

SUMMARY
MET exon 14 alterations are a diverse group of mutations, many of which disrupt splice acceptor or donor sites leading to exon 14 skipping, impaired receptor degradation, and oncogenic transformation. These alterations are clinically targetable with MET-directed therapy.
In this issue of Clinical Cancer Research, Tong and colleagues identified MET mutations that disrupt consensus sequences for exon 14 splicing in 2.6% of treatment-naïve non-small cell lung cancers (1). These alterations co-occurred with MET amplification and copy number gain, and were associated with high MET protein expression.
RNA splicing plays a crucial role in the process of gene regulation. Eukaryotic DNA contains long, non-coding sequences called introns that are interspersed between shorter, coding sequences called exons. DNA is transcribed into pre-mRNA, and intron removal is subsequently achieved through pre-mRNA splicing, forming mRNA. A phenomenon called alternative splicing allows for the exon composition of spliced mRNA to vary significantly. This variation makes it possible for multiple protein isoforms to be expressed from information contained within a single gene, giving rise to a diverse proteome that is much larger than our genome.
The process of splicing is carefully orchestrated. It involves the recognition of specific sequences along the length of an intron: a 5′ splice or donor site, a branch site, a polypyrimidine tract, and a 3′ splice or acceptor site. In addition, cis-acting elements such as splicing enhancers or silencers can influence the recognition of these sites by spliceosomal components. Mutations that disrupt these elements or active cryptic splice sites can lead to aberrant splicing, causing intron retention or exon skipping (2).
Aberrant splicing is strongly associated with the pathogenesis of disease. Up to 20% of genetic disease is caused by mutations that affect pre-mRNA splicing. Duchenne muscular dystrophy can be caused by splice site mutations in the dystrophin gene. These mutations lead to exon skipping and/or cryptic splice site activation, resulting in the loss of dystrophin function and progressive muscle weakness (3). Aberrant splicing is likewise associated with the development of cancer. This most commonly occurs due to dysregulation or alterations involving splicing factors. Recurrent somatic mutations in genes that encode splicing factors, for example, have been identified in samples from patients with myelodysplastic syndrome and several leukemias (4). Mutations that disrupt splice sites represent a less common, but important mechanism of oncogenesis. MET exon 14 alterations have quickly risen in prominence as an example of this biology.
The sequence composition of MET exon 14 alterations is incredibly diverse. Base substitutions or indels (predominantly deletions) that disrupt the branch point of intron 13, the 3′ splice site of intron 13, or the 5′ splice site of intron 14 can effectively result in MET exon 14 skipping (5). Exon 14 encodes a juxtamembrane domain containing the Y1003 residue that serves as a binding site for the E3 ubiquitin ligase CBL (Figure 1A). Exon 14 skipping is thus thought to lead to decreased MET ubiquitination and degradation, increased MET protein stability, and increased ligand-dependent downstream signaling (Figure 1B) (6). It is important to note that genomic alterations that affect the Y1003 residue such as MET Y1003F or MET exon 14 deletion can result in a similar biology without affecting splicing (5, 7).
Figure 1.
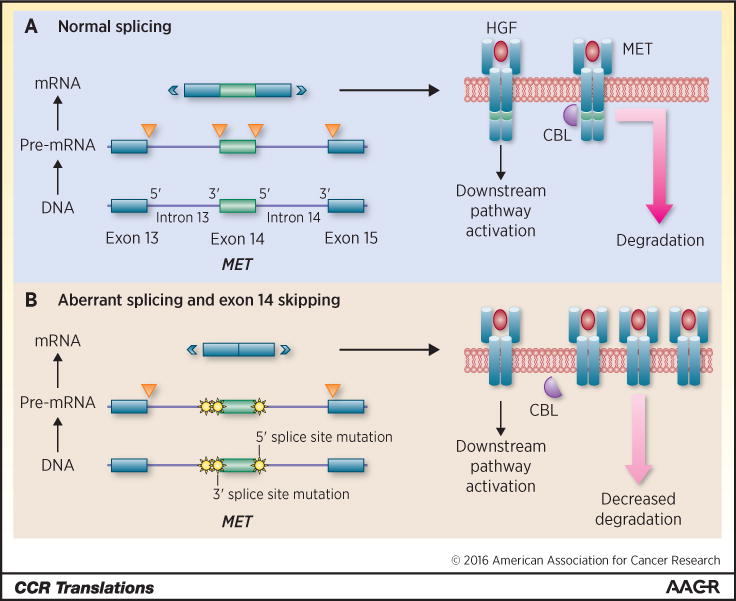
In this diagram, part of the MET gene is depicted on the left in Figure 1A. This portion of the gene includes exons 13, 14, and 15, and introns 13 and 14. DNA is transcribed into pre-mRNA, and introns are spliced out (orange triangles) by normal splicing mechanisms. This process involves the recognition of specific regions along the intron including as 5′ and 3′ splice sites. mRNA is eventually translated into the MET receptor protein. The transmembrane MET receptor is depicted in Figure 1A on the right. Binding of the ligand HGF (red) results in downstream pathway activation and increased cellular proliferation. MET exon 14 encodes a region on the receptor (green) that includes the Y1003 residue. This residue serves as a binding site for the E3 ubiquitin ligase CBL (purple). Ubiquitination tags the MET protein for degradation. In Figure 1B, MET mutations (yellow) that disrupt the branch point and/or 3′ splice site of intron 13, and the 5′ splice site of intron 14 result in aberrant splicing and exon 14 skipping. These mutations normally occur separately (involving a region flanking only one end of exon 14), but are shown here in aggregate for simplicity. MET exon 14 is thus excluded in mRNA that is later translated into a protein product lacking the Y1003 residue. Loss of this region leads to leads to decreased MET receptor ubiquitination by CBL. Decreased degradation results in oncogenesis driven by increased levels of MET. Of note, base substitutions involving Y1003 or exon 14 deletions that span the area encoding this residue can likewise lead to decreased MET degradation without specifically interfering with normal splicing mechanisms.
