

Abstract
Microtubule-targeting agents (MTAs), such as the taxanes and vinca alkaloids, are used to treat a variety of cancers due to their ability to perturb microtubule dynamics. In cell culture, MTAs exert their anticancer effects primarily by causing mitotic arrest and cell death. However, accumulating indirect evidence suggests that MTAs may exert their cytotoxicity in human tumors by interfering with interphase microtubules. In this study, we sought to develop and characterize an experimental system in which to test the hypothesis that MTAs induce cell death during interphase. Primary adult acute lymphoblastic leukemia (ALL) cells treated with vincristine only weakly exhibited colocalization between mitotic and apoptotic markers and major characteristics of mitotic death, such as an increase in cells with 4N DNA content before the appearance of cells with <2N DNA content, suggesting a mixed response. Therefore, we separated ALL cells into distinct phases of the cell cycle by centrifugal elutriation, labeled cells with 5-ethynyl-2'-deoxyuridine (EdU) and then treated each population with vincristine. Cells isolated during G1 underwent cell death without evidence of EdU uptake, indicating that the cytotoxic effects of vincristine took place during G1. Conversely, cells isolated during S or G2/M phases underwent death following mitotic arrest. Thus, vincristine induces distinct death programs in primary ALL cells depending on cell cycle phase, and cells in G1 are particularly susceptible to perturbation of interphase microtubules. Primary ALL cells may therefore provide a powerful model system in which to study the multimodal mechanisms underlying MTA-induced cell death.
Keywords: microtubule inhibitors, vincristine, mitotic arrest, interphase death, acute lymphoblastic leukemia, apoptosis
Introduction
Microtubule targeting agents (MTAs) are clinically important chemotherapeutic agents used in the treatment of various cancers [1]. MTAs act by interfering with microtubule dynamics which plays a major role in mitotic spindle formation. MTAs such as the vinca alkaloids, including vincristine, cause microtubule depolymerization, whereas taxanes stabilize microtubules preventing their depolymerization [2]. Treatment of cultured cells with MTAs often leads to prolonged activation of the mitotic spindle checkpoint leading to mitotic arrest and eventually cell death, providing a rationale for their use as antitumor agents [3,4,5]. Cells may die in mitosis or exit mitosis without proper chromosome segregation or cytokinesis, referred to as mitotic slippage. After slippage, cells may die in interphase, or arrest and survive, or resume cycling [6,7]. Whether cells die in mitosis or undergo mitotic slippage has been postulated to depend on the balance of two pathways, one controlling cyclin B1 levels and the other caspase activation [8]. We have reported that cyclin-dependent kinase 1 (Cdk1)/cyclin B1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and cell death [9], and that cell fate after mitotic arrest is dictated by the degree of Cdk1/cyclin B1-mediated phosphorylation of anti-apoptotic Bcl-2 proteins [10].
Implicit in conventional theories of MTA action is that they induce mitotic arrest prior to cell death. This viewpoint has recently been challenged, on the basis of largely indirect evidence but nonetheless compelling lines of reasoning [11,12]. The major points underlying this paradigm shift include the following: (i) all of the mechanistic data derive from cell culture and xenograft models which may not recapitulate patient tumor conditions; (ii) there is a poor relationship between the status of the spindle assembly checkpoint and cellular sensitivity to MTAs [6]; (iii) MTAs are effective clinically despite the slow doubling times and low mitotic indices of some human tumors [11,12,13]; and (iv) mitosis-specific inhibitors, such as those targeting Aurora kinases and polo-like kinases, are effective in vitro but have shown poor activity in clinical trials [11,12,13]. Such collective evidence has led to the suggestion that the therapeutic efficacy of MTAs in the clinical setting may be attributed to interference with interphase microtubule functions [11,12]. This viewpoint has itself been questioned, resulting in an intense current debate [14,15,16,17]. For example, it has been suggested that the slow doubling time of tumors likely reflects the balance of proliferation and death, and thus cycling cells may be more abundant than predicted based solely on doubling time [14]. In addition, it has been contended that since drugs such as Taxol are concentrated and retained in tumor cells for lengthy periods, their effects may be exerted on quiescent cells that reenter the cycle after a prolonged delay [13,18,19].
Rapid induction of interphase death by vinca alkaloids has been reported in a limited number of cell lines, but generally only with relatively high drug concentrations or in combination with other drugs [20,21,22,23]. A study of breast cancer cell lines treated with microtubule destabilizers reported arrest in G1 and G2 phases, suggesting the drugs may perturb cells in interphase, but the subsequent fate of the treated cells was not examined [24]. Because MTAs typically manifest their effects in mitosis in most cultured cell lines, a major obstacle to progress in this area is the lack of appropriate laboratory models of MTA-induced interphase death.
To develop and characterize a cell system to test the hypothesis that MTAs could also induce interphase death, we assessed the extent of mitotic arrest in several different cell types after MTA treatment. Among these were primary adult acute lymphoblastic leukemia (ALL) cells derived from several individual adult patients, expanded and established in culture using a defined serum-free medium [25,26]. In the present study, to determine whether cells in different cell cycle contexts were differentially affected by vincristine, we used centrifugal elutriation to isolate populations of ALL cells in different cell cycle phases. We show that vincristine can induce cell death directly in G1 phase cells, whereas cells beyond G1 phase transit to M phase before undergoing cell death. Thus vincristine induces distinct cell cycle-dictated pathways of cell death in primary ALL cells. To our knowledge, these results are the first to definitively demonstrate induction of interphase death by relatively low and therapeutically relevant concentrations of an MTA used alone, and, as such, provide a powerful model system to study clinically applicable death mechanisms induced by drugs in this class and to provide hypotheses to explore in vivo.
Materials and Methods
Materials; Cell extraction and immunoblotting; Cell cycle analysis; Analysis of microtubule content; and Statistical analysis: see Supplementary Materials.
Cell culture
KB3 cells (HeLa subline) were maintained in DMEM, and RS4;11 ALL cells were maintained in RPMI-1640 medium, each supplemented with 10% bovine growth serum, 2 mM L-glutamine, 50 units/mL penicillin, and 50 µg/mL streptomycin. ALL cell cultures were maintained in suspension as described [25] in Iscove's modified Dulbecco's medium (IMDM) containing serum-free supplement (10 µg/mL cholesterol, 6 mg/mL human serum albumin, 0.5 µg/mL amphotericin, 1 µg/mL insulin, 200 µg/mL human apo-transferrin, 50 µM 2-mercaptoethanol, 2 mM glutamine and 50 units/mL penicillin). Cells were maintained at 37°C and 5% CO2. The ALL cultures utilized, namely ALL-2 and ALL-5, correspond to their original designations of CM and PH cells, respectively [25]. Doubling time was determined by monitoring viable cell count daily for up to six days. Authentication of the cell lines and ALL cultures was established via short tandem repeat (STR) profiling in September, 2014, by Genetica DNA Laboratories (LabCorp Specialty Testing Group, Burlington, NC). The STR profiles of KB3 and RS4;11 cell lines matched that of reference profiles available in the ATCC database, and, as expected, the two primary ALL cell cultures each gave unique profiles that did not match any in the repository.
