

Abstract
More and more miRNAs have been shown to regulate gene expression in the heart and dysregulation of their expression has been linked to cardiovascular diseases including the miR‐199a/214 cluster. However, the signature of circulating miR‐214 expression and its possible roles during the development of heart failure has been less well studied. In this study, we elucidated the biological and clinical significance of miR‐214 dysregulation in heart failure. Firstly, circulating miR‐214 was measured by quantitative PCR, and we found that miR‐214 was upregulated in the serum of chronic heart failure patients, as well as in hypertrophic and failing hearts of humans and mice. Adeno‐associated virus serotype 9 (AAV9)‐mediated miR‐214 silencing attenuates isoproterenol (ISO) infusion‐induced cardiac dysfunction and impairment of cardiac angiogenesis in mice. Mechanistically, miR‐214 overexpression reduces angiogenesis of HUVECs by targeting XBP1, an important transcription factor of unfolded protein response, and XBP1 silencing decreases HUVECs proliferation and angiogenesis similar to miR‐214 overexpression. Furthermore, ectopic expression of XBP1 enhances endothelial cells proliferation and tube formation, and reverses anti‐angiogenic effect of miR‐214 over expression. All these findings suggest that miR‐214 is an important regulator of angiogenesis in heart in vitro and in vivo, likely via regulating the expression of XBP1, and demonstrate that miR‐214 plays an essential role in the control/inhibition of cardiac angiogenesis. J. Cell. Physiol. 230: 1964–1973, 2015. © 2015 The Authors. Journal of Cellular Physiology published by Wiley Periodicals, Inc.
Abbreviations
- AAV
adeno‐associated virus
- ATF
activating transcription factor
- ER
endoplasmic reticulum
- Grp
glucose‐regulated protein
- IRE
inositol‐requiring kinase
- ISO
isoproterenol
- TAC
thoracic aorta constriction
- UPR
unfolded protein response
- VEGF‐A
vascular endothelial growth factor‐A
- XBP
X‐box binding protein
- CAD
coronary artery disease
- HCC
hepatocellular carcinoma
MicroRNAs (miRNAs) are a class of conserved, short, single‐stranded, noncoding RNAs after maturation into approximately 22 base sequences enter into the RNA interference pathway, bind to identical or similar sequences in the 3′ untranslated region (3′UTR) of genes, resulting in inhibition of translation or cleavage of the target mRNA (Winter et al., 2009). miRNAs are increasingly recognized as master regulators of many processes, including angiogenesis and vascular development, because of their ability to target numerous mRNAs, in particular those with similar functions or within related pathways (van Mil et al., 2012).
miR‐214 was first identified for its role in tumor cell apoptosis(Cheng et al., 2005). Many subsequent reports about miR‐214 and its targets have described its roles in tumor cell survival, muscle cell differentiation, tumor resistance, and T‐cell proliferation, bone formation, and others (Yang et al., 2008; Juan et al., 2009; Wang et al., 2012). Indeed, the biological and clinical significance of miR‐214 was multifunctional and controversial. In contrast to the downregulation of miR‐214 in cervical, breast and hepatocellular cancer (Qiang et al., 2011; Duan et al., 2012; Shih et al., 2012), miR‐214 was usually upregulated in other human cancers including ovarian, stomach, pancreatic, lung, and oral mucosal cancers and malignant melanomas (Yang et al., 2008; Penna et al., 2011; Shih et al., 2012). Namely, miR‐214 suppresses hepatocellular carcinoma (HCC) cell proliferation and viability, but increases human ovarian cancer cell growth and invasion. However, the roles of miR‐214 in different cardiovascular disease remain largely unexplored.
Recently, some studies focused on the effect of miR‐214 in ischemic diseases. For example, miR‐214 was upregulated following renal ischemia/reperfusion (IR) injury (Godwin et al., 2010), showed a striking increase in expression in the border zone of the infarct, both in the murine and human myocardial infarction hearts (van Rooij et al., 2008). miR‐214 also suppressed sodium/calcium exchanger 1 (NCX1) and downstream effectors of Ca2+ signaling and cell death, attenuate Ca2+ overload‐induced cardiomyocyte death, exhibited a protective role against myocardial ischemia/reperfusion (IR) injury (Aurora et al., 2012). Furthermore, miR‐214 level was significantly deceased in the serum of patients with coronary artery disease (CAD) compared with healthy controls (Lu et al., 2013). These findings suggest that miR‐214 may be a protective microRNA for myocardial IR injury. However, these observations raise another important question: what is the role of miR‐214 in the regulation of chronic nonischemic myocardial diseases, particularly for chronic nonischemic heart failure. miRNA microarray analysis showed that expression of miR‐214 is significantly upregulated in thoracic aorta constriction (TAC)‐ and calcineurin A‐induced mouse heart hypertrophy models, as well as in idiopathic end‐stage failing human hearts (van Rooij et al., 2006; Sayed et al., 2007). Surprisingly, miR‐214 appeared to be capable of inducing hypertrophic growth in cardiomyocytes, but cardiac overexpression of miR‐214 had no morphological effect on the heart in miR‐214 transgenic (Tg) mice (van Rooij et al., 2006). These reports indicate that miR‐214 is regulated differentially during cardiac hypertrophy and failure, suggesting the possibility that it might function as a modulator of this process. Nonetheless, its function and molecular mechanisms in heart failure remain largely unknown.
