

Abstract
Segregation of functional organelles during the cell cycle is crucial to generate healthy daughter cells. In S. cerevisiae, ER stress causes an ER inheritance block to ensure cells inherit a functional ER. Here, we report that formation of tubular ER in the mother cell, the first step in ER inheritance, depends on functional symmetry between the cortical ER (cER) and perinuclear ER (pnER). ER stress induces functional asymmetry, blocking tubular ER formation and ER inheritance. Using fluorescence recovery after photobleaching, we show that the ER chaperone Kar2/BiP fused to GFP and an ER membrane reporter, Hmg1-GFP, behave differently in the cER and pnER. The functional asymmetry and tubular ER formation depend on Reticulons/Yop1, which maintain ER structure. Lunapark1 deletion in rtn1Δrtn2Δyop1Δ cells restores the pnER/cER functional asymmetry, tubular ER generation and ER inheritance blocks. Thus, Reticulon/Yop1-dependent changes in ER structure are linked to ER inheritance during the yeast cell cycle.
TOC image
Piña et al. uncover that Reticulons/Yop1 plays a pivotal role in the yeast ERSU pathway, which ensures inheritance of the functional ER into the daughter cell during the cell cycle. In yeast cells, Reticulons/Yop1 contribute to generating ER stress asymmetry that govern the ER inheritance block induced by ER stress.
Introduction
Eukaryotic cells possess many regulatory and cell cycle checkpoints to ensure proper DNA replication and segregation during the cell cycle (Rhind and Russell, 2012; Lara-Gonzalez et al., 2012; Yasutis and Kozminski, 2013). The loss of such control is an underlying cause of many human diseases, including cancer (Abbas et al., 2013). In contrast, less is known about the regulatory pathways governing inheritance of cytoplasmic components, and few studies have investigated how cell cycle checkpoints ensure transmission of functional organelles, such as the endoplasmic reticulum (ER), to daughter cells.
After translation, linear polypeptides of secretory proteins are translocated into the ER lumen for chaperone-assisted folding and post-translational modifications before exiting the ER (Ron and Walter, 2007; Rutkowski and Kaufman, 2004). When folding demand exceeds ER capacity, known as ER stress, three ER transmembrane protein sensors (IRE1, PERK, and ATF6) initiate the unfolded protein response (UPR) (Walter and Ron, 2011). The UPR re-establishes ER homeostasis by upregulating the transcription of genes encoding ER chaperones, protein folding and modifying components, and lipid-generating enzymes (McMaster, 2001). Importantly, the ER cannot be synthesized de novo and arises only from pre-existing ER, implying that regulatory mechanisms must exist to regulate its inheritance during the cell cycle.
We previously identified a cell cycle surveillance mechanism in S. cerevisiae, termed the ERSU (ER Stress Surveillance) pathway that operates during ER stress to ensure daughter cells inherit functional ER (Babour et al., 2010). The ERSU pathway operates independently of the UPR; instead, the ERSU is centrally regulated by the Slt2 MAP kinase. During ER stress, slt2Δ cells fail to relocalize the septin ring away from the bud neck and the stressed ER enters the daughter cell, ultimately causing death. However, slt2Δ cell growth is rescued by preventing stressed ER entry into the daughter cell, showing that inheritance of stressed ER is the major cause of slt2Δ cell death during ER stress.
The yeast ER exists as two major subdomains: the perinuclear ER (pnER), which surrounds the nucleus, and the cortical ER (cER), which is located at the periphery of the cell in close contact with the plasma membrane. Although the two subdomains are contiguous and physically connected by tubules, they adopt different structures. While the pnER is sheet-like and continuous with the nuclear envelope, the cER is a more distinct structure consisting of interconnected tubules (Hu et al., 2011; Friedman and Voeltz, 2011; de Martin et al., 2005). The mammalian ER also contains sheet-like structures (cisternae) and reticular ER. The ER sheets are connected by a network of polygonal tubules generated from three-way junctions of tubular membranes that extend close to the plasma membrane (English et al., 2009; Goyal and Blackstone, 2013). They are covered by abundant ribosomes and play a key role in the production of secretory proteins. In yeast and mammalian cells, the formation and maintenance of tubular ER requires several proteins: the reticulons and DP1/Yop1, which stabilize the highly curved tubular ER structure (Voeltz et al., 2006); members of the dynamin-related GTPase family such as Atlastin/Sey1 (Wang et al., 2013; Anwar et al., 2012): and antagonistic proteins such as Lunapark1 (Chen et al., 2012). How the cell controls the dynamic ratio of sheet-like and tubular ER structures is currently unknown.