Cell viability assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described [27]. Cells (2,000 per well for KB3 and 30,000 per well for others) were seeded in 96-well plates, and vincristine was added in a fixed final concentration of 0.1% DMSO. After incubating for a time period equivalent to twice their doubling time (see Table 1), MTT reagent (50 µg/10 µL/well) was added and incubated overnight at 37°C. The following day, 0.1 mL of 10% SDS in 0.01 M HCl was added, and after overnight incubation, absorbance readings were taken at 540 nm.
Table 1.
Doubling time and vincristine (VCR) sensitivity of KB3, RS4;11, and primary ALL cells. The values for doubling time represent mean ± S.D. (n=6), and IC50 values were derived from data in Fig. S1.
| Doubling Time (d) |
IC50 VCR (nM) |
|
|---|---|---|
| KB3 | 1.0 ± 0.1 | 2.10 ± 0.09 |
| RS4;11 | 1.5 ± 0.3 | 1.05 ± 0.24 |
| ALL-2 | 2.5 ± 0.5 | 3.90 ± 0.07 |
| ALL-5 | 2.8 ± 0.6 | 0.60 ± 0.07 |
Centrifugal elutriation
Centrifugal elutriation was performed using a Beckman JE-6 elutriator rotor as described [28]. Cells (3.5–4 × 108) were suspended in 20 mL elutriation buffer (Hanks buffered salt solution containing 1.6 g/L 2-naphthol-6,8-disulfonic acid dipotassium salt and 2% calf serum), passed through a 25G needle twice, and introduced into the chamber at a flow rate of 0.8–1.1 mL/min with a rotor speed of 3,300 rpm. Rotor speed was reduced in decrements of 80 rpm to collect a total of 20 fractions. Aliquots of individual fractions were fixed and subjected to propidium iodide staining and flow cytometry. Specific fractions were pooled to enrich for cells in defined cell cycle phases. Cells with the smallest diameter (earliest in the cell cycle) eluted first. Pooled fractions were centrifuged and cells resuspended in fresh medium at 1–1.5 × 106 cells/mL. Cell diameter was measured using a Beckman Z2 Coulter Counter and data analyzed with Z2 AccuComp software.
EdU incorporation assay
5-ethynyl-2'-deoxyuridine (EdU) incorporation assay was performed according to manufacturer's instructions (Invitrogen, Grand Island, NY) using the Click-iT® EdU Imaging Kit (Cat. # C10086), with some modifications. Cells isolated after centrifugal elutriation were incubated with 10 µM EdU and either 0.1% DMSO (vehicle) or 100 nM vincristine and harvested at specific time intervals by washing in PBS and fixing with 70% cold ethanol for 15 min. Cells were then washed with wash solution (3% bovine serum albumin in PBS), incubated with permeabilization buffer (0.5% Triton X-100 in PBS) for 20 min, washed again, suspended in 200 µL of Click-iT reaction cocktail (with either Alexa Fluor® 488 or 647), and incubated for 30 min in the dark. Cells were then washed, resuspended, and 20 µL propidium iodide (BD Pharmingen) was added prior to analysis by flow cytometry.
Cell proliferation assay
To monitor individual cell divisions and to determine whether ALL cells harbor a quiescent population, the Cell Proliferation Dye eFluor® 450 kit (eBioscience, San Diego, CA) was used. 1 × 106 ALL cells were washed with PBS thrice and resuspended in PBS. An equal volume of 20 µM eFluor® 450 was added and incubated at room temperature for 20 min according to manufacturer’s instructions. Labeling was stopped by adding 4 and 0.04 volumes of complete media and 20% HSA, respectively, and incubating on ice for 5 min. Cells were washed three times with complete media, resuspended in medium, and returned to culture. Cell aliquots were removed at daily intervals, washed and resuspended in PBS, and propidium iodide was added. Fluorescence was measured using a BD LSRFortessa Flow Cytometer (San Jose, CA) with data analysis using ModFit LT 4 (VSH) software. Non-viable (propidium iodide-positive) cells were excluded from analysis.
Results
Vincristine induces cell death without significant prior mitotic arrest in primary ALL cultures
In initial experiments, we compared the effects of vincristine on mitotic arrest in exponentially growing populations of KB3 (HeLa) and RS4;11 (ALL) cell lines and in two primary ALL cell cultures, namely ALL-2 and ALL-5, derived as described previously [25] and recently characterized with respect to Bcl-2 dependence [26]. DNA content analysis showed that KB3 cells underwent mitotic arrest (determined by 4N DNA content) and subsequent apoptosis (determined by <2N DNA content) in response to 100 nM vincristine (Fig. 1A), similar to the results we had reported previously using vinblastine [29,30]. RS4;11 cells gave a similar response, although the extent of M phase arrest was less than observed for KB3 cells (Fig. 1A). In contrast, when we examined the primary ALL cell cultures, cell death occurred without evidence of a prolonged mitotic arrest (Fig. 1A). Quantitation of the results confirmed that vincristine induced time-dependent death in all cell types as determined by <2N DNA content (Fig. 1B, upper panel), but significant mitotic arrest was only observed in KB3 and RS4;11 cells (Fig. 1B, lower panel). This profound difference in response was not due to major differences in vincristine sensitivity, as IC50 values for vincristine of the ALL cultures were similar to those of the KB3 and RS4;11 cell lines (Fig. S1) (Table 1). Two other MTAs were tested in ALL-5 cells: docetaxel, a microtubule polymerizing agent, and vinblastine, a microtubule depolymerizing agent (Fig. S2). Both drugs induced significant levels of apoptosis without evidence of a prolonged or marked mitotic accumulation, suggesting that they can also induce death from interphase. The lack of a robust mitotic arrest in response to vincristine in the ALL cells could not be attributed to defects in the spindle assembly checkpoint, because robust mitotic arrest was observed in cells treated with the Aurora A kinase inhibitor, MLN8237 (Fig. S3). To further examine the kinetics of cell death and relationship to mitotic arrest, immunoblotting was performed for PARP, which is cleaved during apoptosis, and with MPM2 antibody, which recognizes mitotic phosphoproteins (Fig. 1C, D). PARP cleavage in KB3 cells was coincident with strong MPM2 staining, consistent with Fig. 1A and with the occurrence of mitotic death, whereas PARP cleavage in the primary ALL cultures was incomplete at 24 h and occurred in concert with relatively weak MPM2 staining (Fig. 1C). In RS4;11 cells, PARP cleavage preceded a relatively strong MPM2 signal (Fig. 1D), suggesting that some cells may die via non-mitotic death. Preliminary experiments have indicated that a range of vincristine concentrations, from 10 to 1,000 nM, induces death of ALL cells without significant mitotic arrest, as determined by examination of DNA content by propidium iodide staining and flow cytometry, suggesting that interphase death in these cells is a general response to microtubule inhibition and not a concentration-dependent response.