In this study, we found that upregulation of miR‐214 in the transition of hypertrophic heart to heart failure reduces endothelial cell proliferation and angiogenesis by targeting XBP‐1, a key transcription regulator of endothelial cell proliferation, and blockade of miR‐214 attenuates impairment of cardiac angiogenesis induced by prolonged ISO infusion in mice. These findings further demonstrate that miR‐214 plays an essential role in the control/inhibition of cardiac angiogenesis.
Methods
Materials and reagents
Materials were obtained from the following suppliers: antibodies against XBP‐1, GAPDH, and CD31 were obtained from Santa Cruz biotechnology Inc. (Santa Cruz, CA); BrdU cell proliferation ELISA kit was purchased from Roche (Penzberg, Germany); CCK‐8 kit was obtained from Beyotime Institute of Biotechnology (Nantong, China); pCMV6‐XL5‐XBP‐1s plasmid was purchased from Origene (Rockville, MD); Inhibitor, mimic, and non‐specific negative control oligonucleotides for hsa‐miR‐214, specific siRNA against human XBP‐1 were obtained from RiboBio (Guangzhou, China). All other chemicals and reagents were purchased from Sigma–Aldrich (Shanghai, China) unless otherwise specified.
Animal models
C57BL/6 mice were subjected to thoracic aorta constriction (TAC) or sham operation as described previously (Okada et al., 2004). Pressure overload‐induced cardiac hypertrophy and heart failure was created by TAC for 2 weeks. C57BL/6 mice were continuously infused with isoproterenol hydrochloride (30 mg/kg/d, Sigma–Aldrich China Inc., Shanghai, China) over a period of 14 or 28 days with implanted Mini‐osmotic pumps (Alzet model 1002 and 1004; DURECT Corp., Cupertino, CA) as described previously (Nienaber et al., 2003). Mice were anesthetized with pentobarbital sodium at a dose of 80 mg/kg body weight intraperitoneally. Once anesthesia was induced, it was maintained using a nose cone of halothane. In C57BL/6 mice study, recombinant adeno‐associated virus vector (rAAV9) was used as carrier (Pacak et al., 2006; Suckau et al., 2009; Yang et al., 2009). C57BL/6 mice were injected with rAAV‐GFP and AAV9‐anti‐miR‐214 (n = 5 or 6 per group) through a sublingual vein injection 2 weeks before ISO infusion as described previously (Xie et al., 2012). Two or 4 weeks after ISO infusion, mice were subjected to echocardiography examination followed by morphological and immunohistological tests. Animals were euthanized via an anesthetic overdose (200 mg/kg of pentobarbital sodium delivered by intraperitoneal injection). All animal studies were approved by the Animal Research Committee of Tongji College and were done according to the guidelines of the National Institute of Health (NIH).
Human heart tissue and blood samples
Human heart tissue and blood samples were studied according to the protocol approved by the Clinical Research Committees of Tongji Medical College. The investigation also conforms to the principles outlined in the Declaration of Helsinki. The subjects recruited in the study provided informed consent. We collected heart samples from eight recipients of heart transplantation who suffered end‐stage heart failure, as well as three normal hearts of victims of traffic accidents. Tissue samples were frozen at −80 °C until used for extraction of protein. Plasma was isolated by centrifugation and was maintained at −80 °C until RNA extraction.
Echocardiography
Echocardiography was performed on mice with a Visualsonic Vero 2100 System echocardiograph (Visualsonics, Toronto, CA) using a 35‐MHz probe as described previously (Lin et al., 2009; Wang et al., 2011).
Cell lines
The embryonic rat heart‐derived myogenic cell line H9c2 (2‐1), human umbilical vein endothelial cells (HUVECs) and human kidney 293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). 293T and H9c2 (2‐1) cells were grown in DMEM and HUVECs were grown in DMEM‐F12 supplemented with 10% fetal bovine serum (FBS) (Gibco, Invitrogen, Carlsbad, CA), streptomycin 100 µg/ml, and penicillin 100 U/ml (all obtained from Sigma, St. Louis, MO). The cells were maintained at 37 °C in a humidified incubator containing 5% CO2.
Immunohistochemical analysis
For immunohistochemical examination of CD31, paraffin‐embedded mice heart tissues were cut into 5‐µm sections, and assays were performed as previously described (Jiang et al., 2005). Antibodies against CD31 (Santa Cruz Biotechnology) were used as the primary antibody.
HUVEC transfection
1.5 × 105 HUVEC per 30 mm dish were transfected 24 h after seeding using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. 100 nM miR‐214 mimics or inhibitors or 100 nM miRNA negative control oligonucleotides, 50 nM small interfering RNAs (siRNAs) against XBP‐1 and siRNA control (RiboBio, Guangzhou, China) in a final volume of 2 ml were used. The siRNA sequences for human XBP‐1 were sense 5′‐ACA GCA AGU GGU AGA UUU ATT‐3′ and anti‐sense 5′‐UAA AUC UAC CAC UUG CUG UTT‐3′. Cells were collected at 48 h after transfection for protein and RNA extraction, at 24 h after transfection for wound healing/cell migration assay and tube formation assay, and at 24 or 48 h after transfection for cell counting kit‐8 detection.
Tube formation assay
HUVEC (4 × 104) were seed in a 24‐well plate coated with 100 µl of Matrigel (BD Biosciences, San Jose, CA) 24 h after transfection. Quantification of the average number of tubes per field of view was done after seeding for 6 h. Images of each well were captured using an inverted microscope (Nikon TE 2000; Nikon, Tokyo, Japan).
