

ABSTRACT
Stereotactic ablative radiotherapy (SABR) has emerged as a highly promising treatment for medically inoperable early-stage non-small cell lung cancer patients. Treatment outcomes after SABR have been excellent compared to conventional fractionated radiotherapy (CFRT). However, the biological determinants of the response to ablative doses of radiation remain poorly characterized. Furthermore, there's little data on the cellular and molecular response of genetically distinct NSCLC subtypes to radiation. We assessed the response of 3 genetically distinct lung adenocarcinoma cell lines to ablative and fractionated ionizing radiation (AIR and FIR). We studied clonogenic survival, cell proliferation, migration, invasion, apoptosis and senescence. We also investigated the effect of AIR and FIR on the expression of pro-invasive proteins, epithelial-to-mesenchymal transition (EMT), extracellular signal-regulated kinases (ERK1/2) and the transmembrane receptor cMET. Our findings reveal that AIR significantly reduced cell proliferation and clonogenic survival compared to FIR in A549 cells only. This differential response was not observed in HCC827 or H1975 cells. AIR significantly enhanced the invasiveness of A549 cells, but not HCC827 or H1975 cells compared to FIR. Molecular analysis of pathways involved in cell proliferation and invasion revealed that AIR significantly reduced phosphorylation of ERK1/2 and upregulated cMET expression in A549 cells. Our results show a differential proliferative and invasive response to AIR that is dependent on genetic subtype and independent of intrinsic radioresistance. Further examination of these findings in a larger panel of NSCLC cell lines and in pre-clinical models is warranted for identification of biomarkers of tumor response to AIR.
KEYWORDS: Ablative radiotherapy, adenocarcinoma, cMET, invasion, lung cancer, proliferation
Abbreviations
NSCLC, non-small cell lung cancer; EGFR, epidermal growth factor receptor; KRAS, kirsten rat sarcoma viral oncogene homolog; cMET, mesenchymal epithelial transition factor; EMT, epithelial mesenchymal transition; SABR, stereotactic ablative radiotherapy; AIR, ablative ionizing radiation; FIR, fractionated ionizing radiation; IR, ionizing radiation; ERK, extracellular signal-regulated kinases; Gy, Gray
Introduction
Non-small cell lung cancer (NSCLC) represents the most common type of lung cancer, affecting 75–80% of patients.1,2 Surgical resection is the treatment of choice for stage I NSCLC, resulting in 5-year survival rates of approximately 50–70%.3-6 However, up to a third of early-stage patients are medically inoperable. Historically, these patients were treated with conventional fractionated radiotherapy (CFRT) comprised of 60–66 Gy in 2–3 Gy fractions, which offered poor control with an in-field tumor progression rate of 80–90%.7-10 In recent years, stereotactic ablative radiation therapy (SABR), a technique allowing accurate delivery of very high doses of radiation in a small number of fractions, has evolved as the standard therapy for inoperable patients.2,8,11-14 Typically, a dose of 34–60 Gy is delivered in 1–8 fractions distributed over 1–2 weeks. Reported treatment outcomes with SABR have been excellent, with 3-year local control greater than 90%, and overall survival rates at 3 y that are in excess of 50%.12,13,15
Despite its widespread use, the biology underlying cellular responses and cell fate following SABR remain understudied. It is increasingly appreciated that NSCLC is not a uniform disease but consists of many genotype-defined subtypes. The most common subtypes are defined by oncogenic mutations in KRAS and EGFR, which may confer resistance and sensitivity to radiation, respectively.16-18 Importantly, it is still unknown how these genotypes can affect responses to SABR.
Controversially, a number of studies have demonstrated that SABR-like radiotherapy fractions can promote metastatic features such as migration and invasion in glioma,19-21 breast,22-24 liver,25 pancreas,26 melanoma27 and NSCLC cell lines.28-30 However, these observations were made without comparison to responses to fractionated radiation. Whether SABR has the capacity to enhance metastatic potential in NSCLC remains unknown. Furthermore, it is not known whether this effect is specific to certain genotype-defined tumor subtypes. This study was designed to assess the diversity in cellular and molecular responses to ablative ionizing radiation (AIR) and fractionated ionizing radiation (FIR) in a panel of oncogene driven lung adenocarcinoma cell lines. Our results highlight a differential response to AIR among genetically distinct NSCLC cells.
Materials and methods
Cell culture and irradiation
Human lung adenocarcinoma cells A549, H1975 and HCC827 were obtained from the American type culture collection (ATCC, VA). Cells were cultured at 37°C and 5% CO2 in RPMI 1640 media except for A549, which was cultured in DMEM F/12 media. Media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cell irradiation was performed using a Faxitron X-Ray machine (Faxitron X-ray Corporation, IL). X-ray tube voltage was set to 160 kVp, current of 6.3 mA and dose rate of 1450 R/min. Unless otherwise stated, cells were exposed to 12 Gy for AIR or 4 daily fractions of 3 Gy for FIR.
Cellular proliferation and clonogenic survival
Inhibition of cell proliferation was assessed using the Vybrant® 3-[4,5- dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) assay according to manufacturer instructions (Molecular Probes, OR). Cells were plated in 96-well plates at optimal densities 24 h after irradiation (8 Gy or 12 Gy for AIR and 4 daily fractions of 2 Gy or 3 Gy for FIR) and incubated for 3–5 d The number of fractions in FIR was limited to 4 to maintain optimal cell confluence and tissue culture conditions. Absorbance readings were performed at 560 nm using a standard microplate reader (Thermo Scientific, Multiskan Spectrum). Separate wells were kept for morphological analysis with the fluorescent stain, DAPI.