Fig. 1. Vincristine induces cell death without prior robust mitotic arrest in primary ALL cultures.
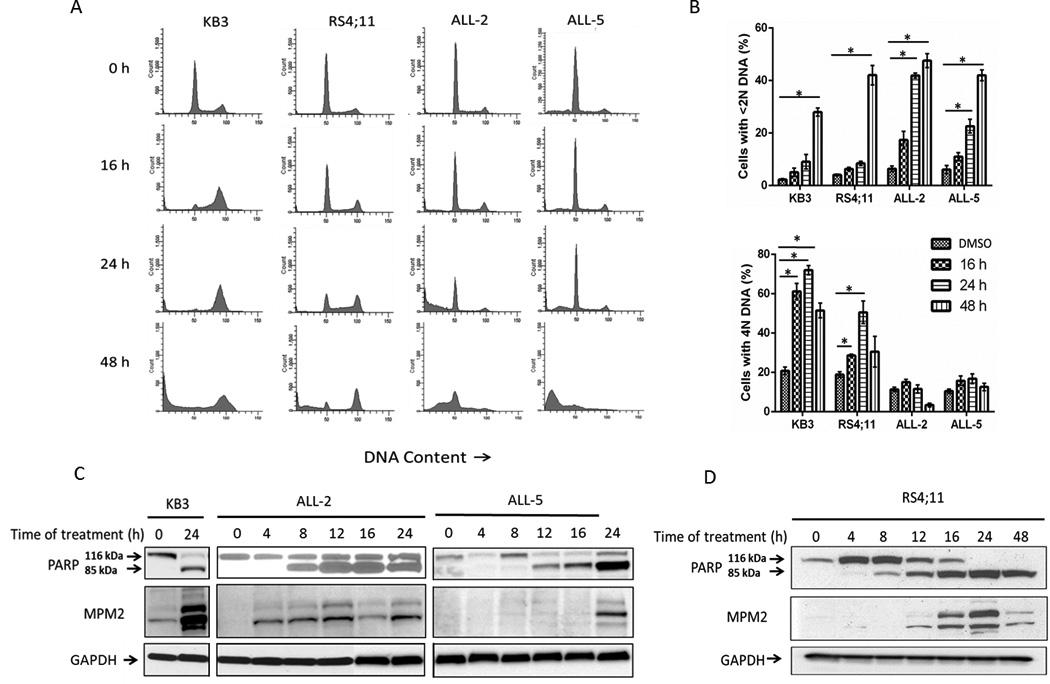
A. Flow cytometric analysis for cell cycle distribution of KB3, RS4;11, ALL-2 and ALL-5 cells, untreated or treated with 100 nM vincristine for the times indicated. Histograms are representative of three independent experiments. B. Quantitation of data from panel A showing the proportion of cells with <2N (sub-G1) or 4N (G2/M) DNA content with respect to time of vincristine treatment. Results are expressed as mean ± S.D. (n = 3). *p ≤ 0.05 (Student’s t test). C, D. Kinetics of PARP cleavage and relationship to the mitotic marker MPM2. Extracts were made from untreated or vincristine-treated (100 nM) KB3, ALL-2, or ALL-5 (C) or RS4;11 (D) cells at the times indicated and subjected to immunoblotting for PARP or MPM2. Intact and cleaved species of PARP are shown. GAPDH was used as a loading control.
Distinct responses of primary ALL cells to vincristine in different phases of the cell cycle
The results of Fig. 1 suggested two possibilities; either that the ALL cells were dying in response to vincristine after a very transient mitotic arrest, such that mitotic markers were only weakly represented due to their rapid reversal, or that they were dying primarily in interphase without transit first to mitosis. To distinguish these possibilities, isolation or synchronization of cells at different cell cycle phases was needed. However, their relatively long doubling times (Table 1) precluded chemical methods, since the cells were unable to survive lengthy treatments with agents such as thymidine or hydroxyurea. Therefore, a physical method of cell synchronization, centrifugal elutriation, which separates cells based on their size which, in turn, increases with advance through the cell cycle, was tested. Flow cytometric analysis of side-scatter, an indicator of cell size, and of DNA content after propidium iodide staining, showed a good correlation between the two parameters (Fig. S4). Initial elutriation experiments indeed established the effectiveness of the technique in isolation of cells in different phases of the cell cycle. As shown in Fig. S5, pools of early fractions were highly enriched (95.4%) in G1 (or G0) phase cells with 2N DNA content, intermediate fractions contained larger diameter cells in G1 and S phases, and later fractions were enriched (61.2%) in cells with 4N DNA content, representing G2/M phases. Measurement of average cell diameter in individual fractions confirmed the relationship between cell diameter and cell cycle phase. For example, specific values in a typical experiment ranged from 8.9 µm for early fractions representing early G1 phase cells, to 11.7 µm for late fractions representing G2/M phase cells.
To determine the response to vincristine of cells in different phases of the cell cycle, elutriated ALL-2 cells in either G1 or G2/M phases were treated with vehicle (0.1% DMSO) or 100 nM vincristine, harvested at specific times, and DNA content analyzed (Fig. 2). When treated with vehicle (Fig. 2A, top row), cells initially in G1 transited into S and G2/M phases over time, with 20.1% exhibiting 4N DNA at 24 h, with a return to approximately normal cell cycle proportions by 48 h. Over this 48 h period, there was essentially no evidence of cell death, with only 2.3% of cells with <2N DNA at 48 h, indicating that the cells suffered no ill-consequences from elutriation and subsequent culture. When cells initially in G1 were treated with vincristine (Fig. 2A, second row), a smaller percentage of the total population transited into S and G2/M phases compared to vehicle-treated cells, and between 24 and 48 h there was a large increase in cells with <2N DNA content indicative of cell death. Similar results were obtained with ALL-5 cells (Fig. S6A). We next analyzed cells enriched for those with 4N DNA content (Fig. 2A, third and fourth rows). When treated with vehicle, the cells completed the cell cycle and divided, mainly returning to G1 phase at 16–24 h, with little evidence of cell death by 48 h (Fig 2A, third row). In contrast, when treated with vincristine, cells underwent M phase arrest, with 86.4% cells having 4N DNA content within 8 h of vincristine treatment, and by 48 h, 50% of cells had <2N DNA content (Fig 2A, fourth row). Similar results were obtained with ALL-5 cells which are represented graphically in Fig. S6B.
Fig. 2. Distinct responses of primary ALL cells to vincristine in different phases of the cell cycle.
