

ABSTRACT
Hepatitis B virus (HBV) infection is a significant cause of liver disease pathogenesis, which results in the development of hepatic dysfunction, cirrhosis and hepatocellular carcinoma (HCC). Our previous studies showed that oncogene STAT3 might be an ideal target for HCC therapy. Here, we investigated whether targeting blockage of STAT3 signaling is efficient for HBV-related HCC. Based on the refractory of HCC and the persistence of HBV, in this study, we designed shRNAs targeting STAT3. The results showed that blocking STAT3 signaling by shRNAs could promote HBV positive HCC cell apoptosis and induce cell cycle arrest, resulting in HCC cell growth inhibition in vitro. Importantly, STAT3-shRNAs efficiently suppressed HBV replication, which would reduce HBV-derived stimulation to STAT3 signaling and augment STAT3-shRNAs-mediated anti-HCC effect. Finally, STAT3-shRNAs-mediated anti-HBV positive HCC effect was confirmed in xenograft nude mice. This study suggested that targeting STAT3 therapies such as STAT3-shRNAs may be an efficacious strategy for HBV-related HCC.
KEYWORDS: Gene targeted therapy, HBV, HCC, STAT3
Abbreviations
- HCC
Hepatocellular carcinoma
- HBV
hepatits B virus
- HBx
Hepatitis B virus X protein
- ODN
decoy oligodeoxynucleotide
- STAT3
Signal transducer and activator of transcription 3
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common solid tumor and one of the most important causes of cancer-related death in the world.1 According to epidemiological studies,2 hepatitis B virus (HBV) infection is the most common underlying cause of HCC worldwide. High HBV load and chronic hepatitis B (CHB) infection increase the risk for HCC, which is thought to be responsible for approximately 50% of all HCC cases and virtually all HCC in childhood.3
HBV is a DNA virus, and its DNA can be integrated into the host genome, causing transactivation of oncogenes of the host and making hepatocytes more susceptible.4,5 Hepatitis B virus X protein (HBx) is well-known to be essential for HCC progression and stimulation.6 Studies revealed that HBx operates as a multifunctional regulator via modulating the activity of host cellular signaling components such as p53,7 NFκB,8 and STAT3.9 Among these transcription factors, signal transducer and activator of transcription (STAT3) is commonly constitutively activated in liver cancers and plays a crucial role for tumor formation.10 Meanwhile, STAT3 is considered as an important factor involved in HBV gene expression,11 which can be promoted by STAT3 activators such as interleukin-6 (IL-6) and epidermal growth factor (EGF). So, a positive feed-back loop was formed between HBV and STAT3 signaling.
Gene therapy targeted STAT3 has been used extensively for the treatment of hematologic malignancies,12 but its application in HBV-related HCC has not been fully explored. In this study, shRNA-based gene therapy was used to silence STAT3 expression in HepG2.2.15 cells, and the efficacy of STAT3-shRNAs against HBV-related HCC was evaluated in vitro and in vivo. The results demonstrated that STAT3-shRNAs exhibited significant suppressive effect on HCC growth, which accompanied with efficient HBV clearance.
Results
HBV activated STAT3 signaling which in turn promoted HBV virus replication
HBx protein, which plays essential roles in viral replication and the occurrence of hepatocellular carcinoma, appears to be tied to tyrosine phosphorylation of STAT3 and STAT5.9 In turn, STAT3 interacts with HNF3 bound to HBV enhancer and stimulates HBV gene expression.11 Consistently, we observed that the tyrosine phosphorylation of STAT3 in HBV positive HCC cells-HepG2.2.15 was much higher than in HBV negative HCC cells-HepG2 (Fig. 1A), and the activation of STAT3 in HepG2 and HL7702 cells could be enhanced by transfection with LMP-HBV vector (Fig. 1B). Furthermore, the expression level of HBx in HepG2.2.15 cells was increased by stimulation of IL-6, an STAT3 activator (Fig. 1Cand1D). These results further confirmed the relationship between HBV and STAT3 in HCC, and suggested that HBV/STAT3 signaling loop could be an ideal target for HBV-related HCC therapy.
Figure 1.