The colony-forming assay was performed as reported previously.31 Exponentially growing cells were irradiated with increasing doses of IR (0–10 Gy) and cultured until colonies appeared (10 days). Colonies with 50 cells or more were counted. The surviving fraction was determined by dividing the number of colonies by the number of cells plated with a correction for plating efficiency. Data was plotted on semi-log graphs and fitted using the linear-quadratic (LQ) model.32
Cell death and senescence
Cell death was assessed in all cell lines after exposure to AIR using Trypan blue exclusion assay. Cells were plated at equal densities in 6-well plates, irradiated and harvested at 0h, 24 h, 48 h, 72 h and 96 h. Harvested cells were counted and analyzed using an automated cell counter (TC-10, Bio-Rad, CA). Cytochemical staining for senescence-associated b-galactosidase (SA-bgal) was performed 5 d after treatment using a commercial kit (Cell Signaling, MA).33 Images were acquired at 200x magnification. The number of stained and unstained cells was counted in at least 5 fields and averaged. Percent senescence was calculated as the fraction of positively staining cells over the total number of cells. Similarly, cell polyploidy was assessed 5 d after treatment as an indicator of cells undergoing mitotic catastrophe. Cells were fixed and stained with DAPI and analyzed under a fluorescent microscope (Zeiss Axio Vert.A1). Images from 5 20x fields were obtained and analyzed for the presence of multinucleated cells. Percent polyploidy was calculated as the fraction of multinucleated cells over the total number of cells.
Cell cycle analysis
Cell cycle analysis was performed on our panel of NSCLC cell lines 24-hours after exposure to 12 Gy. Briefly, cells from treated and untreated plates were collected and fixed in 70% ethanol for 30 minutes. Cells were centrifuged and resuspended in 1mg/mL RNase A (Sigma-Aldrich, St. Louis, MO) and incubated at 37°C for 30 minutes. Cells were stained with propidium iodide (Sigma-Aldrich St. Louis, MO) by incubating for 20 minutes at room temperature. Cell cycle analysis was performed using a BDFACS Canto II Flow cytometer (BD Biosciences, San Jose, USA). The percentage of subG1, G1, S and G2 populations was calculated with FlowJo software (version 9.6.2, FlowJo, LLC, Ashland, OR, USA).
Migration and invasion assays
Cell migration and invasion was assessed using modified Boyden chambers (8-μm pore, BD Biosciences, CA) according to previously established methods.34 Chambers were either non-coated for migration or pre-coated with BD Matrigel basement membrane for invasion studies. Assays were performed 5 d after exposure to AIR or FIR. Invading cells were counted under 200x magnification from at least 5 fields per chamber and averaged. Microscopic images of invading cells were taken using a Zeiss camera attached to a Zeiss Axio Vert.A1 microscope.
Wound-healing assay
A549 cells were incubated for 24 h or 72 h after irradiation. After the incubation period, cells were plated in triplicates in 6-well plates and allowed to adhere overnight. Wounds were made using a sterile 200 uL pipette tip.35 Wells were washed with PBS and cells were replenished with starving media (0.5% FBS). Phase contrast images were acquired from the same region at 0 h, 24 h and 48 h at 100x magnification. Wound closure was measured using ImageJ software (http://imagej.nih.gov/ij) and normalized to the initial wound width (0% closure, 0h).
Western blot analysis
A549 cells were serum starved 16 hours prior to irradiation and cell lysates collected at 3 hr, 24 hr and 48 h time points. Protein concentration was quantified using a BCA protein kit (Thermo Fisher Scientific, MA), according to manufacturer instructions. Equal amounts of protein were separated by SDS-PAGE under reducing conditions and blotted onto polyvinylidene difluoride membranes.36 Membranes were blocked in 5% nonfat milk and probed with primary antibodies directed against p53 (Santa Cruz Biotechnology, TX), p21, poly(ADP-ribose) polymerase-1 (PARP-1), E-cadherin, vimentin, phospho-FAK-tyr397, FAK, c-MET, phospho-ERK1/2, ERK1/2, phospho-AKT, AKT, phospho-mTOR, mTOR or β-actin (Cell Signaling, MA). Densitometric measurements were performed using ImageJ software.
Statistical analysis
All experiments were performed in triplicates and repeated at least in 3 independent settings. Unless otherwise stated, statistical analysis was performed using 2-way ANOVA in Prism Software and all values are expressed as mean ± standard error of the mean (SEM). Statistical significance was set at *p < 0.05; **p < 0.01; ***p < 0.001.
Results
Differential proliferative response to AIR based on molecular subtype
To assess the response to AIR, 3 genetically distinct NSCLC adenocarcinoma cell lines were used. A549 cells harbor mutant KRAS (G12S) whereas HCC827 and H1975 cells harbor mutant EGFR (Del E746-A750 and L858R respectively). In addition, H1975 cells harbor the T790M mutation which has been shown to confer resistance to EGFR tyrosine kinase inhibitors.37 Cells were exposed to 8 Gy or 12 Gy and proliferation was assessed by MTT. Inhibition of proliferation was normalized to untreated control and compared to FIR comprised of 4 fractions of 2 Gy or 3 Gy. Significant differences in the inhibitory capacity of AIR compared to FIR were observed in A549 cells only. Cell proliferation was reduced by 60% and 71% following exposure to single doses of 8 Gy and 12 Gy, respectively. Equivalent fractionated total doses of 8 Gy and 12 Gy resulted in 38% and 36% reduction, respectively (Fig. 1A). In contrast, HCC827 and H1975 cells did not exhibit a significant proliferative difference after exposure to AIR compared to FIR. In HCC827 cells, AIR of 8 Gy and 12 Gy decreased proliferation by 43% and 65%, while fractionated doses reduced cell proliferation by 47% and 72%, respectively (Fig. 1B). Similar findings were also observed in H1975 cells (Fig. 1C).