Despite their complexity, both ER subdomains are present in newly generated cells. In yeast, an initial ER tubule emerges from the mother cell pnER, moves along the mother-daughter axis, enters the daughter cell, and then anchors at the bud tip before spreading around the periphery of the daughter cell (Fehrenbacher et al., 2002). In an elegant study using electron tomography, West et al. also showed that tubular ER can emerge from the mother cell pnER, suggesting that this is the initial event for ER inheritance in S. cerevisiae (West et al., 2011). The distinct origins and activities of the pnER and cER described above raise the possibility that the differential functional status of the two ER subdomains might be critical to ER tubule formation and ER inheritance under both normal and ER stress conditions. Here, we addressed this question by examining differences in pnER and cER function and its relationship to ER tubule formation and the block in ER inheritance during ER stress.
Results
ER stress is induced differentially in the cortical and perinuclear ER
We previously showed that in yeast ER stress blocks cER inheritance, but the pnER is transmitted normally to the daughter cell (Figures 1A and S1A) (Babour et al., 2010). These findings suggested that ER stress inducers might have different effects on the cER and pnER. To investigate this, we analyzed the dynamics of Kar2/BiP-sfGFP, a major ER luminal chaperone, using fluorescence recovery after photobleaching (FRAP) assays (Lajoie et al., 2012; Lai et al., 2010). In response to ER stress, Kar2/BiP binding to unfolded client proteins increases, reducing its mobility within the ER lumen (Snapp et al., 2006). Therefore, the rate of Kar2-sfGFP fluorescence recovery after photobleaching in the pnER or cER is a direct measure of Kar2/BiP mobility, and thus of the ER stress level in that compartment. To induce ER stress, we treated wild type (WT) yeast cells with tunicamycin (Tm), an N-glycosylation inhibitor that causes accumulation of unglycosylated unfolded proteins in the ER, and photobleached discrete regions of the cER or pnER and monitored fluorescence recovery over time. Tm-treated cells showed delayed fluorescence recovery in both the cER and pnER compared with DMSO-treated cells, although the magnitude and kinetics of the effect were markedly different in the two compartments (Figures 1B–1E). The reduced rate of Kar2-sfGFP fluorescence recovery was detected in the cER within 30 min of Tm treatment, and the effect was further increased in cells exposed to Tm for 3 hr (Figures 1B and 1D). In contrast, Tm had little effect on Kar2-sfGFP mobility in the pnER at 30 min, and a small but significant reduction in mobility was noted only after 3 hr incubation with Tm (Figures 1C and 1E). Kar2-sfGFP mobility in the cER and pnER of DMSO-treated cells was essentially identical (Figures 1B–1E). These experiments suggested a fundamental difference in the effects of ER stress on the behavior of the same chaperone protein in the cER and pnER.
Figure 1. ER stress has a greater effect on Kar2-sfGFP mobility in the cER than in the pnER.
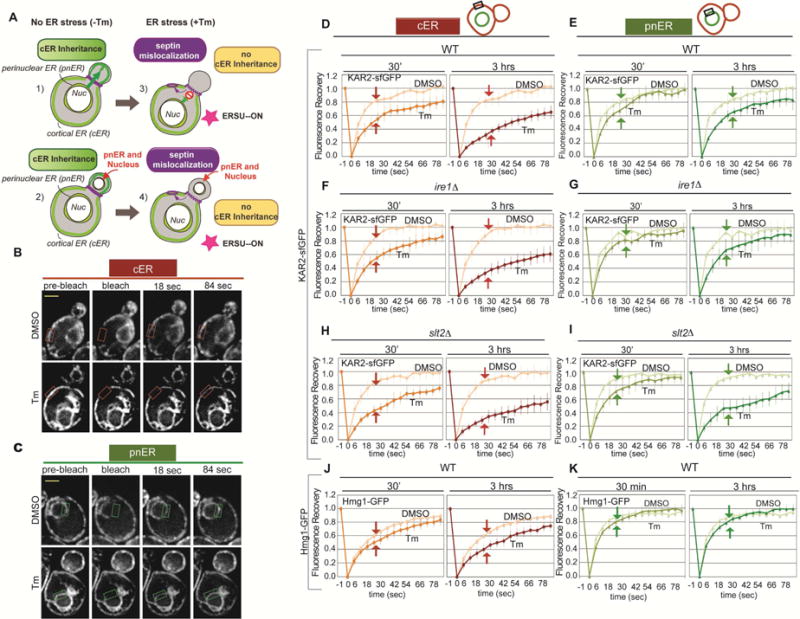
(A) Illustration of the effects of ER stress on tubule formation and cER and pnER inheritance. Under unstressed conditions, an ER tubule formed from the pnER moves from the mother into the bud where it forms the cER (1). This is followed by nuclear migration (2). Under conditions of ER stress, tubule formation is abnormal and the cER is not inherited (3), generating buds that contain the pnER but no cER (4).