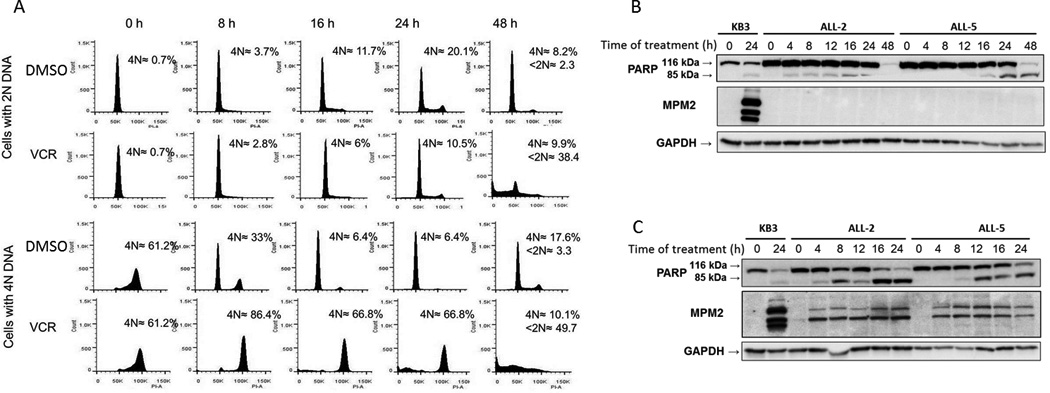
A. ALL-2 cells separated by centrifugal elutriation and enriched for cells in either G1 (2N DNA, top two rows) or G2/M (4N DNA, bottom two rows) were treated with 0.1 % DMSO or 100 nM vincristine (VCR) for the times indicated. Cells were fixed and stained with propidium iodide and analyzed by flow cytometry. The histograms are representative of 3 independent experiments. Inset: Average 4N DNA content and selective <2N DNA content, expressed as percentage of total cells analyzed. B, C. ALL-2 or ALL-5 cells, as indicated, in G1 phase (panel B) or G2/M phase (panel C) were treated with 100 nM vincristine (VCR) for the times indicated and extracts subjected to immunoblotting for PARP or MPM2. Intact and cleaved species of PARP are shown. Untreated or VCR-treated KB3 cells (left two lanes) served as positive control. GAPDH was used as a loading control.
Elutriated ALL-2 or ALL-5 cells initially in G1 (Fig. 2B) or G2/M (Fig. 2C) were treated with 100 nM vincristine for indicated time periods and analyzed by immunoblotting for PARP and MPM2. G1 cells exhibited vincristine-induced PARP cleavage after a delay of 24 h and in the absence of the mitotic marker MPM2 (Fig. 2B). In contrast, G2/M ALL cells exhibited much more rapid vincristine-induced PARP cleavage co-incident with increased MPM2 staining (Fig. 2C). Together, the results of Fig. 2 suggest that vincristine induces distinct pathways of cell death in primary ALL cells, dependent on position in the cell cycle when the drug is encountered. The fact that the majority (typically 70–75%) of ALL cells are in G1 phase with only a small proportion (9–12%) in G2/M phases (Fig. 1A) likely explains why mitotic death signals are not prominent when asynchronous cells were examined (Fig. 1), but become readily detectable when G2/M-enriched cells were used (Fig. 2). As a control for these experiments, elutriated cells were treated with 0.1% DMSO up to 48 h, and extracts subjected to immunoblotting for PARP and MPM2. As shown in Fig. S7, PARP remained intact throughout, consistent with maintenance of cell viability, and MPM2 staining was largely lacking, consistent with an absence of cells undergoing mitotic arrest.
Vincristine causes microtubule depolymerization
Because vincristine appeared to promote death in G1 phase in primary ALL cells, it was particularly important to confirm that microtubules were targeted by the drug. ALL cells were therefore treated with vincristine, or with Taxol or CaCl2 to act as positive and negative controls, respectively, for tubulin polymerization, and depolymerized and polymerized tubulin were separated and analyzed, as described in Materials and Methods. As shown in Fig. 3, tubulin was present in control cells in both depolymerized (soluble) and polymerized forms. Vincristine caused an increase in depolymerized tubulin and a corresponding decrease in polymerized tubulin in both asynchronous (Fig. 3A) and G1-phase (Fig. 3B) ALL cells. The known depolymerizing agent CaCl2 [31] gave very similar results; conversely, the microtubule stabilizing agent Taxol [32] caused an increase in polymerized tubulin and a corresponding decrease in depolymerized tubulin. Quantitation of tubulin expression, performed as described in Materials and Methods, confirmed these observations (see Fig. 3 legend). Total tubulin levels were unaffected. These results confirm that microtubules are a target of vincristine action in ALL cells.
Fig. 3. Vincristine depolymerizes microtubules in both asynchronous and G1-phase ALL cells.
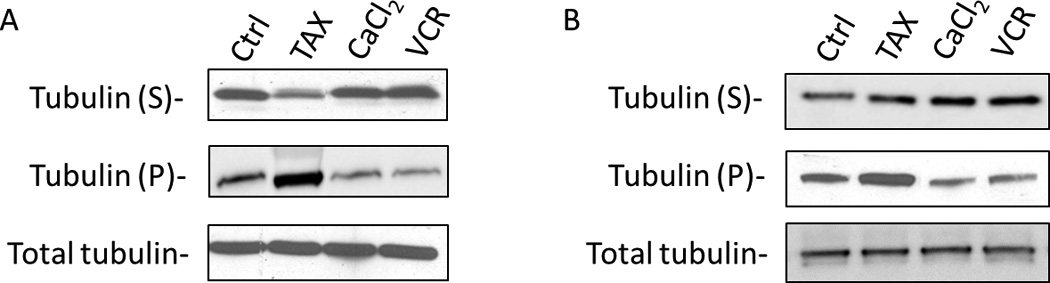
Asynchronous (A) or G1-phase (B) ALL-5 cells were treated with 0.1 % DMSO (Ctrl) or 100 nM vincristine (VCR) for 1 h and harvested. Cells were also treated with 100 nM Taxol (TAX) for 1 h or 2mM CaCl2 for 5 min to serve as polymerization and depolymerization controls, respectively. Soluble tubulin (S) and polymerized tubulin (P) were separated as described in Materials and Methods and subjected to immunoblotting for α-tubulin. Total tubulin, present in cell homogenates prior to centrifugation, was used as a loading control. The relative proportions of α-tubulin present in soluble and pellet fractions, determined as described in Materials and Methods and given as %S/%P, is as follows. Panel A: Cont., 42/58; TAX, 21/79; CaCl2, 50/50; VCR, 56/44. Panel B: Cont., 60/40; TAX, 51/49; CaCl2, 75/25; VCR, 76/24.