Migration/Scratch assay
Migration of HUVEC cells was monitored by an artificial scratch in a monolayer of confluent endothelial cells with a 1000 µl tip 24 h after tranfection and observation of the extent of wound closure after 24 or 48 h. Then the sharp wound in each well was captured using an inverted microscope (Nikon TE 2000; Nikon).
In vivo Matrigel plug assay with transfected HUVECs
HUVECs were transfected with miR‐214 mimics or miRNA negative control or siRNA against XBP‐1 or scrambled oligonucleotides, as described above. In vivo Matrigel plug assays were performed, as described previously (Kuehbacher et al., 2007).
Cell proliferation assay
Twenty‐four hours after transfection, HUVECs were dispensed with trypsin and replated in 96‐well plates. Six hours later, cells in 96‐well plate was labeled with BrdU for 18 h. Then the levels of BrdU incorporation were determined using a BrdU cell proliferation ELISA kit (Roche). At 24 and 48 h after transfection, HUVEC were incubated in 10% CCK‐8 (Beyotime Institute of Biotechnology) diluted in normal culture medium at 37 °C until visual color conversion occurred in 96‐well culture plates. The number of viable cells was assessed by measurement of absorbance at 450 nm using a microplate reader.
Western blot analysis
Proteins from cell lysates (20 µg) were separated by 10% SDS‐polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After blocking in 5% nonfat milk, protein blots were incubated with specific antibodies followed by incubation with a peroxidase‐conjugated secondary antibody in blocking buffer. The bands were visualized with the enhanced chemiluminescence method according to manufacturer's instructions (Beyotime Institute of Biotechnology).
RNA extraction and real‐time quantitative PCR
Total RNAs were extracted using TRIzol according to the manufacture's protocol. Reverse transcription reaction was performed starting from equal amounts of total RNA/sample (1 µg) using EasyScript First‐Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). Human XBP‐1 mRNA levels were determined using the forward primer: for human total XBP‐1, CCC ATG GAT TCT GGC GGT ATT GAC and TCC TTC TGG GTA GAC CTC TGG GAG; for human spliced XBP‐1, GAG TCC GCA GCA GGT G and TCC TTC TGG GTA GAC CTC TGG GAG; for mouse total XBP‐1, AAG AAC ACG CTT GGG AAT GG and ACT CCC CTT GGC CTC CAC; for mouse spliced XBP‐1, GAG TCC GCA GCA GGT G and GTG TCA GAG TCC ATG GGA (Petremand et al., 2012); β‐actin or GAPDH was used as an internal reference. The real‐time quantitative‐PCR results were analyzed and expressed as relative mRNA levels of the CT (cycle threshold) value, which was then converted to fold change.
Vector construction and luciferase reporter assay
The 3′UTR of the human XBP‐1 gene was PCR amplified using the XBP‐1 primers: 5′‐GCG CGA GCT CTT CTC TGT CAG TGG GGA CGT CAT‐3′ and 3′‐GCG CAA GCT TAG AAG AAA TCA AAC AAG GAT GCT GC‐5′. Vector construction and luciferase reporter assay were performed as described previously. The rAAV viral particles were prepared and purified according to a published protocol (Xie et al., 2012). The plasmid constructs were verified by sequencing.
Statistical analysis
The data are expressed as mean values ± SD. Difference between groups were evaluated for significance using Student t‐test of unpaired data or Mann–Whitney Rank Sum Test or one‐way analysis of variance (ANOVA) and Bonferroni post‐test. P < 0.05 was considered significant.
Results
miR‐214 is upregulated in the serum of chronic heart failure patients
To identify whether miR‐214 is deregulated in the serum of patients with chronic heart failure, we measured the expression of miR‐214 in two groups of subjects. One group consisted of subjects who were diagnosed with HF (HF cases, n = 104), and another group consisted of healthy controls (n = 72). Baseline characteristics of these groups are displayed in the Table 1. We measured miR‐214 levels in serum by real‐time PCR, and found that miR‐214 was specifically enriched in the sera of HF patients (Figs. 1 and S1).
Table 1.
Characteristics for the study population
| Characteristics | HF cases (n = 104) | Normal (n = 72) | P‐value |
|---|---|---|---|
| Age (years) | 62.2 ± 15 | 60 ± 10 | 0.86 |
| Male/female (n/n) | 72/32 | 40/32 | >0.05 |
| Current smoking, n (%) | 32 (30.7%) | 18 (25%) | >0.05 |
| Diabetes mellitus, n (%) | 0 (0%) | 0 (0%) | – |
| Hypertension, n (%) | 59 (56.7%) | 15 (20.8%) | >0.05 |
| Fasting glucose (mmol/L) | 6.608 ± 1.35 | 5.2 ± 0.68 | 0.18 |
| SBP (mmHg) | 125.8 ± 24 | 115.2 ± 18 | 0.15 |
| DBP (mmHg) | 74.9 ± 12 | 65.5 ± 7 | 0.24 |
| Ejection Fraction (%) | 45.8 ± 9.2 | 62 ± 7.8 | 0.10 |
Mean ± standard deviation. SBP, systolic blood pressure; DBP, diastolic blood pressure. P, comparison between patients with healthy adults.
Figure 1.