HBV activates STAT3 signaling which backs to promote HBV virus replication. (A) Western blotting analysis of STAT3 and p-STAT3 levels in HepG2.2.15 and HepG2 cells. (B) Western blotting analysis of STAT3 and p-STAT3 levels in HBV-stably transfected HepG2 and HL7702 cells. (C) Western blotting analysis of STAT3 and p-STAT3 levels in HepG2.2.15 cells incubated with 100 ng/mL IL-6. (D) After 6 h or 24 h, mRNA levels of HBx was analyzed by qRT-PCR in HepG2.2.15 cells incubated with 100 ng/mL IL-6. Data are expressed as the mean ± SEM from at least 3 separate experiments. *p < 0.05, **p < 0.01.
HBV virus replication could be efficiently inhibited by oligonucleotide and shRNA targeting STAT3
In order to identify whether inhibition of STAT3 signaling could disturb HBV replication, firstly Decoy ODN strategy, which was previously used to treat human HCC cells and lung cancer cells in our studies,13,14 was performed in HepG2.2.15 cells. As shown in Fig 2A, HBx mRNA and HBV replication were declined about 47.5% and 50% by STAT3 Decoy-ODN, respectively. Meanwhile, the production of HBsAg and HBeAg were decreased from 80.3 IU/ml to 49.6 IU/ml and from 44.0 NCU/ml to 12.1 NCU/ml, respectively (Fig. 2B). These data indicated HBV replication in HCC could be suppressed by blocking STAT3 signaling. However, for persistent HCC, STAT3 Decoy-ODN remains some disadvantages in clinic application, especially its durability. To overcome this defect, we used shRNA technique to targeting silence STAT3 in HBV positive HCC. A retrovirus vector-LMP was selected as siRNA delivery vehicle, because of the property of that LMP vector can infect both dividing and nondividing cells efficiently as well as sustain long-term gene expression by integrating into the host genome. As shown in Fig. 3A, compared with LMP control group, the mRNA and protein levels of STAT3 were both significantly restrained by shRNA1 and shRNA2. Furthermore, STAT3-shRNAs inhibited HBx mRNA and HBV replication about 54% and 59%, respectively (Fig. 3B), as well as HBsAg and HBeAg production from 99.3 IU/ml to 55.6 IU/ml and from 52.0 NCU/ml to 18 NCU/ml, respectively (Fig. 3C). These results demonstrated STAT3 specific shRNAs designed was effective for silencing the expression of STAT3, and then inhibiting HBV replication.
Figure 2.
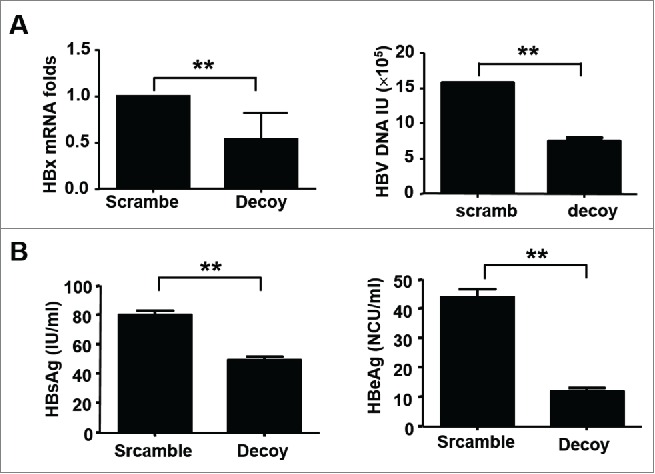
HBV virus replication was inhibited by STAT3-Decoy ODN. After transfected with STAT3-Decoy ODN for 48 h, HBx mRNA in HepG2.2.15 cells and supernatant HBV DNA were detected by qRT-PCR (A), and HBsAg and HBeAg protein levels were detected by ELISA (B). Data are expressed as the mean ± SEM from at least 3 separate experiments. *p < 0.05, **p < 0.01.
Figure 3.

HBV virus replication was inhibited by STAT3-shNRAs. After HepG2.2.15 cells transfected with STAT3-shRNA or LMP control plasmid for 48 h, the expression of STAT3 was detected by qRT-PCR (left) and western blotting (right) (A), HBx mRNA in HepG2.2.15 cells and supernatant HBV DNA were detected by qRT-PCR (B), and HBsAg and HBeAg protein expression were detected by ELISA (C). Data are expressed as the mean ± SEM from at least 3 separate experiments. *p < 0.05, **p < 0.01.