Figure 1.
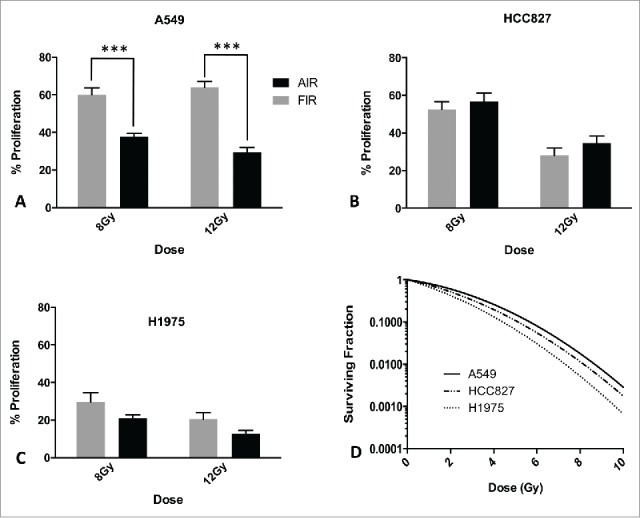
Proliferation and clonogenic surival of NSCLC cells treated with AIR or FIR. (A-C) MTT proliferation assay 5 d after exposure of cells to AIR and FIR of 8Gy and 12Gy. Data is normalized to respective untreated controls. Error bars represent SEM of 3 independent experiments. (D) Clonogenic cell survival of NSCLC cells exposed to doses of 2Gy-10Gy. Data was fitted using the linear-quadratic model (α/β = 10).
To determine whether the difference in inhibition of proliferation between AIR and FIR in A549 cells is due to differences in the biologically equivalent dose (BED), we compared a single-dose of 8 Gy to 4 fractions of 3 Gy (BED of 14.4 Gy vs 15.6 Gy, respectively). Our findings show similar inhibition of proliferation between 8 Gy and 12 Gy single-doses. Therefore, for subsequent experiments a dose of 12 Gy was used and compared to an equivalent dose in 4 fractions.
To determine whether the differences in proliferation were related to intrinsic radiosensitivity of the individual cell lines, we performed clonogenic cell survival assays and assessed the survival fraction at 2 Gy (SF2). Consistent with literature values,38 our results show that A549 and HCC827 cells are relatively less radiosensitive (SF2 0.74 and 0.67 respectively) than H1975 cells (SF2 0.34) (Fig. 1D). Collectively, these results show a differential proliferative response to AIR based on genetic subtype and independent of intrinsic radioresistance as measured by SF2. AIR differentially regulated cell proliferation of A549 cells compared to FIR. In contrast, AIR and FIR were equally effective in inhibiting proliferation in HCC827 and H1975 cells.
Differential cell death response of genetically distinct NSCLC subtypes to AIR
We sought to examine the effects of AIR on cell fate in our panel of cell lines. Following exposure to AIR, A549 cells acquired a distinct phenotype with large cell cytoplasm and formation of numerous cell protrusions. In contrast, H1975 and HCC827 cells exposed to AIR became enlarged and developed fragmented and floating bodies. Time-course analysis of cell death, up to 96h after AIR, using Trypan blue exclusion revealed significant increase in cell death in HCC827 and H1975 cells, but not in A549 cells compared to untreated cells (Fig. 2A). Analysis of SA-bgal staining identified the presence of similar levels of senescent cells in all cell lines, 5 d after exposure to AIR (Fig. 2B). Further analysis, revealed that all senescent HCC827 and H1975 cells were highly multinucleated and contained micronuclei indicative of cells undergoing mitotic catastrophe.39 In contrast, less than 5% of senescent A549 cells were multinucleated (Fig. 2C). Since senescence did not dominate the entire population of AIR-treated A549 cells (less than 40%), in separate experiments we observed that A549 cells regained their full proliferative capacity after 28 d (data not shown).
Figure 2.

Analysis of modes of cell death after AIR. (A) Time course analysis of Trypan blue in NSCLC cell lines after exposure to 12Gy. (B) Quantitative analysis of senescence-associated β-galactosidase activity 5 d after exposure to AIR. (C) Analysis of polyploidy after exposure to AIR. Bars represent SEM of 3 independent experiments. Captions show representative images from AIR-treated cells (magnification x100).
Cell cycle analysis of our panel of lung adenocarcinoma cell lines 24 hours following exposure to 12 Gy using flow cytometry showed substantial increase in subG1 cells in HCC827 and H1975 cells, indicative of apoptosis (Fig. 3B, C). The reduction in S-phase population in HCC827 and H1975 cells was at the expense of an elevated G1-phase population and a drastic reduction in the G2-phase population. These results indicate that AIR of 12 Gy induces apoptosis and G1/S cell cycle arrest in HCC827 and H1975 cells. Conversely, A549 cells demonstrated a lack of G1 cell cycle arrest, a reduction in S-phase population and elevated G2-phase population indicating G2/M arrest with no evidence of apoptosis or cell death (Fig. 3A). Consistent with the observed G1 cell-cycle arrest in A549 cells, AIR induced significantly higher levels of p53 and downstream p21 compared to control as shown in Fig. 3.
Figure 3.

Cell cycle analysis of NSCLC cells 24 hours after exposure to AIR as assessed using flow cytometry. Right panels show Western blot expression of proteins involved in cell cycle (p53 and p21) and apoptosis (PARP). Actin was used as loading control.