miR‐214 is upregulated in hypertrophic and failing heart and the circulating level. (A) Real‐time PCR of miR‐214 expression in plasma of 72 healthy controls and 104 heart failure patients. (B–D) dynamic levels of miR‐214 in failing human heart (n = 5 or 8), in mouse heart model after ISO infusion at 2 weeks and 4 weeks (n = 6), and in TAC‐treated mouse heart (n = 6), respectively. n = 5 or 8 in B; n = 6 for C and D. Three independent experiments were carried out, and each experiment was done in triplicate. *P < 0.05 compared with control (Mann–Whitney rank sum test).
miR‐214 is upregulated in hypertrophic and failing heart tissue
Importantly, to determine whether miR‐214 expression is also modulated in human failing heart tissue, we performed real time PCR and found that the expression level of miR‐214 was increased by ∼2 fold in all failing human hearts when compared to those from control hearts (Fig. 1B). Next, we established two mouse models of cardiac hypertrophy and heart failure by using isoproterenol (ISO) infusion and transverse aortic constriction (TAC). Real‐time PCR showed that miR‐214 was consistently and significantly increased in both ISO‐ and TAC‐induced cardiac hypertrophy (Fig. 1C and D). These data support the notion that miR‐214 may act as a novel regulator of cardiac hypertrophy and development. Thus, we decided to further investigate its roles in heart failure.
Blockade of miR‐214 by AAV9‐anti‐miR‐214 reversed cardiac dysfunction induced by prolonged ISO infusion in vivo
To investigate the pathophysiological significance of miR‐214 in maladaptive hypertrophy, we tested whether silencing miR‐214 can influence cardiac remodeling. rAAV vectors with capsids of rAAV serotype 9 enable an efficient transduction of the heart after systemic delivery, and therefore, we constructed plasmids and rAAV9 vectors harboring anti‐miR‐214 (rAAV‐anti‐miR‐214). Mice were first treated with rAAV‐anti‐miR‐214 for 2 weeks, and then subjected to ISO infusion for additional 4 weeks. As expected, rAAV‐anti‐miR‐214 efficiently repressed cardiac miR‐214 expression (Fig. 2A). Saline‐pumped mice treated with GFP or anti‐miR‐214 showed no signs of histopathological abnormalities. (Fig. S2). In contrast, rAAV‐anti‐miR‐214 treatment inhibited the increase in myofiber size, interstitial fibrosis, and the activation of fetal cardiac genes, ANP and β‐MHC, induced by ISO infusion (Figs. 2 and S3–S6). Furthermore, echocardiographic measurements revealed that LV dimension was significantly increased, and percentage of fractional shortening (%FS) and ejection fraction (%EF) were significantly decreased in the mice treated by ISO infusion compared with saline treated mice. In contrast, silencing miR‐214 treatment reduced the cardiac remodeling and improved cardiac function in ISO‐treated mice (Fig. 2E and F). All these results further suggest the essential roles of miR‐214 in the progression of heart failure.
Figure 2.

Inhibition of miR‐214 attenuates impairment of cardiac function induced by prolonged ISO infusion in vivo. (A) Real‐time PCR of miR‐214 expression in ISO treated mice after anti‐miR‐214 treatment. (B) Representative pictures of myocardium with HE (hematoxylin and eosin) stain and quantitative analysis of cardiac myocyte cross‐sectional area. (C) Representative pictures of myocardium with Masson trichrome stain in ISO treated mice after AAV‐anti‐214 injection. (D–F), Echocardiographic analysis of ISO treated mice after AAV‐ anti‐miR‐214 injection. Error bars indicate s.e.m. n = 6 for A, E, and F; n = 3 for B and C. *P < 0.05 compared with GFP or #P < 0.05 compared with ISO + GFP.
Silencing of miR‐214 attenuates impairment of cardiac angiogenesis in vivo and in vitro
Angiogenesis is a critical component of many pathological conditions such as heart failure and myocardial infarction, and is impaired in the maladaptive phase of cardiac hypertrophy (Shiojima et al., 2005; Sano et al., 2007). So, we measured cardiac angiogenesis in mice with AAV9‐anti‐miR‐214 after ISO treatment. Immunohistochemical studies showed that anti‐miR‐214 treatment significantly upregulated the number of microvessels per cardiomyocyte in prolonged ISO‐treated mice hearts, and attenuated the impairment of cardiac angiogenesis (Figs. 3 and S7).
Figure 3.

miR‐214 reduces angiogenesis of HUVECs. (A) Inhibitory effect of miR‐214 on tube formation in HUVECs. HUVECs, 24 h after transfection, were seeded on BD Matrigel and the average number of tubes per field of view were counted after 6 h. (B) Hemoglobin concentration of HUVEC transfected with miR‐214 mimics or miRNA negative control oligonucleotides (100 nM) confirmed by in vivo Matrigel plug assays. n = 5 per group. (C) miR‐214 inhibitor enhances HUVECs tube formation. (D) Immunohistochemical analysis revealed that the number of vessels per cardiomyocyte was increased in ISO treated mice after anti‐miR‐214 treatment. Three independent experiments were carried out, and each experiment was done in triplicate. *P < 0.05 compared with control.