The growth of HBV positive HCC cells was inhibited by blocking STAT3 signaling
To confirm whether these STAT3-shRNAs could influence HBV positive HCC cell proliferation, HepG2.2.15 cells were transfected with STAT3-shRNAs followed by MTT assay at different time points. Compared to LMP control, the proliferation of HepG2.2.15 cells treated with STAT3-shRNAs was suppressed in a time-dependent manner (Fig. 4A). Meanwhile, STAT3-shRNAs promoted HepG2.2.15 cell apoptosis from 24.8% to 38.5% (Fig. 4B). In addition, HepG2.2.15 cell cycle was arrested at S phase by STAT3-shRNAs (Fig. 4C). These data indicated STAT3-shRNAs could effectively inhibit the growth of HBV-related HCC cells.
Figure 4.

The growth of HBV positive HCC cells was inhibited by blocking STAT3 signaling. HepG2.2.15 cells were transfected with STAT3-shRNA for 48 h, (A) the proliferation of HepG2.2.15 cells was measured by MTT assay, (B) the apoptosis of HepG2.2.15 cells were analyzed by Annexin V and PtdIns staining method, and (C) the cell cycle analysis was subjected to flow cytometry. Data are expressed as the mean ± SEM from at least 3 separate experiments. *p < 0.05, **p < 0.01.
HBV- induced angiogenesis was inhibited by STAT3-shRNAs
Angiogenesis is a key survival factor for HCC tissue, especially in HBV associated HCC patients.15 Reports indicate that HBx protein increases vascular endothelial growth factor (VEGF) production in hepatocellular carcinoma,16 which results in the sprouting of new blood vessels. As shown in Fig. 5A, the expression of molecules associated with sprouting new vasculature in HepG2.2.15 cells, including VEGF, MMP-9 and TGF-β, was higher than in HepG2 cells. And, the supernatant from HepG2.2.15 cells augmented HUVEC tube formation ((Fig. 5B). However, blocking STAT3 in HepG2.2.15 cells by shRNAs significantly inhibited the transcription of VEGF, MMP-9 and TGF-β (Fig. 5C), as well as tube formation (Fig. 5D). These data indicated that blocking STAT3 could suppress HBV-induced angiogenesis, which could enhance STAT3-shRNA-mediated anti-HCC effects.
Figure 5.

HBV- induced angiogenesis was inhibited by STAT3-shRNAs. (A) VEGF, MMP-9 and TGF-β mRNA in HepG2.2.15 and HepG2 cells were detected by qRT-PCR. (B) HUVECs seeded on matrigel were treated with medium, supernatant of HepG2 and HepG2 2.15 cells for 4 h, and then the network growth area was examined using an inverted fluorescence microscope (original magnification = 200×). Tube formation was digitally quantified by assessing the number of tubes (n). (C) After transfected with STAT3-shRNA for 48h, the relative expression of VEGF, MMP-9 and TGF-β was analyzed in HepG2.2.15 cells. (D) HUVECs seeded on matrigel were treated with supernatant of HepG2.2.15 cells transfected with STAT3-shRNAs or control LMP, and then tube formation was digitally quantified by assessing the number of tubes (n). Representative images and statistical data are expressed as the mean ± SEM from at least 3 separate experiments. *p < 0.05, **p < 0.01.
STAT3-SHRNAS exhibited anti-HBV positive HCC role in xenograft mice
To evaluate the anti-HBV associated HCC efficacy of STAT3-shRNAs in vivo, nude mice bearing xenografted human HepG2.2.15 cells were used. As the tumors were established, the mice were randomly assigned and treated with STAT3-shRNAs by intratumoral injection. At the same time, control plasmid (LMP)- and transfection reagent Lipofectamine 2000 (Lipo)- treated mice were used as control groups. As shown in Figs. 6A&B, after 34 days, the tumor volumes in Lipo and LMP control groups were 1180 ± 439 mm3 and 1107 ± 411 mm3,respectively, whereas the tumor volume in STAT3-shRNA1 and -shRNA2 treatment groups were 560± 75 mm3 and 574 ± 153 mm3 (Fig. 6C), respectively. Meanwhile, the tumor weight in Lipo and LMP control groups reached to 0.79 ± 0.24 g and 0.72 ± 0.22 g, respectively, whereas that in STAT3-shRNA1 and -shRNA2 treatment groups was 0.38± 0.10 g and 0.40 ± 0.23 g (Fig. 6D), respectively. These results showed that the tumor growth could be inhibited about 50% by STAT3-shRNA treatment, indicating STAT3-shRNAs could control HBV positive HCC efficiently.
Figure 6.