AIR modulates the invasive response of genetically distinct NSCLC Subtypes
We investigated the effect of AIR on cell migration and invasion in NSCLC cells using modified Boyden chambers. A549 cells exposed to AIR, but not FIR showed a statistically significant increase (fold2-) in cell migration and invasion (Fig. 4A, D) compared to untreated cells. HCC827 cells exposed to either AIR or FIR showed significantly lower migration and invasion compared to untreated cells (Fig. 4B,E). In H1975 cells, AIR did not significantly impact migration levels, but significantly reduced invasion, while FIR significantly reduced migration and invasion compared to untreated cells (Fig. 4C, F). These results clearly demonstrate that AIR-induced invasion is dependent on the cell's molecular subtype.
Figure 4.

Migration and invasion of NSCLC cells in modified Boyden chambers 5 d after exposure to AIR and FIR. (A-C) Migration of NSCLC cells through uncoated Boyden chambers. (D-F) Invasion of NSCLC cells through matrigel-coated Boyden chambers. Data is normalized to respective controls. Bars represent SEM from 3 independent experiments. Representative images of invaded cells are shown at the bottom (magnification x200).
AIR increases wound healing capacity of A549 cells
To gain better visualization of morphological alterations associated with cell migration of A549 cells after exposure to IR, we performed wound-healing assays. In A549 cells exposed to FIR, no significant difference in wound-healing capacity was observed compared to untreated cells. In contrast, A549 cells exposed to AIR showed a significant increase in motility and wound closure at 24h and 48h relative to untreated as well as FIR-treated cells (Fig. 5).
Figure 5.

Wound healing assay of A549 cells 3 d after exposure to AIR and FIR. (A) Representative wound-healing images taken at 0h, 24h and 48h with respective wound-closure ratios. Magnification x100. (B) Wound-closure analysis after AIR and FIR normalized to 0% at 0h. Experiments were repeated 3 times. Bars represent standard deviation from one representative experiment.
Modulation of pro-invasive proteins by AIR
Exposure to IR can induce epithelial-to-mesenchymal transition (EMT) and activate pro-invasive pathways involving focal adhesion kinas (FAK), extracellular signal-regulated kinases (ERK1/2), mechanistic target of rapamycin (mTOR) and the transmembrane receptor c-MET.21,26,40-42 However, the effect of radiation regimen (AIR versus FIR) on radiation-induced invasion has not been previously investigated. We did not observe a significant difference in the expression of vimentin and E-cadherin at 24h following AIR compared to FIR (Fig. 6). However, AIR upregulated c-MET expression by 2.fold3- in A549 cells compared to FIR (Fig. 6). Phosphorylation of ERK1/2 was significantly reduced after AIR compared to FIR. This is consistent with the role of ERK1/2 in cell-cycle progression and cell proliferation.43 No significant difference was observed in total and phosphorylated levels of FAK, AKT and mTOR after AIR compared to FIR (Fig. 6).
Figure 6.
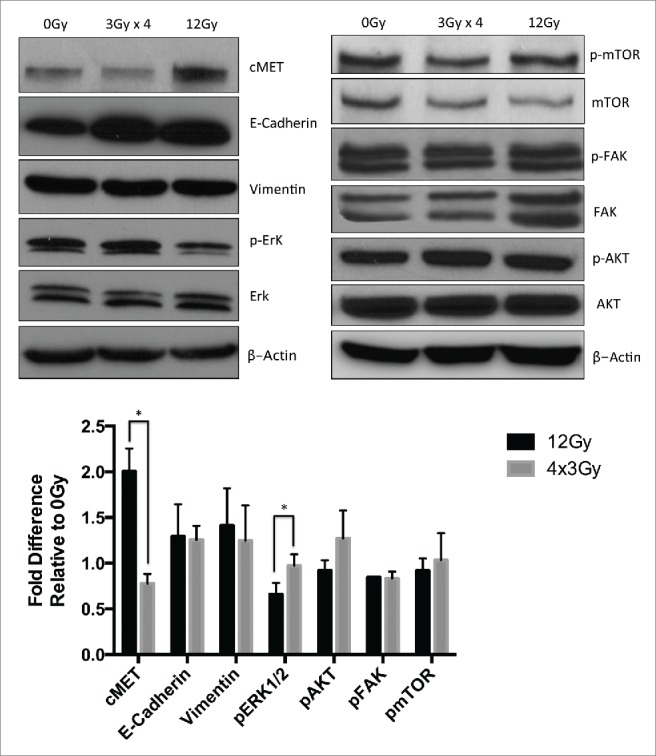
Western blot analysis of pro-invasive proteins in A549 cells treated with AIR or FIR. (A) Representative blots of membranes probed with antibodies directed against c-MET, ERK1/2, phosphor ERK1/2, vimentin, E-cadherin, FAK, phospho FAK, AKT, phospho AKT, mTOR and phospho mTOR. Actin was used as loading control. (B) Quantification of protein expression relative to baseline expression from untreated A549 cells. Bars represent SEM from 3 independent experiments.
Discussion
NSCLC consists of many genotype-defined subtypes. The common subtypes are defined by oncogenic mutations in KRAS or EGFR.44 SABR is increasingly used in the clinic as a curative treatment for inoperable early-stage NSCLC patients. The molecular and cellular characterization of the response to ablative-doses of radiation compared to conventional fractionation in genetically distinct NSCLC cells remain poorly investigated. Our study is the first attempt to examine the heterogeneity of response to AIR across genetically distinct adenocarcinoma cell lines. Our results identify significant differences in the proliferative, clonogenic, invasive and cell death response to AIR across 3 lung adenocarcinoma cells lines harboring KRAS and EGFR mutations.