On the other hand, recent studies have reported high expression of miR‐214 in the main vascular cell types, especially in HUVECs (van Mil et al., 2012). We further examined effect of miR‐214 on angiogenesis in cultured endothelial cells. Overexpression of miR‐214 in endothelial cells by transfection reduced tube formation, cell migration (Figs. 3 and S8), and reduced Matrigel plug hemoglobin concentrations (Fig. 3B). Conversely, miR‐214 inhibitor increased endothelial cell tube formation and angiogenesis (Fig. 3C), indicating the anti‐angiogenic effects of miR‐214. These results suggest the potential roles of miR‐214 in the transition of hypertrophic hearts and heart failure by inhibiting cardiac angiogenesis. Since cardiac angiogenesis was negatively regulated by miR‐214 in vivo, we focused our subsequent studies on the molecular mechanisms by which miR‐214 modulates cardiac angiogenesis.
miR‐214 targets XBP1 in HUVECs
In previous study, we found that miR‐214 suppresses XBP1 expression in human hepatoma cells (Duan et al., 2012). Here, we further examined the effect of miR‐214 on the endogenous expression of XBP1 in HUVECs. As expected, overexpression of miR‐214 caused a significant decrease in protein level of XBP1 in HUVECs (Fig. 4B). Similar results were obtained in rat heart‐derived myogenic cell line H9c2 (2‐1) cells (Fig. 4C). XBP1 3′UTR luciferase reporter experiments showed that miR‐214 transfection significantly enhanced luciferase activity in XBP1 3′UTR but not in mutated XBP1 3′UTR (Fig. S9). All these data suggest that miR‐214 can inhibit the expression of XBP1 at transcriptional level by directly targeting the 3′UTR of XBP1 mRNA in HUVECs. Finally, to investigate the role of miR‐214 on XBP1 in AAV9‐anti‐miR‐214 treated mice, real‐time PCR was performed which showed that anti‐miR‐214 efficiently prevented repression of XBP1 expression (Fig. 4D). Taken together, these findings showed a direct interaction between miR‐214 and XBP1 mRNA in HUVEC cell line.
Figure 4.

XBP‐1 is experimentally validated as a direct target of miR‐214 in HUVECs. (A) The sequence of the miR‐214 binding sites within the human XBP‐1 3′UTR and a schematic diagram of the reporter constructs showing the entire XBP‐1 3′UTR sequence. (B) XBP‐1 protein expression in the presence of candidate miRNAs. 100 nM of individual miRNA was introduced into cells. Western blot was performed 48 h after transfection. Blank control culture medium (mock); non‐specific negative control oligonucleotides (negative control); miR‐34c, miR‐214mimics; miR‐34c, miR‐214 inhibitors. (C) XBP‐1 expression in the presence of 100 nM of miR‐214 mimics or miRNA negative control oligonucleotides in H9C2 (2‐1) cells. (D) Real‐time PCR of miR‐214 expression in ISO treated mice after anti‐miR‐214 treatment. Blank control culture medium (mock); non‐specific negative control oligonucleotides (negative control); hsa‐miR‐214 mimics (miR‐214); hsa‐miR‐214 inhibitors (miR‐214 inhibitor).
XBP1 is essential for HUVECs tube formation and angiogenesis
We also examined the effect of XBP1 on angiogenesis of HUVECs, and results showed that XBP1 knockdown by siRNA (Fig. S10) significantly reduced the tube‐forming ability of HUVECs (Fig. 5A) and cell migration as well as the percentage of wound recovery and Matrigel plug hemoglobin concentrations compared to the control group (Fig. 5B and C). We next determined if ectopic expression of XBP1 by transient transfection of pCMV6‐XL5‐XBP1s plasmid altered the tube formation in HUVECs. Compared to those transfected with control vectors, overexpression of XBP1s markedly enhanced tube formation in endothelial cells compared with vector control (Fig. 5D). All these data indicate that XBP1 is essential for tube formation and angiogenesis in vitro.
Figure 5.

XBP‐1 silencing suppresses HUVEC proliferation and angiogenesis. (A) Transfection of XBP‐1 siRNA oligonucleotide suppresses tube formation of HUVECs. HUVECs transfected with 50 nM siRNA XBP1 or siRNA control oligonucleotide for 24 h were seeded on BD Matrigel and quantification of the average number of tubes per field of view, 6 h after seeding. (B) Transfection of XBP‐1 siRNA oligonucleotide reduces migration of HUVECs by wound closure. HUVECs were transfected with siRNA XBP1 (50 nM) or siRNA control oligonucleotide and the extent of wound closure was observed at 0 and 48 h. (C) Hemoglobin concentration of HUVEC transfected with siRNA XBP‐1 or siRNA control, confirmed by in vivo Matrigel plug assays. n = 5 per group. (D) Ectopic expression of XBP‐1 by transient transfection of pCMV6‐XL5‐XBP‐1s (XBP‐1s) plasmid enhances tube formation in endothelial cells compared with vector control (pCMV). Three independent experiments were carried out, and each experiment was performed in triplicate. *P < 0.05 compared with control.
Restored expression of XBP‐1 reverses anti‐angiogenic effect of miR‐214 overexpression
Proliferation of the endothelial cells is essential for the activation of angiogenesis. Herein, two proliferation assays using the CCK‐8 method and BrdU incorporation revealed that blocking XBP1 decreased cell proliferation (Figs. 6 and S11). Conversely, pCMV6‐XL5‐XBP1s transfection enhanced HUVEC cell proliferation as evaluated by BrdU incorporation assay (Fig. 6B). To demonstrate that HUVECs proliferation is also negatively regulated by miR‐214 through targeting XBP1, we introduced the synthesized miRNA mimics of miR‐214 into HUVECs. Results showed that overexpression of miR‐214 decreased cell proliferation, while silencing of miR‐214 dramatically increased cell proliferation (Figs. 6 and S11 and S12). As expected, the effect of miR‐214 mimics on proliferation of HUVECs was similar to transfection of XBP‐1 siRNA.
Figure 6.