STAT3-shRNAs exhibited anti-HBV positive HCC role in xenograft mice. Following subcutaneously (s.c.) injection of HepG2.2.15 cells, these nude mice bearing palpable tumor were intratumorally treated with STAT3-shRNAs on day 10,2 and 15, and tumor tissue (A) and statistical analysis of tumor volume (B) were shown. (C-D) The volume of tumor (C) and the weight of tumor (D) were measured at the indicated time. Results were shown as means ± s.e.m., n = 4. *p < 0.05, **p < 0.01.
Discussion
HBV infection is a major risk for hepatocellular carcinoma, and the relationship between HBV and the development of hepatocellular carcinoma has been well established.17,18 Generally, HBV triggers tumorigenesis through 2 different aspects.18 The one is a direct mechanism, by which HBV DNA and its main product HBx induce host DNA instability; and another one is HBV infection-induced abnormalities of host immune system.
As an oncogene, STAT3 is a point of convergence for numerous oncogenic signaling pathways,19 promoting the generation of the pro-tumorigenic inflammatory microenvironment,20 inducing cell survival and proliferation as well as angiogenesis to promote tumor development. Studies suggest that HBx can activate JAK/STAT3 signaling pathway.9, 21 Especially, STAT3 signaling pathway was found involved in both aspects of HBV infection-induced tumorigenesis, and displayed important roles during HCC development.13,22,23 In this study, we also observed the relationship between HBV infection and STAT3 activation (Fig. 1). Therefore, STAT3 may be an ideal target for HBV-related HCC therapy.
To identify this hypothesis, firstly, STAT3 signaling in HepG2.2.15 cells was blocked by STAT3 Decoy-ODN, which was commonly used in our previous studies,13,14 and found HBV replication was inhibited by STAT3 blockage (Fig. 2). Considering the refractory of HCC and the persistence of HBV, shRNAs were designed and delivered by LMP vector. Subsequently, anti-HBV positive HCC effects of these STAT3-shRNAs were evaluated in vitro and in vivo. We found these STAT3-shRNAs could eliminate HBV significantly (Fig. 3), and suppress HBV positive HCC cell proliferation via cell cycle arresting as well as cell apoptosis (Fig. 4).
Angiogenesis inhibition is a natural therapeutic target for all solid tumors. Further investigates showed that STAT3-shRNAs inhibited the angiogenesis induced by HBV infection (Fig. 5). Finally, we used human HepG2.2.15 cell xenograft mice to assay the efficacy of STAT3-shRNAs therapy. Compared to control groups, STAT3-shRNAs significantly inhibited the tumor growth in vivo (Fig. 6).
Studies showed that effective antiviral therapies for the cure of HCC are important and necessary, which is conducive to restrain HBV reactivation, promote liver function recovery and reduce complication.24,25 At present, nucleoside analogs and IFN-α or its derivates are major drugs for HBV treatment. But the limits of these drugs are very apparent, including HBV resistance, poor tolerance and only a subgroup of patients response.26,27 Different from these traditional antiviral drugs, RNA interference (RNAi) technology provide a potential therapeutic tool with several advantages, including precision, high efficacy and no apparent toxicity as down-regulating gene expression.28 To ensure STAT3-shRNA efficiency, in our study, we selected microRNA-30-adapted shRNAmir retroviral vector (MSCV/LTRmiR30-PIGΔRI/LMP). This vector seems exhibits its role via natural mechanisms of RNA-dependent gene inhibition and produces potent, stable and regulatable gene knock-down in vitro and in vivo, even target gene presents at a single copy in the genome.29 In addition, improving liver tumor-targeting would enhance the efficiency and the safety of STAT3-shRNAs through liver specific promoters such as α-fetoprotein (AFP)30 and liver-targeting release gene delivery systems.31,32
With the potential of considerable crosstalk and redundancy in signaling pathways, HCC has a complex pathogenesis, and targeting single molecule or pathway may have a limited benefit in HCC treatment.33 So, based on the phase I study of the combination of the JAK1/2 inhibitor Ruxolitinib and EGFR inhibitor Afatinib for patients with treatment-refractory non-small cell lung cancer (NSCLC) (NCT02145637), the combination of Ruxolitinib and Afatinib was used to treat HBV-positive HCC in vitro. However, the results showed that the EGFR inhibitor Afatinib didn't aggravate Ruxolitinib-mediated the suppression of STAT3 phosphorylation (Fig. S1), suggesting the role of EGFR on HCC was complex and should be clarified by further studies.