ALL cells in G1 phase can undergo death directly in response to vincristine without cell cycle transit
To more rigorously test whether primary ALL cells in G1 phase die directly in response to vincristine without cell cycle transit, ALL-5 cells in G1 phase were incubated with 5-ethynyl-2’-deoxyuridine (EdU) and either vehicle or 100 nM vincristine and harvested at specific intervals up to 48 h. After fluorescently labeling the EdU incorporated into DNA, cells were stained with propidium iodide and subjected to two-parameter flow cytometry. The resulting cytograms are shown in Fig. 4A, and quantitation of the proportion of EdU-positive versus EdU-negative cells that were either alive (>2N DNA) or dead (<2N DNA) shown in Figs. 4B and 4C, respectively. Cells initially gated to the left side of the lower right quadrant of the cytogram, reflecting a DNA content (x-axis) of 2N and an absence of EdU labeling (y-axis) (Fig. 4A). When treated with vehicle (Fig. 4A, top row), the cells became EdU labeled and increased their DNA content, reflecting transit through S phase and beyond. By 48 h, 43% of live cells were EdU labeled (Fig. 4B), and only a minor population had <2N DNA content (Fig. 4A, C). When treated with vincristine (Fig. 4A, bottom row), a majority of cells in G1 phase shifted from 2N DNA content to <2N DNA content without becoming EdU labeled, representing 43.5% of total cells by 48 h, and only a minor proportion (2.4%) of dead cells were EdU positive (Fig. 4C). These results indicate that ALL cells treated with vincristine can die directly from G1 without further cell cycle transit. Essentially identical results were obtained in two independent experiments with ALL-5 cells and the same pattern was obtained with ALL-2 cells (Fig. S8).
Fig. 4. Primary ALL cells in G1 phase can undergo death directly without transit to S phase.
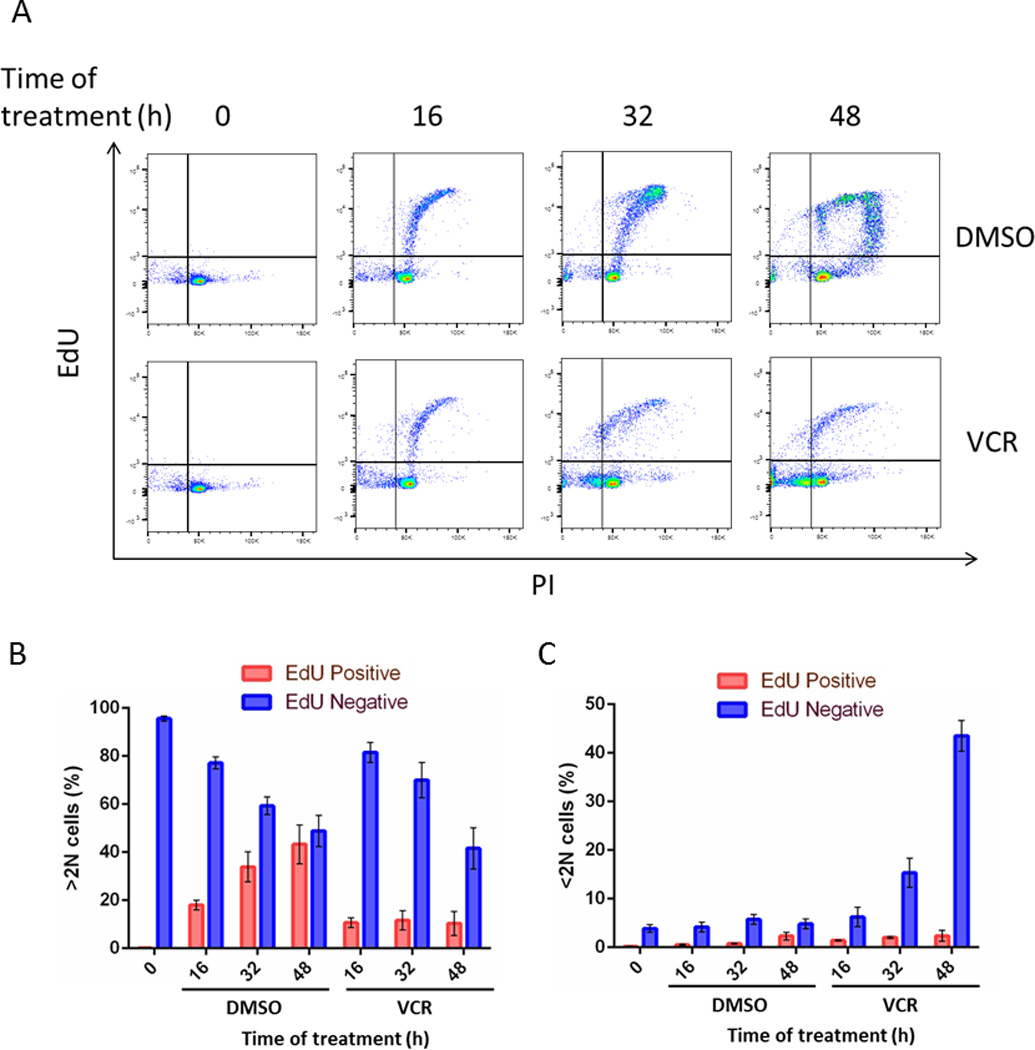
ALL-5 cells in G1 phase obtained by centrifugal elutriation were incubated with EdU and either 0.1% DMSO or 100 nM vincristine (VCR) for the times indicated. Cells were harvested, fixed and stained for EdU incorporation and with PI and analyzed by flow cytometry, with PI staining represented on the x-axis and EdU staining on the y-axis. B, C. Graphical representation of data from panel A. Panel B shows distribution between EdU-negative and EdU-positive for live cells (>2N DNA) and panel C shows distribution between EdU-negative and EdU-positive for dead cells (<2N DNA), expressed as percentage of total cells. Results are expressed as mean ± S.D. (n = 3).
Validation of EdU labeling through use of a mixed population
To rule out the possibility that the presence of vincristine affected the efficiency of EdU labeling, ALL-5 cells were subjected to centrifugal elutriation, and intermediate fractions pooled such that both G1 (82.4%) and S (17.6%) phases were represented (see Fig. S5B). Cells were incubated with EdU and 100 nM vincristine or 0.1% DMSO, harvested at intervals up to 48 h and subjected to flow cytometry (Fig. 5). On treatment with vehicle (Fig. 5A, top row), by 48 h, about half the population of cells had become EdU labeled (Fig. 5B) with only a low incidence of dead cells (Fig. 5C). When treated with vincristine (Fig. 5A, bottom row), by 48 h, dead cells were approximately evenly divided between EdU-positive and EdU-negative, with about 21% of the total appearing in the upper left quadrant, i.e. dead and EdU-positive (Fig. 5A, 5C). This corresponds well with the initial S phase population (17% of total cells), and shows that vincristine does not interfere with EdU labeling. Essentially the same pattern was obtained with ALL-2 cells. These data also emphasize and reinforce the conclusion that G1 cells treated with vincristine are highly susceptible to death without further cell cycle transit.
Fig. 5. Vincristine does not affect EdU labeling of ALL cells.