It is generally established that ablative-doses (above 8 Gy) of radiation provide superior tumor growth inhibition compared to equivalent fractionated doses.45 Our results confirm this finding in A549 cells, but we demonstrate that in HCC827 and H1975 cells, the response to AIR and FIR is similar, irrespective of intrinsic radioresistance. Radioresistance is traditionally defined by the survival fraction at 2 Gy (SF2) derived from cell survival curves of clonogenic assays.45 The similar response to AIR and FIR observed in HCC827 and H1975 cells can be explained partly by the presence of tyrosine kinase mutations in EGF receptors of both cell lines. HCC827 cells harbor a deletion at the E746-A750 of exon 19 while H1975 cells harbor 2 mutations resulting in amino acid change in the tyrosine kinase domain of EGFR (L858R and T790M). Previous studies have shown that EGFR can translocate to the nucleus upon activation with IR where it binds to DNA-dependent protein kinase catalytic subunits (DNA-PKcs), leading to activation of the nonhomologous end-joining DNA repair pathway.46,47 Mutations in EGFR block its translocation and binding to DNA-PKcs.48 This may result in comparable levels of DNA damage after AIR and FIR, resulting in similar inhibition of proliferation as observed in our study.
Cell fate is an important consideration when assessing the response to AIR. The decrease in cell proliferation observed in HCC827 and H1975 cells following AIR, was associated with increased apoptosis whereas in A549 cells, inhibition of proliferation was attributed to cell-cycle arrest leading to senescence. The exact mechanism controlling cell fate following AIR remains unknown. The p53 tumor suppressor protein is a key mediator of the DNA damage response that is activated following exposure to ionizing radiation.49,50 Wild type p53 is known to induce cell cycle arrest through the induction of cyclin-dependent kinase inhibitor, p21CIP1/WAF151. p53-dependent activation of p21 is also a key pathway in stress-induced senescence and promotes apoptosis resistance in several cancer cell lines.43,52 A549 cells harbor wild-type p53, which may have contributed to the induction of senescence and decreased proliferation in A549 cell exposed to AIR. However, stress-induced senescence is not the most desirable outcome of cancer treatment as senescent cells remain metabolically active and can secrete growth factors, cytokines and matrix-degrading enzymes which result in increased migration and invasion of nearby tumor cells.53-56 Converting senescence to apoptosis is an active area of research and represents the most desirable outcome of anti-neoplastic treatment.17,52
Radiation-induced invasion has been reported by a number of investigators. Canazza et al compared a single-dose of 8 Gy to 4 fractions of 2 Gy in a glioblastoma cell line and reported increased invasion after single-dose, but not fractionated doses.20 In Lewis lung tumor cells, irradiation with 7.5 Gy increased invasion in vitro while in tumor-bearing rats irradiation with 50 Gy in 5 fractions was shown to promote pulmonary metastasis.57 Our findings support these reports, as we demonstrate that A549 cells that survived AIR develop a more invasive phenotype compared to FIR. Studies have also shown that radiation can activate pro-invasive pathways and induce epithelial-to-mesenchymal transition in vitro.30,41 Our analysis did not reveal significant changes in EMT marker expression after AIR compared to FIR. However, we showed increased expression of cMET after exposure to AIR but not FIR, which may have contributed to the development of more invasive profile in AIR-treated A549 cells. Activity of the cMET tyrosine kinase receptor has been shown to promote invasion and metastasis in NSCLC.58,59 Furthermore, cMET overexpression was shown to promote survival and inhibit apoptosis in NSCLC cells.60 More studies are needed to define the potential role of c-MET in AIR-induced invasiveness. This is of clinical significance as c-MET inhibition was shown as an effective strategy to radiosensitize NSCLC tumors and is currently being tested in clinical trials of NSCLC.61,62
Current radiotherapy schedules do not account for a tumor's genetic composition. In particular, knowledge of EGFR and K-RAS status may not only impact patient prognosis, but also the choice of radiotherapy treatment regimen in NSCLC. Our findings underscore the need for genetic characterization of tumors and elucidation of radiation-induced pathways in order to provide better treatment with SABR. Combining SABR with targeted therapy may provide an effective regimen to circumvent the pro-invasive effects of radiation. Identification of biomarkers that predict tumor response to ablative radiotherapy will help identify patients who may derive the most benefit from SABR, and will allow tailoring of ablative radiation doses to genetically distinct NSCLC subtypes.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was done with grant support from the Montreal General Hospital foundation. AO was funded by Fonds de recherche du Quebec - Santé (FRQS).