In conclusion, HBV infection is one of the major causes of HCC development. Targeting the key elements involved in HBV infection-activated signaling pathways would be contribute to develop a feasible and effectual theraputics for HCC. STAT3-shRNA silenced STAT3 expression directly in HBV positive HCC, and suppressed HBV replication, which broke HBV-STAT3 signaling loop and decreased HBV-derived stimulation to STAT3 signaling, augmenting STAT3-shRNAs-mediated anti-HCC effect. Therefore, STAT3-shRNA might be a potential therapeutic approach for the treatment of HBV-related HCC. In addition, in order to improve the effectiveness and usefulness for the treatment of HCC, we will investigate the combination therapies of STAT3-shRNA with appropriate chemotherapy, radiotherapy as well as targeted therapy in the future.
Materials and methods
Cell lines and reagents
HepG2.2.15 cells, an HBV genomic DNA transfected cell line,34 were cultured in MEM medium (Gibco/BRL, Grand Island, NY, USA); HepG2 and HL7702 cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco); Human umbilical vein endothelial cells (HUVEC) were cultured in RPMI1640 (Gibco). All the cell lines were maintained in our lab and cultured in appropriate medium supplemented with10% fetal bovine serum (FBS, Gibco), 100 U/mL penicillin and 100 mg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
MSCV/LTRmiR30-PIGΔRI (LMP) plasmid was kindly provided by Yuekang Xu (Walter and Eliza Hall Institute of Medical Research, Australia). pAAV/HBV1.2 plasmid expressing 1.2 copies of full-length HBV genome35 was kindly provided by professor Pei-Jer Chen (National Taiwan University, College of Medicine and National Taiwan University Hospital, Taiwan).
Oligonucleotides and shRNA
As previous report,13 STAT3-decoy ODN was synthesized by using phosphorothioate chemistry, and purified by HPLC (Takara, Dalian, PR China). Human STAT3 shRNAs were designed using BLOCK-iTTM RNAi Designer (Invitrogen). The sequences of STAT3 (GenBank accession no. NM_003150) siRNAs correspond to the coding region 1663–1681 (5′-TGCTGACCAACAATCCCAA-3′) and 989–1007 (5′-CATCTGCCTAGATCGGCTA-3′) were cloned into LMP plasmid, respectively, and named as STAT3-shRNA1 and STAT3-shRNA2.
Transfection
According to the manufacturer's instructions, HepG2.2.15 cells were transfected with 100 nM ODN or 2 μg plasmids by Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA) for 6 h in 12-well plates. HepG2 and HL7702 cells were transferred with LMP-HBV, LMP containing HBV genome element from pAAV/HBV1.2 plasmid, by using Lipofectamine 2000, and then selected by puromycin.
Cell proliferation
Cell proliferation was determined using the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) as described previously.36 In brief, cells were seeded in a 96-well plate at a concentration of 2×104/well (200 µL/well). Then MTT was added at indicated time points, and cultured for another 4 h. The optical density (OD) readings were obtained at 490 nm using a microplate reader (Synergy 2; BioTek, Winooski, Vermont, USA).
Cell cycle analysis
For cell cycle analysis, cells were fixed in the presence of 70% ethanol at 4°C overnight. After washing with phosphate-buffered saline (PBS), these cells were incubated in PBS containing 20 μg/mL of propidium iodide (PI), 200 μg/mL of RNase A, and 0.1% Triton X-100 (BD Biosciences, Bedford, MA, USA) at 37°C for 30 minutes. The stained cells were then analyzed for cell cycle distribution using a FACSCalibur flow cytometer (BD Biosciences).
Apoptosis assay
Following the manufacturers instructions, the apoptosis was performed by apoptosis kit (BestBio, Shanghai, China) and analysised by FACSCalibur flow cytometer (BD Biosciences).
Western blot analysis
Cells were washed twice with ice-cold PBS and lysed in RIPA buffer (Beyotime, Beijing, China) containing protease inhibitors. Lysates (20 μg/lane) were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA). After that, the membranes were blocked with 7% nonfat dry milk, followed by hybridization with anti-p-STAT3 (tyr-705), anti-STAT3 and anti-β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Then appropriate antibody conjugated with horseradish peroxidase was used as the secondary antibody, and membranes were visualized by enhanced chemiluminescence (Millipore, Billerica, MA, USA) and analyzed by ImageLab software (Version 3.0, Bio-Rad).