The diversity of MET exon 14 alterations poses a challenge to assay selection for diagnostic and clinical trial selection purposes. Molecular profiling will quickly need to move towards comprehensive platforms such as broad, hybrid-capture next-generation sequencing in an effort to both capture these mutations and detect concurrent genomic alterations (5). In this study, Tong et al demonstrate a significant correlation between these mutations and strong MET protein expression via immunohistochemistry (IHC), likely reflecting the biology of these tumors (1). Only a fraction of IHC-positive tumors harbored MET exon 14 alterations, however, making IHC an adjunctive tool at best, and a poor primary selection biomarker for clinical trial enrollment.
MET exon 14 alterations are most commonly found in lung cancers. These mutations have been identified in approximately 3–4% of lung adenocarcinomas. Tong and colleagues demonstrate that about 32% of pulmonary sarcomatoid carcinomas harbor MET exon 14 alterations (1), consistent with the results of other series. Interestingly, pulmonary sarcomatoid carcinomas with an adenocarcinoma component were more likely to harbor these mutations than those without an adenocarcinoma component in one report (8).
In patients with lung cancer, mutations that disrupt MET exon 14 splicing tend to occur in older individuals (median age of 73), with a lower proportion of never-smokers relative to patients with other oncogene-driven lung cancers (9). In the current report from Tong et al, MET exon 14 alterations were identified independent of stage as poor prognostic factors for overall disease-specific survival (1). MET exon 14 alterations have also been detected in a variety of other tumors such as brain gliomas, gastrointestinal cancers, sarcomas, and cancers of unknown primary origin (5, 10).
Preclinical data support the role of MET inhibition as a viable therapeutic strategy for these tumors (11). Both MET-directed monoclonal antibody therapy (onartuzumab and SAIT301) and tyrosine kinase inhibitor therapy (crizotinib, cabozantinib, and capmatinib) are active in vitro against patient-derived and non-patient derived cell lines containing MET exon 14 alterations (6, 8, 10). Reports of clinical responses to MET inhibition in patients with MET exon 14-altered cancers have also quickly begun to emerge (5). In a series from Paik and colleagues, durable responses were achieved with single-agent crizotinib or cabozantinib therapy (12).
The presence of these alterations in pulmonary sarcomatoid carcinomas is of particular therapeutic interest. Whereas these tumors were previously thought to be relatively refractory to systemic therapies, a response to crizotinib has been reported in a patient with pulmonary sarcomatoid carcinoma harboring a MET splice site mutation concurrent with MET amplification (8). Prospective clinical trials of MET inhibitors that enrich for lung cancers with MET exon 14 alterations are currently underway, including an expansion cohort of an ongoing phase I trial of crizotinib (NCT00585195).
The presence of concurrent genomic alterations as potential modifiers of response to MET inhibition in MET exon 14-altered lung cancers will require exploration. MET exon 14 alterations harbor concurrent high-level MET copy gain in about 20% of patients with lung cancer (9), and frequently co-occur with MET amplification across a variety of tumors (5). As responses to crizotinib have been reported in patients with MET-amplified lung cancers, the relative contribution of concurrent MET amplification to response in a MET exon 14-altered tumor remains unclear.
Finally, prospective targeted therapy trials should focus on the identification of mechanisms of innate and acquired resistance to MET inhibition in MET exon 14-altered lung cancers. In terms of innate resistance, concurrent genomic alterations such as PIK3CA mutations have been speculated to modify response (5, 8). In H596, a lung cancer cell line harboring a MET exon 14 alteration concurrent with a PIK3CA E545K mutation, MET inhibition alone showed limited effects. In contrast, combined MET- and PI3K-directed therapy with crizotinib and GDC0941 was synergistic in decreasing cell proliferation and downstream activation (8). Whenever safe and feasible, paired pre- and post-treatment biopsies should be considered for patients with MET exon 14-altered tumors that initially respond to therapy and later develop acquired resistance. Understanding mechanisms that drive resistance will allow for the development of therapeutic strategies that are active in this space.
Acknowledgments
Grant Support:
A. Drilon is supported in part by the NIH under a P30CA008748 Core Grant.
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Tong JHM, Yeung SF, Chan AWH, Chung LY, Chau SL, Lung RWM, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016 Feb 4; doi: 10.1158/1078-0432.CCR-15-2061. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuffery-Giraud S, Chambert S, Demaille J, Claustres M. Point mutations in the dystrophin gene: evidence for frequent use of cryptic splice sites as a result of splicing defects. Hum Mutat. 1999;14:359–68. doi: 10.1002/(SICI)1098-1004(199911)14:5<359::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 4.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119:2635–41. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 5.Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–9. doi: 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
- 6.Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283–9. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 7.Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001;8:995–1004. doi: 10.1016/s1097-2765(01)00378-1. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, Chen J, et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34:794–802. doi: 10.1200/JCO.2015.62.0674. [DOI] [PubMed] [Google Scholar]
- 9.Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J Clin Oncol. 2016;34:721–30. doi: 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Ou SH, Lee JM, Kim HC, Hong M, Kim SY, et al. Gastrointestinal malignancies harbor actionable MET exon 14 deletions. Oncotarget. 2015;6:28211–22. doi: 10.18632/oncotarget.4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 12.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–9. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]