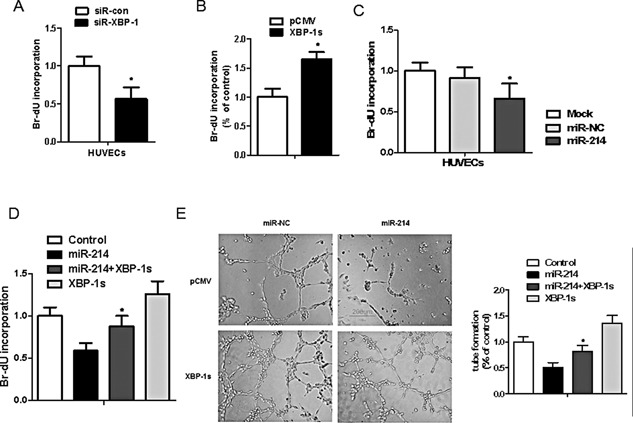
Restored expression of XBP‐1 reverses the suppression of miR‐214 on endothelial cells angiogenesis. (A) XBP‐1 silencing inhibits endothelial cells proliferation. (B) Overexpression of XBP‐1 promotes endothelial cells proliferation. (C) miR‐214 mimics increases HUVECs cell proliferation assayed by BrdU incorporation assay. HUVEC growth was measured by BrdU cell proliferation assay after transfection with XBP1 siRNA (50 nM) or siRNA control oligonucleotide. Three independent experiments were carried out, and each experiment was performed in triplicate. *P < 0.05 compared with control. (D) Overexpression of XBP‐1 by transient transfection of pCMV6‐XL5‐XBP‐1s plasmid reverses the suppression of miR‐214 on tube formation in cultured endothelial cells. (E) XBP‐1 reintroduction reverses the suppression of miR‐214 in endothelial cells proliferation. HUVECs were transfected with pCMV6‐XL5‐XBP‐1s (XBP‐1s) plasmid or/and miR‐214 mimics at 24 h as indicated. n = 6; *P < 0.05 compared with miR‐214.
To further verify if miR‐214 targets XBP1, we examined if XBP1s upregulation in Matrigels can alter the suppressive effects of miR‐214 transfection on capillary tube formation. XBP1 reintroduction by transfection of pCMV6‐XL5‐XBP1s plasmid into miR‐214 mimics‐transfected HUVECs ablated the inhibitory effects of miR‐214 on both tube formation and endothelial cell proliferation (Fig. 6D and E). These findings suggest that XBP1 is an authentic target of miR‐214, and further verify a key role for the miR‐214/XBP1 pathway in mediating endothelial cell proliferation, and in regulating cardiac angiogenesis.
Discussion
In this study, we present comprehensive evidence supporting anti‐angiogenesis activity of miR‐214 in the heart. Ectopic expression of miR‐214 decreases HUVECs proliferation and angiogenesis in vitro, and AAV9‐mediated miR‐214 downregulation attenuates isoproterenol (ISO) infusion‐induced impairment of cardiac angiogenesis in vivo. Importantly, we further found that ER stress transcription factor XBP1 is a direct target of miR‐214 in endothelial cells during heart failure, and identified a novel “cross‐talk” mechanism between ER stress and miRNAs in cardiovascular system.
XBP‐1, a key unfolded protein response transcription factor, induced by ATF6 and spliced by IRE1, is essential for cell survival under hypoxic conditions, and is required for tumor growth (Romero‐Ramirez et al., 2004). Recently, XBP‐1‐mediated tumor angiogenesis has been found in human pancreatic adenocarcinomas and breast cancer (Romero‐Ramirez et al., 2009; Ruan et al., 2013). Interestingly, transient activation of XBP1 splicing lead to EC proliferation, while sustained activation induced EC apoptosis, cell loss from vessel walls, and atherosclerotic lesion development in aorta isograft model (Zeng et al., 2009). In this study, we also found that blocking XBP1 inhibited the proliferation of ECs, while upregulation of XBP1 promoted ECs proliferation. In the search of the possible mechanism(s) of XBP1‐mediated ECs angiogenesis, we further found that VEGF expression was downregulated in XBP1 siRNA experiments in HUVECs and H9C2 cells (data not shown). As a transcription factor, the spliced XBP1 is not only involved in the transcriptional regulation of genes essential for cell survival or apoptosis in response to stress stimuli, but is also involved in other physiological processes, e.g., spliced XBP‐1 directly binds to the ACGT core of the VEGFA promoter to regulate VEGFA mRNA expression especially in response to ER stress (Ghosh et al., 2010; Pereira et al., 2010; Miyagi et al., 2013). On the other hand, a recent study discovered that XBP1 splicing was activated by VEGF through KDR/XBP1u/IRE1α interaction, and regulates endothelial cell proliferation in a PI3K/Akt/GSK3β/β‐catenin/E2F2‐dependent manner and modulates the cell size increase in a PI3K/Akt/GSK3β/β‐catenin/E2F2‐independent manner (Zeng et al., 2013). Our data demonstrated that XBP1 was essential for ECs survival and growth and was an important regulator of vascular function and angiogenesis. Additionally, we revealed a novel VEGF/XBP1s/VEGF signaling pathway regulating ECs angiogenesis. However, possible effects of XBP1 on other proangiogenic and anti‐angiogenic factors deserve further investigation.