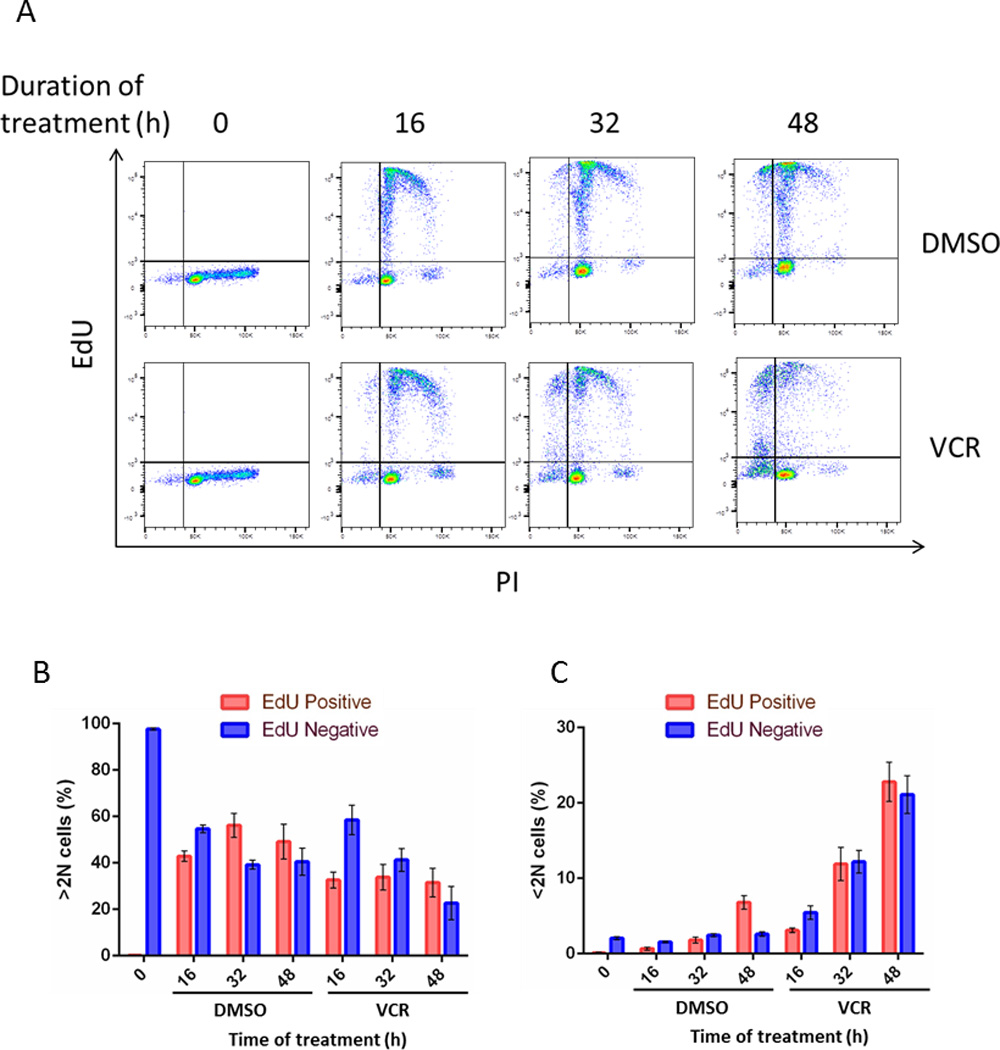
A. ALL-5 cells, separated by centrifugal elutriation and representing cells in both G1 (82.4%) and S (17.6%) phases (see Fig. S5B), were incubated with EdU and either 0.1% DMSO or 100 nM vincristine (VCR) for the time indicated, stained for incorporation of EdU and with PI, and subjected to flow cytometry, as in Fig. 4. B,C. Graphical representation of data from panel A. Panel B shows distribution between EdU-negative and EdU-positive for live cells (>2N DNA) and panel C shows distribution between EdU-negative and EdU-positive for dead cells (<2N DNA), expressed as percentage of total cells. Results are expressed as mean ± S.D. (n = 3).
All cells within the primary ALL cultures are actively dividing
We considered the possibility that ALL cells with 2N DNA may be comprised of two sub-populations, namely quiescent (or non-cycling, G0) and cycling (G1), and that G0 cells may be the population most susceptible to vincristine-induced death. To determine if quiescent cells were present, a cell proliferation assay using the violet fluorescent dye, eFluor 450®, was performed, as described in Materials and Methods. The dye binds to primary amines in proteins, and the label is expected to dilute with each cell division but be maintained at original levels in non-dividing cells. ALL-5 cells were labeled with the dye and after washing, fluorescence was determined at daily intervals. As shown in Fig. S9, at day-zero, a single parent peak of fluorescence was observed, and over three days the fluorescent intensity reduced to approximately one-half with no residual signal at the day-zero position. These results indicate that ALL cells are in an actively dividing state with no evidence of a quiescent population, and are also consistent with a doubling time of 2.8 days determined independently (Table 1). Similar results were obtained when carboxyfluorescein succinimidyl ester (CFSE), another proliferation marker similar to eFluor™450, was used. These results are also consistent with a previous study which inferred that all cells within the ALL cultures were in a proliferative state [25].
Discussion
In this study, we provide evidence that primary ALL cell cultures treated with vincristine undergo two distinct pathways of cell death. Using centrifugal elutriation to separate cells into different cell cycle phases together with EdU labeling to monitor subsequent S phase progression, we showed that cells in G1 phase can die directly, whereas cells in later phases transit to M phase first and then die. A model summarizing these findings is presented in Fig. 6. We propose that primary ALL cells in G1 phase are susceptible to death directly, whereas cells that have passed a putative “microtubule sensitivity checkpoint” in late G1/early S phase continue to cycle and undergo M phase arrest and cell death. Docetaxel and vinblastine appear to act similarly (Fig. S2), suggesting that this model applies to MTAs in general. Further studies will be needed to elucidate the cellular and molecular basis for G1 phase sensitivity to MTAs, and the mechanisms which allow many cell lines to bypass such susceptibility.
Fig. 6. Proposed model for mode of action of vincristine in ALL cells.
