peltier GC, Lee RH. Pre-activated human mesenchymal stromal cells in combination with doxorubicin synergistically enhance tumor-suppressive activity in mice. Cytotherapy 2015; 17(10):1332-41; PMID:26227206; http://dx.doi.org/ 10.1016/j.jcyt.2015.06.009 [DOI] [PubMed] [Google Scholar]
- 28.Vaculova A, Kaminskyy V, Jalalvand E, Surova O, Zhivotovsky B. Doxorubicin and etoposide sensitize small cell lung carcinoma cells expressing caspase-8 to TRAIL. Mol Cancer 2010; 9:87; PMID:20416058; http://dx.doi.org/ 10.1186/1476-4598-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayasooriya RGPT, Choi YH, Hyun JW, Kim G-Y. Camptothecin sensitizes human hepatoma Hep3B cells to TRAIL-mediated apoptosis via ROS-dependent death receptor 5 upregulation with the involvement of MAPKs. Environ Toxicol Pharmacol 2014; 38:959-67; PMID:25461556; http://dx.doi.org/ 10.1016/j.etap.2014.10.012 [DOI] [PubMed] [Google Scholar]
- 30.Brooks AD, Sayers TJ. Reduction of the antiapoptotic protein cFLIP enhances the susceptibility of human renal cancer cells to TRAIL apoptosis. Cancer Immunol Immunother 2005; 54:499-505; PMID:15614529; http://dx.doi.org/ 10.1007/s00262-004-0595-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frew AJ, Lindemann RK, Martin BP, Clarke CJP, Sharkey J, Anthony DA, Banks K-M, Haynes NM, Gangatirkar P, Stanley K, et al.. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A 2008; 105:11317-22; PMID:18685088; http://dx.doi.org/ 10.1073/pnas.0801868105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiland T, Weiller M, Künstle G, Wendel A. Sensitization by 5-azacytidine toward death receptor-induced hepatic apoptosis. J Pharmacol Exp Ther 2009; 328:107-15; PMID:18829727; http://dx.doi.org/ 10.1124/jpet.108.143560 [DOI] [PubMed] [Google Scholar]
- 33.Ramp U, Mahotka C, Krieg A, Walczak H. Sensitivity to TRAIL / APO-2L-mediated apoptosis in human renal cell carcinomas and its enhancement by topotecan. 2000; 1127-36. [DOI] [PubMed] [Google Scholar]
- 34.Kang MR, Kang JS, Yang JW, Kim BG, Kim J-A, Jo YN, Lee K, Lee CW, Lee KH, Yun J, et al.. Gene expression profiling of KBH-A42, a novel histone deacetylase inhibitor, in human leukemia and bladder cancer cell lines. Oncol Lett 2012; 3:113-8; PMID:22740865; http://dx.doi.org/ 10.3892/ol.2011.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Taylor D, Agrawal N, Wang H, Kim H, Han A, Rege K, Jayaraman A. A programmable microfluidic cell array for combinatorial drug screening. Lab Chip 2012; 12:1813-22; PMID:22456798; http://dx.doi.org/ 10.1039/c2lc21202a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Gallant J-N, Katz SI, Dolloff NG, Smith CD, Abdulghani J, Allen JE, Dicker DT, Hong B, Navaraj A, et al.. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL and chemotherapeutic agents. Cancer Biol Ther 2011; 12:229-38; PMID:21725212; http://dx.doi.org/ 10.4161/cbt.12.3.17033 [DOI] [PubMed] [Google Scholar]
- 37.Badr CE, Wurdinger T, Tannous BA. Functional drug screening assay reveals potential glioma therapeutics. Assay Drug Dev Technol 2011; 9:281-9; PMID:21184646; http://dx.doi.org/ 10.1089/adt.2010.0324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apoptosis F-AL, Krishnamoorthy B, Darnay B, Aggarwal B, Apoptosis AL, Dinh DH, Kouraklis G, Olivero WC, Gujrati M, Rao JS. Glioma Cells Deficient in Urokinase Plaminogen Activator Receptor Expression Are Susceptible to Tumor Necrosis Glioma Cells Deficient in Urokinase Plaminogen Activator Receptor Expression Are Susceptible to Tumor Necrosis Factor- ␣ -related. 2001; 4195-201. [PubMed] [Google Scholar]
- 39.Yoon MJ, Kang YJ, Kim IY, Kim EH, Lee JA, Lim JH, Kwon TK, Choi KS. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis 2013; 34:1918-28; PMID:23615398; http://dx.doi.org/ 10.1093/carcin/bgt137 [DOI] [PubMed] [Google Scholar]
- 40.Guo L, Fan L, Pang Z, Ren J, Ren Y, Li J, Chen J, Wen Z, Jiang X. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J Control Release 2011; 154:93-102; PMID:21609741; http://dx.doi.org/ 10.1016/j.jconrel.2011.05.008 [DOI] [PubMed] [Google Scholar]