Recently, more and more miRNAs have been shown to regulate angiogenesis in vitro and in vivo, such as miR‐126, miR‐591c, miR‐378, miR‐296, miR‐101, miR‐223, and the miR‐17∼92 cluster (Matkovich et al., 2009; Small et al., 2010). Now, in this study we were excited to find that miR‐214 directly targets XBP‐1 expression to regulate endothelial cells angiogenesis. In 2009, one of the well‐known Chinese medicines, Rg1, was reported to down‐regulate miR‐214 and promote angiogenesis (Leung et al., 2006; Chan et al., 2009), which indicated a possible relationship between miR‐214 and angiogenesis. Recent studies have also indicated that in vivo silencing of miR‐214 enhanced retinal developmental angiogenesis in mice (van Mil et al., 2012), ectopic expression of miR‐214 in HCC cells significantly suppressed angiogenesis in vitro and in vivo and xenograft tumor growth in nude mice (Shih et al., 2012), which support our conclusion that miR‐214 is an important negative regulator of angiogenesis, likely via its regulation of XBP‐1.
Cardiac hypertrophy occurs as an adaptive response to increased workload to maintain cardiac function, but prolonged cardiac hypertrophy causes heart failure (Sano et al., 2007). Very recently, impaired cardiac angiogenesis was considered as one of the most important mechanisms for the transition from cardiac hypertrophy to cardiac failure (Shiojima et al., 2005; Izumiya et al., 2006; Sano et al., 2007), but little is known concerning the effects of key regulators of angiogenesis on heart failure. Excitingly, the present study has for the first time demonstrated that aberrant expression of miR‐214 plays an important role in the down regulation of XBP‐1, suppression of cardiac angiogenesis in heart failure. Moreover, miR‐214 expression was gradually increased not only in TAC and ISO‐induced cardiac hypertrophy in mice, but also in human failing heart tissue and blood samples, suggesting miR‐214 may be a circulating biomarker and a diagnostic predictor for heart failure in future. Importantly, we tested the hypothesis in vivo and found that blockade of endogenous miR‐214 expression by AAV9‐mediated gene therapy regulated cardiac angiogenesis and cardiac function. Interestingly, cardiac‐specific transgenic miR‐214 mice exhibit no phenotypic changes (van Rooij et al., 2006), which also support our observation that XBP‐1 is the target of miR‐214, and miR‐214 regulates vascular angiogenesis and cardiac function only under the conditions in which XBP‐1 is dysregulated. Certainly, as XBP1 is a multifunctional transcription factor involved in cell survival (Romero‐Ramirez et al., 2004), inflammation (Martinon et al., 2010), and glucose and lipid metabolism (Zhou et al., 2011), miR‐214 also may target other genes in heart tissue (el Azzouzi et al., 2013). The other physiological influence of miR‐214/XBP1 on myocyte hypertrophy, cardiac fibrosis, cardiomyocyte apoptosis, cardiac inflammation, and myocardial energy metabolism needs further investigation.
In summary, we demonstrated that XBP1 is essential for HUVECs proliferation and angiogenesis, and miR‐214 plays an important suppressive role in intracellular XBP1 signaling‐mediated angiogenesis and endothelial function by directly binding the XBP1 3′UTR that leads to downregulation of XBP1 expression level. These results revealed a mechanism by which miR‐214 modulates gene and protein expressions and cell proliferation in endothelial cells, providing a new target for modulating vascular formation and function, as well in the progression of heart failure.
Supporting information
Supporting Information.
Quanlu Duan and Lei Yang contributed equally to this work.
Conflict of interest: None.
Literature Cited
- Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel‐Duby R, Sadek HA, Olson EN. 2012. MicroRNA‐214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest 122:1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LS, Yue PY, Mak NK, Wong RN. 2009. Role of microRNA‐214 in ginsenoside‐Rg1‐induced angiogenesis. Eur J Pharm Sci 38:370–377. [DOI] [PubMed] [Google Scholar]
- Cheng AM, Byrom MW, Shelton J, Ford LP. 2005. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q, Wang X, Gong W, Ni L, Chen C, He X, Chen F, Yang L, Wang P, Wang DW. 2012. ER stress negatively modulates the expression of the miR‐199a/214 cluster to regulates tumor survival and progression in human hepatocellular cancer. PLoS ONE 7:e31518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MM, Armand AS, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, da Costa Martins PA, Schrauwen P, De Windt LJ. 2013. The hypoxia‐inducible microRNA cluster miR‐199a approximately 214 targets myocardial PPARdelta and impairs mitochondrial fatty acid oxidation. Cell Metab 18:341–354. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F. 2010. Transcriptional regulation of VEGF‐A by the unfolded protein response pathway. PLoS ONE 5:e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. 2010. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci U S A 107:14339–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. 2006. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 47:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW. 2005. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up‐regulated in human tumors. Cancer Res 65:4707–4715. [DOI] [PubMed] [Google Scholar]
- Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. 2009. Mir‐214‐dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell 36:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. 2007. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res 101:59–68. [DOI] [PubMed] [Google Scholar]
- Leung KW, Pon YL, Wong RN, Wong AS. 2006. Ginsenoside‐Rg1 induces vascular endothelial growth factor expression through the glucocorticoid receptor‐related phosphatidylinositol 3‐kinase/Akt and beta‐catenin/T‐cell factor‐dependent pathway in human endothelial cells. J Biol Chem 281:36280–36288. [DOI] [PubMed] [Google Scholar]
- Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. 2009. miR‐23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A 106:12103–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HQ, Liang C, He ZQ, Fan M, Wu ZG. 2013. Circulating miR‐214 is associated with the severity of coronary artery disease. J Geriatr Cardiol 10:34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich SJ, Van Booven DJ, Youker KA, Torre‐Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW 2nd. 2009. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support Circulation 119:1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi H, Kanemoto S, Saito A, Asada R, Iwamoto H, Izumi S, Kido M, Gomi F, Nishida K, Kiuchi Y, Imaizumi K. 2013. Transcriptional regulation of VEGFA by the endoplasmic reticulum stress transducer OASIS in ARPE‐19 cells. PLOS ONE 8:e55155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA. 2003. Inhibition of receptor‐localized PI3K preserves cardiac beta‐adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest 112:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. 2004. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 110:705–712. [DOI] [PubMed] [Google Scholar]
- Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, Zolotukhin I, Tarantal AF, Byrne BJ. 2006. Recombinant adeno‐associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res 99:e3–9. [DOI] [PubMed] [Google Scholar]
- Penna E, Orso F, Cimino D, Tenaglia E, Lembo A, Quaglino E, Poliseno L, Haimovic A, Osella‐Abate S, De Pitta C, Pinatel E, Stadler MB, Provero P, Bernengo MG, Osman I, Taverna D. 2011. microRNA‐214 contributes to melanoma tumour progression through suppression of TFAP2C. EMBO J 30:1990–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Liao N, Neale GA, Hendershot LM. 2010. Transcriptional and post‐transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One 5:e12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petremand J, Puyal J, Chatton JY, Duprez J, Allagnat F, Frias M, James RW, Waeber G, Jonas JC, Widmann C. 2012. HDLs protect pancreatic beta‐cells against ER stress by restoring protein folding and trafficking. Diabetes 61:1100–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang R, Wang F, Shi LY, Liu M, Chen S, Wan HY, Li YX, Li X, Gao SY, Sun BC, Tang H. 2011. Plexin‐B1 is a target of miR‐214 in cervical cancer and promotes the growth and invasion of HeLa cells. Int J Biochem Cell Biol 43:632–641. [DOI] [PubMed] [Google Scholar]
- Romero‐Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. 2004. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 64:5943–5947. [DOI] [PubMed] [Google Scholar]
- Romero‐Ramirez L, Cao H, Regalado MP, Kambham N, Siemann D, Kim JJ, Le QT, Koong AC. 2009. X box‐binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl Oncol 2:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Xi L, Boye SL, Han S, Chen ZJ, Hauswirth WW, Lewin AS, Boulton ME, Law BK, Jiang WG, Jiang H, Cai J. 2013. Development of an anti‐angiogenic therapeutic model combining scAAV2‐delivered siRNAs and noninvasive photoacoustic imaging of tumor vasculature development. Cancer Lett 332:120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. 2007. p53‐induced inhibition of Hif‐1 causes cardiac dysfunction during pressure overload. Nature 446:444–448. [DOI] [PubMed] [Google Scholar]
- Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. 2007. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100:416–424. [DOI] [PubMed] [Google Scholar]
- Shih TC, Tien YJ, Wen CJ, Yeh TS, Yu MC, Huang CH, Lee YS, Yen TC, Hsieh SY. 2012. MicroRNA‐214 downregulation contributes to tumor angiogenesis by inducing secretion of the hepatoma‐derived growth factor in human hepatoma. J Hepatol 57:584–591. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. 2005. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EM, Frost RJ, Olson EN. 2010. MicroRNAs add a new dimension to cardiovascular disease. Circulation 121:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, Kawase Y, Chen J, Liang L, Sipo I, Vetter R, Weger S, Kurreck J, Erdmann V, Tschope C, Pieske B, Lebeche D, Schultheiss HP, Hajjar RJ, Poller WC. 2009. Long‐term cardiac‐targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation 119:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mil A, Grundmann S, Goumans MJ, Lei Z, Oerlemans MI, Jaksani S, Doevendans PA, Sluijter JP. 2012. MicroRNA‐214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc Res [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. 2006. A signature pattern of stress‐responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A 103:18255–18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. 2008. Dysregulation of microRNAs after myocardial infarction reveals a role of miR‐29 in cardiac fibrosis. Proc Natl Acad Sci U S A 105:13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. 2011. miR‐499 regulates mitochondrial dynamics by targeting calcineurin and dynamin‐related protein‐1. Nat Med 17:71–78. [DOI] [PubMed] [Google Scholar]
- Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, Li D, Hou Z, Lv K, Kan G, Cao H, Wu H, Song J, Pan X, Sun Q, Ling S, Li Y, Zhu M, Zhang P, Peng S, Xie X, Tang T, Hong A, Bian Z, Bai Y, Lu A, He F, Zhang G. 2012. miR‐214 targets ATF4 to inhibit bone formation. Nat Med [DOI] [PubMed] [Google Scholar]
- Winter J, Jung S, Keller S, Gregory RI, Diederichs S. 2009. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234. [DOI] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, Li H, Mu X, Zhang H, Broderick JA, Kim JK, Weng Z, Flotte TR, Zamore PD, Gao G. 2012. Long‐term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. 2008. MicroRNA expression profiling in human ovarian cancer: miR‐214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 68:425–433. [DOI] [PubMed] [Google Scholar]
- Yang L, Jiang J, Drouin LM, Agbandje‐McKenna M, Chen C, Qiao C, Pu D, Hu X, Wang DZ, Li J, Xiao X. 2009. A myocardium tropic adeno‐associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A 106:3946–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q. 2009. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A 106:8326–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Xiao Q, Chen M, Margariti A, Martin D, Ivetic A, Xu H, Mason J, Wang W, Cockerill G, Mori K, Li JY, Chien S, Hu Y, Xu Q. 2013. Vascular endothelial cell growth‐activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 127:1712–1722. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Fisher SJ, White MF, Biddinger SB, Ozcan U. 2011. Regulation of glucose homeostasis through a XBP‐1‐FoxO1 interaction. Nat Med 17:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.