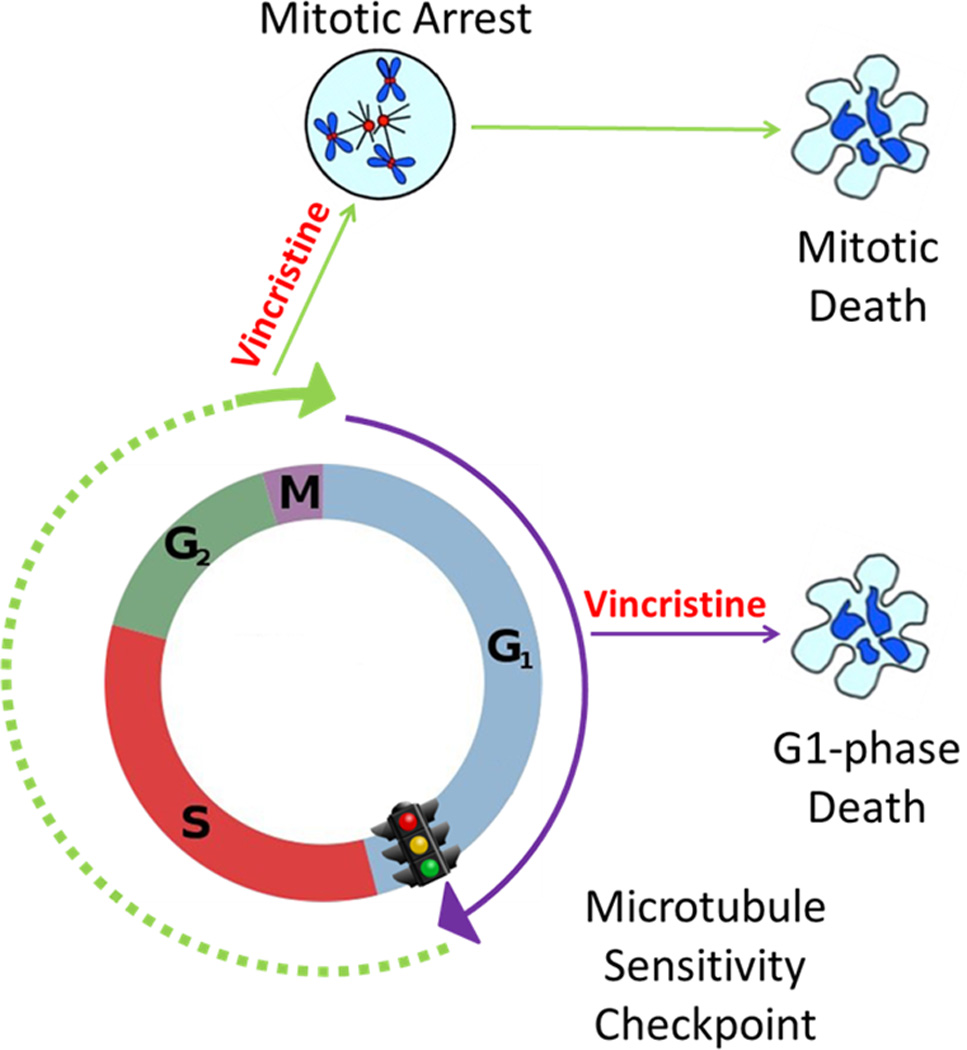
Cells in G1 phase are susceptible to death directly, whereas cells that have passed a putative “microtubule sensitivity checkpoint” in late G1/early S phase continue to cycle and die following mitotic arrest.
When G1 phase cells were subjected to EdU labeling, only about half the population migrated out of their starting position, and many remained EdU-negative with 2N DNA (Fig. 4A). However, when G1 phase cells were subjected to the cell proliferation dye assay in the absence of drug treatment, all the cells showed the expected dye dilution within two days of culture, indicating they had progressed through the cell cycle. Thus isolated G1 cells appear to be fully cell-cycle competent. The reason for the apparent inability of many G1 phase cells to progress into subsequent cell cycle phases during the EdU assay may therefore be related to the specific conditions of the assay. Exhaustion of the pool of EdU does not seem to be a factor, because cells would still be expected to enter S phase and show an increase in DNA content from 2N to 4N. Since this fraction of the population remained EdU-negative with 2N DNA content, it appears they failed to enter S phase, perhaps because EdU incorporation limits DNA replication in certain cells. Further work will be required to clarify the basis for this observation.
Previous studies have provided indirect evidence for interphase death or cell cycle-independent death in response to vinca alkaloids [20,21,22,23,24]. In most instances, significant levels of interphase death were only observed in conjunction with other agents, such as that seen in ML-1 cells co-treated with vinblastine plus inhibitors of either ERK or CDKs [21,23]. In a more recent study of a panel of leukemia cell lines, one cell line, OC1-AML1, underwent very rapid death, with 100% of the cells affected within 4 h after being treated with vincristine alone [22]. In another study, rapid cell death was observed in U2OS osteosarcoma cells treated with vinorelbine [20]. However, in both cases, relatively high drug concentrations, in the low- to mid-micromolar range, were used, and it remains unclear whether such acute effects would have been observed at lower concentrations. Importantly, in these and related studies, evidence for interphase death has often been inferred based on the rapid appearance of death signals such as cleaved PARP or activated caspase-3 in asynchronous cells. We also noted PARP cleavage at early time points in vincristine-treated asynchronous ALL cell cultures (Fig. 1C), but examination of cells enriched in different cell cycle phases revealed that those in G2 and/or M phase were the source of these signals (Fig. 2C). In contrast, vincristine-induced cell death in G1 phase cells was considerably delayed (Fig. 2B). Thus, the early appearance of death signals in cells treated with an MTA does not necessarily imply death originated in interphase cells, as cells advancing to and entering M phase can be rapidly susceptible, especially if death ensues without a prolonged mitotic arrest. The use of cells enriched in different phases as exploited here eliminates interpretational problems which can result from the study of asynchronous populations undergoing mixed responses.
Our data imply that microtubules play some essential role in G1 phase in primary ALL cells. Although the exact mechanism of interphase cell death induced by MTAs is still unclear, other studies have implicated rapid events such as the induction of the pro-apoptotic protein Noxa in CLL cells [33] or the activation of JNK in ML-1 cells [21]. However, in preliminary studies, we have failed to observe any acute effects on Bcl-2 family protein expression or JNK pathway activation (unpublished observations). Recently, interference with intracellular protein trafficking has been suggested as a possible mechanism to explain the interphase effects of MTAs. One study showed that in prostate cancer cells, taxanes act, at least in part, by inhibiting nuclear transport of the androgen receptor (AR), and hence block AR-mediated signaling [34]. It was also recently reported that MTAs interfere with the trafficking of DNA repair proteins on interphase microtubules, providing a mechanistic explanation for the long-standing observation that MTAs and DNA damaging agents act synergistically [35]. We observed that the induction of apoptosis in primary ALL cells in G1 phase was not initiated until 24 h after vincristine treatment (Fig. 2B). Thus the response is delayed rather than acute, and as such is more compatible with a mechanism involving interference with protein trafficking than one involving rapid induction of a death stimulus.
The results of this study have several important implications. Firstly, MTAs are well established as an effective therapy for many cancer types, many of which are relatively slow-growing with low mitotic indices [12,36]. Our finding that vincristine also targets cells in G1 phase provides a plausible explanation for these clinical observations and a potential solution to this paradox. Secondly, the results suggest that primary ALL cell cultures, and perhaps other primary or non-immortalized tumor cell types, represent unique tools to uncover novel mechanisms of MTA action with clinical relevance. Thirdly, our observation that relatively low and clinically relevant concentrations of vincristine can kill cells in G1 phase adds to a growing body of evidence that microtubules contribute to fundamental cellular processes outside of mitosis [37]. Finally, understanding drug mechanism is critically important in rational drug design and discovery, in the testing and development of novel drug combinations, and in patient stratification. Conversely, a lack of understanding of clinical mechanisms can lead to the development of ineffective drugs [12], resulting in poor utilization of available resources. As we enter an era where customized therapies are under development and may replace standardized regimens, studies designed to elucidate the molecular mechanisms of action of antitumor agents are even more vital.