- 41.Ciusani E, Croci D, Gelati M, Calatozzolo C, Sciacca F, Fumagalli L, Balzarotti M, Fariselli L, Boiardi A, Salmaggi A. In vitro effects of topotecan and ionizing radiation on TRAIL/Apo2L-mediated apoptosis in malignant glioma. J Neurooncol 2005; 71:19-25; PMID:15719269; http://dx.doi.org/ 10.1007/s11060-004-9180-4 [DOI] [PubMed] [Google Scholar]
- 42.Badr CE, Wurdinger T, Nilsson J, Niers JM, Whalen M, Degterev A, Tannous BA. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro Oncol 2011; 13:1213-24; PMID:21757445; http://dx.doi.org/ 10.1093/neuonc/nor067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morales JC, Ruiz-Magaña MJ, Carranza D, Ortiz-Ferrón G, Ruiz-Ruiz C. HDAC inhibitors with different gene regulation activities depend on the mitochondrial pathway for the sensitization of leukemic T cells to TRAIL-induced apoptosis. Cancer Lett 2010; 297:91-100; PMID:20580868; http://dx.doi.org/ 10.1016/j.canlet.2010.04.029 [DOI] [PubMed] [Google Scholar]
- 44.Bagci-Onder T, Agarwal A, Flusberg D, Wanningen S, Sorger P, Shah K. Real-time imaging of the dynamics of death receptors and therapeutics that overcome TRAIL resistance in tumors. Oncogene 2012; 32:2818-2827; PMID:22824792; http://dx.doi.org/ 10.1038/onc.2012.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim DR, Park M-Y, Lee C-S, Shim S-H, Yoon H-I, Lee JH, Sung M-W, Kim Y-S, Lee C-T. Combination of vorinostat and adenovirus-TRAIL exhibits a synergistic antitumor effect by increasing transduction and transcription of TRAIL in lung cancer cells. Cancer Gene Ther 2011; 18:467-77; PMID:21455254; http://dx.doi.org/ 10.1038/cgt.2011.11 [DOI] [PubMed] [Google Scholar]
- 46.Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai Y-Y, Sarcar B, Kahali S, Brem S, Yu HM, Rojiani A, et al.. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol 2012; 14:93-100; PMID:22028388; http://dx.doi.org/ 10.1093/neuonc/nor187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov 2008; 7:926-35; PMID:18948999; http://dx.doi.org/ 10.1038/nrd2682 [DOI] [PubMed] [Google Scholar]
- 48.Lee D-H, Lee CS, Kim D-W, Ae JE, Lee T-H. Digitoxin sensitizes glioma cells to TRAIL-mediated apoptosis by upregulation of death receptor 5 and downregulation of survivin. Anticancer Drugs 2014; 25:44-52; PMID:24045365; http://dx.doi.org/ 10.1097/CAD.0000000000000015 [DOI] [PubMed] [Google Scholar]
- 49.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833:3448-59; PMID:23770045; http://dx.doi.org/ 10.1016/j.bbamcr.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Atsina K-B, Himes BT, Strohbehn GW, Saltzman WM. Novel delivery strategies for glioblastoma. Cancer J 2012; 18:89-99; PMID:22290262; http://dx.doi.org/ 10.1097/PPO.0b013e318244d8ae [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fox EJ. Mechanism of action of mitoxantrone. Neurology 2004; 63:S15-8; PMID:15623664; http://dx.doi.org/ 10.1212/WNL.63.12_suppl_6.S15 [DOI] [PubMed] [Google Scholar]
- 52.Gomez-Manzano C, Fueyo J, Kyritsis AP, Steck PA, Levin VA, Alfred Yung WK, McDonnell TJ. Characterization of p53 and p21 Functional Interactions in Glioma Cells en Route to Apoptosis. JNCI J Natl Cancer Inst 1997; 89:1036-44; PMID:9230885; http://dx.doi.org/ 10.1093/jnci/89.14.1036 [DOI] [PubMed] [Google Scholar]
- 53.Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 1997; 94:12479-84; PMID:9356475; http://dx.doi.org/ 10.1073/pnas.94.23.12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hingtgen S, Ren X, Terwilliger E, Classon M, Weissleder R, Shah K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol Cancer Ther 2008; 7:3575-85; PMID:19001440; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adiguzel Z, Baykal AT, Kacar O, Yilmaz VT, Ulukaya E, Acilan C. Biochemical and proteomic analysis of a potential anticancer agent: Palladium(II) Saccharinate complex of terpyridine acting through double strand break formation. J Proteome Res 2014; 13:5240-9; PMID:25210790; http://dx.doi.org/ 10.1021/pr5006718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.