RNA quantification
According to the manufacturers instructions, total RNA was isolated using TRIzol reagent (Invitrogen), and cDNA was synthesized using M-MLV reverse transcriptase (Invitrogen). The mRNA level of genes was determined by real-time RT-PCR (qPCR) using a SYBR green Mix (Roche, Indianapolis, IN, USA ) and a MyiQ thermocycler (Bio-Rad). The expression levels of the target genes were normalized to GAPDH. PCR primer sequences were provided in Table 1.
Table 1.
Primer sequences used in the real-time PCR assays.
| Target sequence | Sequence (5′-3′) | Size (bp) |
|---|---|---|
| GAPDH | Forward: GAA GGT GAA GGT CGG AGT Reverse: CAT GGG TGG AAT CT ATT GGA A | 155 |
| STAT3 | Forward: GAG GAC TGA GCA TCG AGC A Reverse: CAT GTG ATC TGA CAC CCT GAA | 85 |
| VEGF | Forward: GAG ACC CTG GTG GAC ATC TT Reverse: TTG ATC CGC ATA ATC TGC AT | 165 |
| TGF-β | Forward: GCC CTG GAC ACC AAC TAT TGC T Reverse: AGG CTC CAA ATG TAG GGG CAG G | 162 |
| HBx | Forward: CCG TCT GTG CCT TCT CT CTG C Reverse: ACC AAT TTA TGC CTA CAG CCT CC | 256 |
| MMP-9 | Forward: TTG GTC CAC CTG GTT CAA CT Reverse: ACG ACG TCT TCC AGT ACC GA | 95 |
ELISA assay
Following the manufacturers instructions, HBsAg and HBeAg levels in supernatants were detected by ELISA kits from Autobio (Zhengzhou, China).
HBV DNA analysis
According to the instructions, HBV viral particles in supernatants were quantified by qPCR by the Diagnostic Kit for Quantification of HBV DNA (DA AN GENE CO., Guangzhou, China). Primers detecting the HBV S region were 5′-ATCCTGCTGCTATGCCTCATCTT-3′ and 5′-ACAGGGGGAAAGCCCTACGAA-3′ as well as the 5′-FAM-TGGCTAGTTTACAGTGCCATTTG-TAMRA fluorescent probe.
Angiogenesis assay in vitro
The tube formation assay was performed using HUVEC as described in report.37 Briefly, 50μL matrigel basement membrane matrix growth factors reduced (GFR) (BD, Bioscienses) was added to each well of 96 well plate and incubated at 37 °C for 60 min to allow gel formation. HUVEC were cultured in RPMI1640 full medium or supernatants of hepatoma carcinoma cells. Tube formation was assessed at 4 h and photographs were taken using an inverted fluorescence microscope (Olympus, Japan).
Nude mouse xenograft assay
Five-week-old female nude BALB/c mice were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, PR China), and kept in an animal facility under pathogen-free conditions. The handing of mice and experimental procedures were conducted in accordance with guidelines for experimental animals approved by Ethical Committee of Shandong University. To evaluate tumor growth, 5×106 HepG2.2.15 cells were subcutaneously (s.c.) injected into the right flank of nude mice. When the tumor volumes reached about 100 mm3, the mice were divided randomly into 4 groups with 5 mice per group. The mice were injected intratumorally with Lipofectamine 2000 delivered shRNA every other day for 3 times. On day 34 post of inoculation, these mice were sacrificed and the tumors were taken and weighed.
Statistical analysis
The results were analyzed using Student's t-test, ANOVA and Pearson correlation. All data are expressed as a mean ± standard deviation. A P-value of 0.05 or less was considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Natural Science Foundation of China (81172789, 81373222), National Basic Research Program of China (No. 2013CB531503), and National Mega Project on Major Infectious Diseases Prevention and Treatment (2012ZX10002006).
Author's contribution
Y.-L.Y. designed, performed, and analyzed experiments and wrote the manuscript; B.-Q.Z. has performed RT-PCR analyses; Z.-G.T, C.Z., and J.Z. provided reagent and technique support; J.Z. designed and analyzed experiments and edited the manuscript.