Supplementary Material
Acknowledgments
We thank Dr. Fred Falkenburg for providing the ALL cell cultures, Andrea Harris and Debopam Ghosh for their assistance with optimization of the EdU assay, Dr. Gang Lee for advice on the cell proliferation assay, Dr. Gwen Childs for advice on centrifugal elutriation, Sarah Hardin and Katie Hart for technical assistance, Dr. Joshua Eichhorn for critical review of the manuscript, and Megan Reed for help with the figures.
Grant support: This work was supported by National Institutes of Health Grant CA-109821 from the National Cancer Institute (to T.C. Chambers) and in part by internal funds from UAMS College of Medicine Research Council, UAMS Department of Biochemistry and Molecular Biology, Arkansas Biosciences Institute, and the Center for Microbial Pathogenesis and Host Inflammatory Responses grant P20GM103625 through the NIH National Institute of General Medical Sciences Centers of Biomedical Research Excellence.
Abbreviations
- ALL
acute lymphoblastic leukemia
- DMSO
dimethyl sulfoxide
- EdU
5-ethynyl-2'-deoxyuridine
- IMDM
Iscove's modified Dulbecco's medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSA
human serum albumin
- PARP
poly(ADP-ribose) polymerase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Footnotes
Conflicts of interest: No potential conflicts of interest were disclosed.
References
- 1.Rowinsky EK, Donehower RC. The Chemotherapy Source Book. Baltimore, MD: Williams and Wilkins; 1998. pp. 387–423. [Google Scholar]
- 2.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 3.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- 5.Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012;19:369–377. doi: 10.1038/cdd.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5:2963–2969. doi: 10.1158/1535-7163.MCT-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 8.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–656. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakurikar N, Eichhorn JM, Chambers TC. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem. 2012;287:39193–39204. doi: 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8:244–250. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- 12.Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18:51–63. doi: 10.1158/1078-0432.CCR-11-0999. [DOI] [PubMed] [Google Scholar]
- 13.Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitagawa K. Too early to say, "no targeting of mitosis!". Nat Rev Clin Oncol. 2011;8:444. doi: 10.1038/nrclinonc.2010.228-c1. author reply 444. [DOI] [PubMed] [Google Scholar]
- 15.Tunquist BJ, Wood KW, Walker DH. Tales of how great drugs were brought down by a flawed rationale--letter. Clin Cancer Res. 2013;19:1302. doi: 10.1158/1078-0432.CCR-12-1041. [DOI] [PubMed] [Google Scholar]
- 16.Wissing MD, Carducci MA, Gelderblom H, van Diest PJ. Tales of how great drugs were brought down by a flawed rationale--letter. Clin Cancer Res. 2013;19:1303. doi: 10.1158/1078-0432.CCR-12-2695. [DOI] [PubMed] [Google Scholar]
- 17.Komlodi-Pasztor E, Sackett DL, Fojo T. Tales of how great drugs were brought down by a flawed rationale--response. Clin Cancer Res. 2013;19:1304. doi: 10.1158/1078-0432.CCR-12-2058. [DOI] [PubMed] [Google Scholar]
- 18.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:229ra243. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klotz DM, Nelson SA, Kroboth K, Newton IP, Radulescu S, Ridgway RA, Sansom OJ, Appleton PL, Nathke IS. The microtubule poison vinorelbine kills cells independently of mitotic arrest and targets cells lacking the APC tumour suppressor more effectively. J Cell Sci. 2012;125:887–895. doi: 10.1242/jcs.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stadheim TA, Xiao H, Eastman A. Inhibition of extracellular signal-regulated kinase (ERK) mediates cell cycle phase independent apoptosis in vinblastine-treated ML-1 cells. Cancer Res. 2001;61:1533–1540. [PubMed] [Google Scholar]
- 22.Salerni BL, Bates DJ, Albershardt TC, Lowrey CH, Eastman A. Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed. Mol Cancer Ther. 2010;9:791–802. doi: 10.1158/1535-7163.MCT-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bates DJ, Salerni BL, Lowrey CH, Eastman A. Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute cell cycle phase-independent apoptosis. Cancer Biol Ther. 2011;12:314–325. doi: 10.4161/cbt.12.4.16909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blajeski AL, Phan VA, Kottke TJ, Kaufmann SH. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J Clin Invest. 2002;110:91–99. doi: 10.1172/JCI13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijmeijer BA, Szuhai K, Goselink HM, van Schie ML, van der Burg M, de Jong D, Marijt EW, Ottmann OG, Willemze R, Falkenburg JH. Long-term culture of primary human lymphoblastic leukemia cells in the absence of serum or hematopoietic growth factors. Exp Hematol. 2009;37:376–385. doi: 10.1016/j.exphem.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Alford SE, Kothari A, Loeff FC, Eichhorn JM, Sakurikar N, Goselink HM, Saylors RL, Jedema I, Falkenburg JH, Chambers TC. BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 2015;75:1366–1375. doi: 10.1158/0008-5472.CAN-14-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eichhorn JM, Alford SE, Sakurikar N, Chambers TC. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res. 2014;322:415–424. doi: 10.1016/j.yexcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grdina DJ, Hittelman WN, White RA, Meistrich ML. Relevance of density, size and DNA content of tumour cells to the lung colony assay. Br J Cancer. 1977;36:659–669. doi: 10.1038/bjc.1977.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan M, Goodwin ME, Birrer MJ, Chambers TC. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001;61:4450–4458. [PubMed] [Google Scholar]
- 30.Sakurikar N, Eichhorn JM, Alford SE, Chambers TC. Identification of a mitotic death signature in cancer cell lines. Cancer Lett. 2014;343:232–238. doi: 10.1016/j.canlet.2013.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karr TL, Kristofferson D, Purich DL. Calcium ion induces endwise depolymerization of bovine brain microtubules. J Biol Chem. 1980;255:11853–11856. [PubMed] [Google Scholar]
- 32.Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 33.Bates DJ, Danilov AV, Lowrey CH, Eastman A. Vinblastine rapidly induces NOXA and acutely sensitizes primary chronic lymphocytic leukemia cells to ABT-737. Mol Cancer Ther. 2013;12:1504–1514. doi: 10.1158/1535-7163.MCT-12-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa ST, Bander NH, Nanus DM, Giannakakou P. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71:6019–6029. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poruchynsky MS, Komlodi-Pasztor E, Trostel S, Wilkerson J, Regairaz M, Pommier Y, Zhang X, Kumar Maity T, Robey R, Burotto M, Sackett D, Guha U, Fojo AT. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A. 2015;112:1571–1576. doi: 10.1073/pnas.1416418112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan KS, Koh CG, Li HY. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 2012;3:e411. doi: 10.1038/cddis.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4:153. doi: 10.3389/fonc.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.