References
- 1.Soliman H, Cheung P, Yeung L, Poon I, Balogh J, Barbera L, Spayne J, Danjoux C, Dahele M, Ung Y. Accelerated hypofractionated radiotherapy for early-stage non-small-cell lung cancer: long-term results. Int J Radiat Oncol Biol Phys 2011; 79:459-65; PMID:20385455; http://dx.doi.org/ 10.1016/j.ijrobp.2009.11.003 [DOI] [PubMed] [Google Scholar]
- 2.Timmerman R, Paulus R, Galvin J, Michalski J, Straube W, Bradley J, Fakiris A, Bezjak A, Videtic G, Johnstone D, et al.. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA 2010; 303:1070-6; http://dx.doi.org/ 10.1001/jama.2010.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asamura H. Treatment of choice for stage I non-small cell lung cancer: surgery or radiotherapy? J Thorac Oncol 2006; 1:766-7; PMID:17409957; http://dx.doi.org/ 10.1016/S1556-0864(15)30403-2 [DOI] [PubMed] [Google Scholar]
- 4.Chang MY, Sugarbaker DJ. Surgery for early stage non-small cell lung cancer. Semin Surg Oncol 2003; 21:74-84; PMID:14508857; http://dx.doi.org/ 10.1002/ssu.10024 [DOI] [PubMed] [Google Scholar]
- 5.Graham PH, Vinod SK, Hui AC. Stage I non-small cell lung cancer: results for surgery in a patterns-of-care study in Sydney and for high-dose concurrent end-phase boost accelerated radiotherapy. J Thorac Oncol 2006; 1:796-801; PMID:17409962; http://dx.doi.org/ 10.1016/S1556-0864(15)30408-1 [DOI] [PubMed] [Google Scholar]
- 6.Ost D, Goldberg J, Rolnitzky L, Rom WN. Survival after surgery in stage IA and IB non-small cell lung cancer. Am J Respir Crit Care Med 2008; 177:516-23; PMID:18006887; http://dx.doi.org/ 10.1164/rccm.200706-815OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belderbos JS, Heemsbergen WD, De Jaeger K, Baas P, Lebesque JV. Final results of a Phase I/II dose escalation trial in non-small-cell lung cancer using three-dimensional conformal radiotherapy. Int J Radiat Oncol Biol Phys 2006; 66:126-34; PMID:16904518; http://dx.doi.org/ 10.1016/j.ijrobp.2006.04.034 [DOI] [PubMed] [Google Scholar]
- 8.Timmerman RD, Park C, Kavanagh BD. The North American experience with stereotactic body radiation therapy in non-small cell lung cancer. J Thorac Oncol 2007; 2:S101-12; PMID:17603304; http://dx.doi.org/ 10.1097/JTO.0b013e318074e4fa [DOI] [PubMed] [Google Scholar]
- 9.Carlson DJ, Keall PJ, Loo BW Jr., Chen ZJ, Brown JM. Hypofractionation results in reduced tumor cell kill compared to conventional fractionation for tumors with regions of hypoxia. Int J Radiat Oncol Biol Phys 2011; 79:1188-95; PMID:21183291; http://dx.doi.org/ 10.1016/j.ijrobp.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumann M, Appold S, Petersen C, Zips D, Herrmann T. Dose and fractionation concepts in the primary radiotherapy of non-small cell lung cancer. Lung Cancer 2001; 33 Suppl 1:S35-45; PMID:11576706; http://dx.doi.org/ 10.1016/S0169-5002(01)00301-4 [DOI] [PubMed] [Google Scholar]
- 11.Chang BK, Timmerman RD. Stereotactic body radiation therapy: a comprehensive review. Am J Clin Oncol 2007; 30:637-44; PMID:18091059; http://dx.doi.org/ 10.1097/COC.0b013e3180ca7cb1 [DOI] [PubMed] [Google Scholar]
- 12.Fakiris AJ, McGarry RC, Yiannoutsos CT, Papiez L, Williams M, Henderson MA, Timmerman R. Stereotactic body radiation therapy for early-stage non-small-cell lung carcinoma: four-year results of a prospective phase II study. Int J Radiat Oncol Biol Phys 2009; 75:677-82; PMID:19251380; http://dx.doi.org/ 10.1016/j.ijrobp.2008.11.042 [DOI] [PubMed] [Google Scholar]
- 13.Rusthoven KE, Pugh TJ. Stereotactic body radiation therapy for inoperable lung cancer. JAMA 2010; 303:2354-5; author reply 5; http://dx.doi.org/ 10.1001/jama.2010.777 [DOI] [PubMed] [Google Scholar]
- 14.Iyengar P, Timmerman RD. Stereotactic ablative radiotherapy for non-small cell lung cancer: rationale and outcomes. J Natl Compr Canc Netw 2012; 10:1514-20; PMID:23221789 [DOI] [PubMed] [Google Scholar]
- 15.Timmerman R, Bastasch M, Saha D, Abdulrahman R, Hittson W, Story M. Stereotactic body radiation therapy: normal tissue and tumor control effects with large dose per fraction. Front Radiat Ther Oncol 2011; 43:382-94; PMID:21625164; http://dx.doi.org/ 10.1159/000322494 [DOI] [PubMed] [Google Scholar]
- 16.Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res 2005; 65:7902-10; PMID:16140961; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0513 [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Kern AM, Hulskotter M, Greninger P, Singh A, Pan Y, Chowdhury D, Krause M, Baumann M, Benes CH, et al.. EGFR-mediated chromatin condensation protects KRAS-mutant cancer cells against ionizing radiation. Cancer Res 2014; 74:2825-34; PMID:24648348; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, Muschel RJ, McKenna WG. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res 2000; 60:6597-600; PMID:11118040 [PubMed] [Google Scholar]
- 19.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res 2001; 61:2744-50; PMID:11289157 [PubMed] [Google Scholar]
- 20.Canazza A, Calatozzolo C, Fumagalli L, Bergantin A, Ghielmetti F, Fariselli L, Croci D, Salmaggi A, Ciusani E. Increased migration of a human glioma cell line after in vitro CyberKnife irradiation. Cancer Biol Ther 2011; 12:629-33; PMID:21775821; http://dx.doi.org/ 10.4161/cbt.12.7.16862 [DOI] [PubMed] [Google Scholar]
- 21.Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, Park IC, Rhee CH, Hong SI. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res 2006; 66:8511-9; PMID:16951163; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4340 [DOI] [PubMed] [Google Scholar]