References
- 1.Center MM, Jemal A. International trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers Prev 2011; 20:2362-8; PMID: 21921256; http://dx.doi.org/17440771 10.1158/1055-9965.EPI-11-0643 [DOI] [PubMed] [Google Scholar]
- 2.Barazani Y, Hiatt JR, Tong MJ, Busuttil RW. Chronic viral hepatitis and hepatocellular carcinoma. World J Surg 2007; 31:1243-8; PMID:17440771; http://dx.doi.org/ 10.1007/s00268-007-9041-3 [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012; 142:1264-73.e1; PMID:22537432; http://dx.doi.org/ 10.1053/j.gastro.2011.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tu H, Bonura C, Giannini C, Mouly H, Soussan P, Kew M, Paterlini-Brechot P, Brechot C, Kremsdorf D. Biological impact of natural COOH-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res 2001; 61:7803-10; PMID:11691796 [PubMed] [Google Scholar]
- 5.Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, et al.. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 2012; 22:593-601; PMID:22267523; http://dx.doi.org/ 10.1101/gr.133926.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benhenda S, Cougot D, Buendia MA, Neuveut C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv Cancer Res 2009; 103:75-109; PMID:19854353; http://dx.doi.org/ 10.1016/S0065-230X(09)03004-8 [DOI] [PubMed] [Google Scholar]
- 7.Dewantoro O, Gani RA, Akbar N. Hepatocarcinogenesis in viral Hepatitis B infection: the role of HBx and p53. Acta Med Indones 2006; 38:154-9; PMID:16953033 [PubMed] [Google Scholar]
- 8.Lucito R, Schneider RJ. Hepatitis B virus X protein activates transcription factor NF-kappa B without a requirement for protein kinase C. J Virol 1992; 66:983-91; PMID:1309924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee YH, Yun Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J Biol Chem 1998; 273:25510-5; PMID:9738022; http://dx.doi.org/ 10.1074/jbc.273.39.25510 [DOI] [PubMed] [Google Scholar]
- 10.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 2005; 41:2502-12; PMID:16199153; http://dx.doi.org/ 10.1016/j.ejca.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 11.Waris G, Siddiqui A. Interaction between STAT-3 and HNF-3 leads to the activation of liver-specific hepatitis B virus enhancer 1 function. J Virol 2002; 76:2721-9; PMID:11861839; http://dx.doi.org/ 10.1128/JVI.76.6.2721-2729.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz J, Dhillon N, Janku F, Watowich SS, Hong DS. STAT3 inhibitors: finding a home in lymphoma and leukemia. Oncologist 2014; 19:536-44; PMID:24705981; http://dx.doi.org/ 10.1634/theoncologist.2013-0407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun X, Zhang J, Wang L, Tian Z. Growth inhibition of human hepatocellular carcinoma cells by blocking STAT3 activation with decoy-ODN. Cancer Lett 2008; 262:201-13; PMID:18248786; http://dx.doi.org/ 10.1016/j.canlet.2007.12.009 [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Zhang J, Wei H, Tian Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol Rep 2007; 17:1377-82; PMID:17487394; http://dx.doi.org/ 10.3892/or.17.6.1377 [DOI] [PubMed] [Google Scholar]
- 15.Yang P, Markowitz GJ, Wang XF. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl Sci Rev 2014; 1:396-412; PMID:25741453; http://dx.doi.org/ 10.1093/nsr/nwu038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yen CJ, Lin YJ, Yen CS, Tsai HW, Tsai TF, Chang KY, Huang WC, Lin PW, Chiang CW, Chang TT. Hepatitis B virus X protein upregulates mTOR signaling through IKKbeta to increase cell proliferation and VEGF production in hepatocellular carcinoma. PloS One 2012; 7:e41931; PMID:22848663; http://dx.doi.org/ 10.1371/journal.pone.0041931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. BioMed Res Int 2013; 2013:810461; PMID:23991421; http://dx.doi.org/ 10.1155/2013/810461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan ZM, Sun BC. Effects of antiviral therapy on preventing liver tumorigenesis and hepatocellular carcinoma recurrence. World J Gastroenterol 2013; 19:8895-901; PMID:24379613; http://dx.doi.org/17610223 10.3748/wjg.v19.i47.8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DJ, Chan KS, Sano S, Digiovanni J. Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol Carcinogen 2007; 46:725-31; PMID:17610223; http://dx.doi.org/ 10.1002/mc.20342 [DOI] [PubMed] [Google Scholar]
- 20.Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011; 19:429-31; PMID:21481782; http://dx.doi.org/ 10.1016/j.ccr.2011.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quetier I, Brezillon N, Duriez M, Massinet H, Giang E, Ahodantin J, Lamant C, Brunelle MN, Soussan P, Kremsdorf D. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol 2013; 59:285-91; PMID:23542345; http://dx.doi.org/ 10.1016/j.jhep.2013.03.021 [DOI] [PubMed] [Google Scholar]
- 22.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7:41-51; PMID:17186030; http://dx.doi.org/ 10.1038/nri1995 [DOI] [PubMed] [Google Scholar]
- 23.Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther 2013; 12:2885-96; PMID:24107450; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-1087 [DOI] [PubMed] [Google Scholar]
- 24.Koda M, Nagahara T, Matono T, Sugihara T, Mandai M, Ueki M, Ohyama K, Hosho K, Okano J, Kishimoto Y, et al.. Nucleotide analogs for patients with HBV-related hepatocellular carcinoma increase the survival rate through improved liver function. Inter Med 2009; 48:11-7; PMID:19122351; http://dx.doi.org/ 10.2169/internalmedicine.48.1534 [DOI] [PubMed] [Google Scholar]
- 25.Ke Y, Ma L, You XM, Huang SX, Liang YR, Xiang BD, Li LQ, Zhong JH. Antiviral therapy for hepatitis B virus-related hepatocellular carcinoma after radical hepatectomy. Cancer Biol Med 2013; 10:158-64; PMID:24379991; http://dx,doi.org/ 10.7497/j.issn.2095-3941.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dienstag JL. Hepatitis B virus infection. N Engl J Med 2008; 359:1486-500; PMID:18832247; http://dx.doi.org/ 10.1056/NEJMra0801644 [DOI] [PubMed] [Google Scholar]
- 27.Dusheiko G. Treatment of HBeAg positive chronic hepatitis B: interferon or nucleoside analogues. Liver Int 2013; 33(Suppl 1):137-50; PMID:23286858; http://dx.doi.org/ 10.1111/liv.12078 [DOI] [PubMed] [Google Scholar]
- 28.Uprichard SL. The therapeutic potential of RNA interference. FEBS Lett 2005; 579:5996-6007; PMID:16115631; http://dx.doi.org/ 10.1016/j.febslet.2005.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 2005; 37:1289-95; PMID:16200064; http://dx,doi.org/ 10.1038/ng1651 [DOI] [PubMed] [Google Scholar]
- 30.Ohguchi S, Nakatsukasa H, Higashi T, Ashida K, Nouso K, Ishizaki M, Hino N, Kobayashi Y, Uematsu S, Tsuji T. Expression of alpha-fetoprotein and albumin genes in human hepatocellular carcinomas: limitations in the application of the genes for targeting human hepatocellular carcinoma in gene therapy. Hepatology 1998; 27:599-607; PMID:9462663; http://dx.doi.org/ 10.1002/hep.510270239 [DOI] [PubMed] [Google Scholar]
- 31.Nguyen TH, Pages JC, Farge D, Briand P, Weber A. Amphotropic retroviral vectors displaying hepatocyte growth factor-envelope fusion proteins improve transduction efficiency of primary hepatocytes. Hum Gene Ther 1998; 9:2469-79; PMID:9853514; http://dx.doi.org/ 10.1089/hum.1998.9.17-2469 [DOI] [PubMed] [Google Scholar]
- 32.Nguyen TH, Loux N, Dagher I, Vons C, Carey K, Briand P, Hadchouel M, Franco D, Jouanneau J, Schwall R, et al.. Improved gene transfer selectivity to hepatocarcinoma cells by retrovirus vector displaying single-chain variable fragment antibody against c-Met. Cancer Gene Ther 2003; 10:840-9; PMID:14605670; http://dx.doi.org/ 10.1038/sj.cgt.7700640 [DOI] [PubMed] [Google Scholar]
- 33.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010; 29:4989-5005; PMID:20639898; http://dx.doi.org/ 10.1038/onc.2010.236 [DOI] [PubMed] [Google Scholar]
- 34.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 1987; 84:1005-9; PMID:3029758; http://dx.doi.org/ 10.1073/pnas.84.4.1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang LR, Wu HL, Chen PJ, Chen DS. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc Natl Acad Sci U S A 2006; 103:17862-7; PMID:17095599; http://dx.doi.org/ 10.1073/pnas.0608578103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang C, Yang G, Jiang T, Cao J, Huang KJ, Qiu ZJ. Down-regulation of STAT3 expression by vector-based small interfering RNA inhibits pancreatic cancer growth. World J Gastroenterol 2011; 17:2992-3001; PMID: 21799645; http://dx.doi.org/11073829 10.3748/wjg.v17.i25.2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salani D, Taraboletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 2000; 157:1703-11; PMID:11073829; http://dx.doi.org/ 10.1016/S0002-9440(10)64807-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.