- 22.Paquette B, Therriault H, Wagner JR. Role of interleukin-1beta in radiation-enhancement of MDA-MB-231 breast cancer cell invasion. Radiation Res 2013; 180:292-8; PMID:23927563; http://dx.doi.org/ 10.1667/RR3240.1 [DOI] [PubMed] [Google Scholar]
- 23.Paquette B, Therriault H, Desmarais G, Wagner R, Royer R, Bujold R. Radiation-enhancement of MDA-MB-231 breast cancer cell invasion prevented by a cyclooxygenase-2 inhibitor. Br J Cancer 2011; 105:534-41; PMID:21792195; http://dx.doi.org/ 10.1038/bjc.2011.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilalta M, Rafat M, Giaccia AJ, Graves EE. Recruitment of circulating breast cancer cells is stimulated by radiotherapy. Cell Rep 2014; 8:402-9; PMID:25017065; http://dx.doi.org/ 10.1016/j.celrep.2014.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou LY, Wang ZM, Gao YB, Wang LY, Zeng ZC. Stimulation of hepatoma cell invasiveness and metastatic potential by proteins secreted from irradiated nonparenchymal cells. Int J Radiat Oncol Biol Phys 2012; 84:822-8; PMID:22420973; http://dx.doi.org/ 10.1016/j.ijrobp.2012.01.011 [DOI] [PubMed] [Google Scholar]
- 26.Qian LW, Mizumoto K, Urashima T, Nagai E, Maehara N, Sato N, Nakajima M, Tanaka M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin Cancer Res 2002; 8:1223-7; PMID:11948136 [PubMed] [Google Scholar]
- 27.Rofstad EK, Mathiesen B, Galappathi K. Increased metastatic dissemination in human melanoma xenografts after subcurative radiation treatment: radiation-induced increase in fraction of hypoxic cells and hypoxia-induced up-regulation of urokinase-type plasminogen activator receptor. Cancer Res 2004; 64:13-8; PMID:14729600; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-2658 [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Huang YJ, Liu C, Yang YY, Liu H, Cui JG, Cheng Y, Gao F, Cai JM, Li BL. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab Invest 2014; 94:362-70; PMID:24468793; http://dx.doi.org/ 10.1038/labinvest.2013.153 [DOI] [PubMed] [Google Scholar]
- 29.Li X, Ishihara S, Yasuda M, Nishioka T, Mizutani T, Ishikawa M, Kawabata K, Shirato H, Haga H. Lung cancer cells that survive ionizing radiation show increased integrin alpha2beta1- and EGFR-dependent invasiveness. PloS One 2013; 8:e70905; PMID:23951036; http://dx.doi.org/ 10.1371/journal.pone.0070905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, Epperly M, Levina V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer 2013; 12:94; PMID:23947765; http://dx.doi.org/ 10.1186/1476-4598-12-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdulkarim B, Sabri S, Zelenika D, Deutsch E, Frascogna V, Klijanienko J, Vainchenker W, Joab I, Bourhis J. Antiviral agent cidofovir decreases Epstein-Barr virus (EBV) oncoproteins and enhances the radiosensitivity in EBV-related malignancies. Oncogene 2003; 22:2260-71; PMID:12700662; http://dx.doi.org/ 10.1038/sj.onc.1206402 [DOI] [PubMed] [Google Scholar]
- 32.Barendsen GW. Dose fractionation, dose rate and iso-effect relationships for normal tissue responses. Int J Radiat Oncol Biol Phys 1982; 8:1981-97; PMID:6759484; http://dx.doi.org/ 10.1016/0360-3016(82)90459-X [DOI] [PubMed] [Google Scholar]
- 33.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated β-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:1798-806; PMID:20010931; http://dx.doi.org/ 10.1038/nprot.2009.191 [DOI] [PubMed] [Google Scholar]
- 34.Chahal M, Abdulkarim B, Xu Y, Guiot MC, Easaw JC, Stifani N, Sabri S. O6-Methylguanine-DNA methyltransferase is a novel negative effector of invasion in glioblastoma multiforme. Mol Cancer Ther 2012; 11:2440-50; PMID:22986464; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0977 [DOI] [PubMed] [Google Scholar]
- 35.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007; 2:329-33; PMID:17406593; http://dx.doi.org/ 10.1038/nprot.2007.30 [DOI] [PubMed] [Google Scholar]
- 36.Chahal M, Xu Y, Lesniak D, Graham K, Famulski K, Christensen JG, Aghi M, Jacques A, Murray D, Sabri S, et al.. MGMT modulates glioblastoma angiogenesis and response to the tyrosine kinase inhibitor sunitinib. Neuro Oncol 2010; 12:822-33; PMID:20179017; http://dx.doi.org/ 10.1093/neuonc/noq017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG, Halmos B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res 2005; 65:7096-101; PMID:16103058; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1346 [DOI] [PubMed] [Google Scholar]
- 38.Das AK, Sato M, Story MD, Peyton M, Graves R, Redpath S, Girard L, Gazdar AF, Shay JW, Minna JD, et al.. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res 2006; 66:9601-8; PMID:17018617; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2627 [DOI] [PubMed] [Google Scholar]
- 39.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4:303-13; PMID:11991684; http://dx.doi.org/ 10.1054/drup.2001.0213 [DOI] [PubMed] [Google Scholar]
- 40.Guerra LE, Smith RM, Kaminski A, Lagios MD, Silverstein MJ. Invasive local recurrence increased after radiation therapy for ductal carcinoma in situ. Am J Surg 2008; 196:552-5; PMID:18809062; http://dx.doi.org/ 10.1016/j.amjsurg.2008.06.008 [DOI] [PubMed] [Google Scholar]
- 41.Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, Qiu LB, Ding GR, Su XM, Mei S, et al.. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-β-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys 2011; 81:1530-7; PMID:22115555; http://dx.doi.org/ 10.1016/j.ijrobp.2011.06.1956 [DOI] [PubMed] [Google Scholar]
- 42.Nilsson M, Giri U, Gudikote J, Tang X, Lu W, Tran HT, Fan YH, Koo A, Diao L, Tong P, et al.. KDR AMPLIFICATION IS ASSOCIATED WITH VEGF-INDUCED ACTIVATION OF mTOR AND INVASION PATHWAYS BUT DOES NOT PREDICT CLINICAL BENEFIT TO THE VEGFR TKI VANDETANIB. Clin Cancer Res 2015; http://dx.doi.org/ 10.1158/1078-0432.CCR-15-1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007; 26:3227-39; PMID:17496918; http://dx.doi.org/ 10.1038/sj.onc.1210414 [DOI] [PubMed] [Google Scholar]
- 44.Clinical Lung Cancer Genome P, Network Genomic M . A genomics-based classification of human lung tumors. Sci Transl Med 2013; 5:209ra153; PMID:24174329; http://dx.doi.org/ 10.1126/scitranslmed.3006802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horsman MR, Nielsen T, Ostergaard L, Overgaard J. Radiation administered as a large single dose or in a fractionated schedule: Role of the tumour vasculature as a target for influencing response. Acta Oncol 2006; 45:876-80; PMID:16982553; http://dx.doi.org/ 10.1080/02841860600900068 [DOI] [PubMed] [Google Scholar]
- 46.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Kehlbach R, Rodemann HP. Nuclear epidermal growth factor receptor modulates cellular radio-sensitivity by regulation of chromatin access. Radiother Oncol 2011; 99:317-22; PMID:21704408; http://dx.doi.org/ 10.1016/j.radonc.2011.06.001 [DOI] [PubMed] [Google Scholar]
- 47.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem 2005; 280:31182-9; PMID:16000298; http://dx.doi.org/ 10.1074/jbc.M506591200 [DOI] [PubMed] [Google Scholar]
- 48.Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, Nirodi CS. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res 2007; 67:5267-74; PMID:17545606; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0242 [DOI] [PubMed] [Google Scholar]
- 49.Willers H, McCarthy EE, Alberti W, Dahm-Daphi J, Powell SN. Loss of wild-type p53 function is responsible for upregulated homologous recombination in immortal rodent fibroblasts. Int J Radiat Biol 2000; 76:1055-62; PMID:10947118; http://dx.doi.org/ 10.1080/09553000050111523 [DOI] [PubMed] [Google Scholar]
- 50.Bohnke A, Westphal F, Schmidt A, El-Awady RA, Dahm-Daphi J. Role of p53 mutations, protein function and DNA damage for the radiosensitivity of human tumour cells. Int J Radiat Biol 2004; 80:53-63; PMID:14761850; http://dx.doi.org/ 10.1080/09553000310001642902 [DOI] [PubMed] [Google Scholar]
- 51.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- 52.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al.. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012; 21:793-806; PMID:22698404; http://dx.doi.org/ 10.1016/j.ccr.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saretzki G. Cellular senescence in the development and treatment of cancer. Curr Pharm Des 2010; 16:79-100; PMID:20214620; http://dx.doi.org/ 10.2174/138161210789941874 [DOI] [PubMed] [Google Scholar]
- 54.Yu YC, Yang PM, Chuah QY, Huang YH, Peng CW, Lee YJ, Chiu SJ. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci Rep 2013; 3:1675; PMID:23591770; http://dx.doi.org/ 10.1038/srep01675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han NK, Kim BC, Lee HC, Lee YJ, Park MJ, Chi SG, Ko YG, Lee JS. Secretome analysis of ionizing radiation-induced senescent cancer cells reveals that secreted RKIP plays a critical role in neighboring cell migration. Proteomics 2012; 12:2822-32; PMID:22833545; http://dx.doi.org/ 10.1002/pmic.201100419 [DOI] [PubMed] [Google Scholar]
- 56.Weihua Z, Lin Q, Ramoth AJ, Fan D, Fidler IJ. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011; 117:4092-9; PMID:21365635; http://dx.doi.org/ 10.1002/cncr.26021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chou CH, Teng CM, Tzen KY, Chang YC, Chen JH, Cheng JC. MMP-9 from sublethally irradiated tumor promotes Lewis lung carcinoma cell invasiveness and pulmonary metastasis. Oncogene 2012; 31:458-68; PMID:21706046; http://dx.doi.org/ 10.1038/onc.2011.240 [DOI] [PubMed] [Google Scholar]
- 58.Johnson H, Lescarbeau RS, Gutierrez JA, White FM. Phosphotyrosine profiling of NSCLC cells in response to EGF and HGF reveals network specific mediators of invasion. J Proteome Res 2013; 12:1856-67; PMID:23438512; http://dx.doi.org/ 10.1021/pr301192t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Preusser M, Streubel B, Berghoff AS, Hainfellner JA, von Deimling A, Widhalm G, Dieckmann K, Wohrer A, Hackl M, Zielinski C, et al.. Amplification and overexpression of CMET is a common event in brain metastases of non-small cell lung cancer. Histopathology 2014; 65(5):684-92. [DOI] [PubMed] [Google Scholar]
- 60.Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res 2007; 67:2081-8; PMID:17332337; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3495 [DOI] [PubMed] [Google Scholar]
- 61.Bhardwaj V, Cascone T, Cortez MA, Amini A, Evans J, Komaki RU, Heymach JV, Welsh JW. Modulation of c-Met signaling and cellular sensitivity to radiation: potential implications for therapy. Cancer 2013; 119:1768-75; PMID:23423860; http://dx.doi.org/ 10.1002/cncr.27965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhardwaj V, Zhan Y, Cortez MA, Ang KK, Molkentine D, Munshi A, Raju U, Komaki R, Heymach JV, Welsh J. C-Met inhibitor MK-8003 radiosensitizes c-Met-expressing non-small-cell lung cancer cells with radiation-induced c-Met-expression. J Thorac Oncol 2012; 7:1211-7; PMID:22617250; http://dx.doi.org/ 10.1097/JTO.0b013e318257cc89 [DOI] [PMC free article] [PubMed] [Google Scholar]