(B and C) Representative image of WT cells expressing Kar2-sfGFP. FRAP analysis monitored Kar2-sfGFP mobility in the cER (B) or pnER (C) of DMSO or Tm (1 μg/ml, 3hr) treated cells. Images were acquired before (pre-bleach), at the same time as (bleach), and at 18 or 84 sec after photobleaching. The boxed area shows the cER (orange box) and pnER (green box) photobleached. Scale bar is 2 μm.
(D and E) Fluorescence intensity was normalized to the pre-bleach signal and recovery was plotted over time for the cER (D) and pnER (E) of WT cells treated with DMSO or Tm for 30 min or 3 hr. Graphs are the mean ± SD of 3 experiments, each examining ≥7 cells.
(F and G) Kar2-sfGFP mobility in the cER and pnER of ire1Δ cells is similar to WT cells. (H and I) slt2 deletion reduces Kar2-sfGFP mobility in the cER and pnER of stressed cells. (J and K) As described for D and E except the experiments were performed with WT cells expressing Hmg1-GFP. Experiments in F–K were performed as in D and E. See also Figures S1 and S2.
Slt2, but not Ire1, is required to maintain Kar2/BiP immobility in the pnER during ER stress
Previously, we reported that the ERSU and UPR pathways are distinct and that the UPR was not involved in the ER stress-induced ER inheritance block (Babour et al., 2010). Activation of the UPR requires the ER transmembrane receptor kinase/endoribonuclease Ire1 (Cox et al, 1993; Mori et al, 1993), whereas the MAP kinase Slt2 activates the ERSU pathway, which does not require Ire1 (Babour et al., 2010). To test whether Kar2-sfGFP mobility in the cER and pnER is differentially regulated by the ERSU and UPR pathways, we performed FRAP assays in ire1Δ and slt2Δ cells (Figures 1F–1I; Figures S1B and S1C). There were no differences in the cER or pnER Kar2-sfGFP fluorescence recovery of unstressed WT, slt2Δ, and ire1Δ cells (Figures 1D–1I, DMSO). Kar2-sfGFP mobility was also similar in the cER of Tm-treated WT, slt2Δ, and ire1Δ cells (Figures 1D, 1F, and 1H). However, a striking difference was noted in the pnER analyses: Tm treatment for 3 hr caused a marked reduction in Kar2-sfGFP mobility in the pnER of slt2Δ cells but not WT or ire1Δ cells (Figures 1E, 1G, and 1I). In slt2Δ cells, Tm had very similar effects on Kar2/BiP-sfGFP mobility in the cER and pnER (Figures 1H and 1I). Since slt2Δ cells do not block ER inheritance under ER stress conditions, unlike WT and ire1Δ cells, these data point to a potential link between the functional state of the pnER and the ability to halt ER inheritance during ER stress.
To ensure that Kar2-sfGFP fluorescence recovery faithfully reflects its association with unfolded proteins, we performed similar FRAP experiments with cells expressing kar2-1, a temperature-sensitive KAR2 mutation that disrupts its ability to bind unfolded proteins (Kabani et al., 2003) (Figures S2A and S2B). Indeed, there was little difference in kar2-1-sfGFP fluorescence recovery in the pnER of control and ER-stressed cells, but there was a small but notable delay in recovery detected in the cER of stressed cells (Figures S2A and S2B). This observation suggested that at least a portion of the delay in Kar2-sfGFP fluorescence recovery was due to an unknown, chaperone-independent effect of ER stress on mobility. To further test this idea, we examined the mobility of Hmg1-GFP, a fusion protein carrying a single ER transmembrane domain of the non-chaperone ER protein Hmg1 (Hampton et al., 1996). As we observed with the mutant kar2-1-sfGFP protein, ER stress (Tm) caused a small delay in Hmg1-GFP fluorescence recovery in the cER, but not in the pnER (Figures 1J, and 1K; Figure S2C). While the relationship between the FRAP response and functional status of ER chaperones has been well-documented, the kar2-1-sfGFP and Hmg1-GFP results suggest that a small portion of the delay in Kar2-sfGFP fluorescence recovery in the stressed cER is independent of its chaperone activity and instead reflects the ER status. Collectively, these experiments demonstrate that ER stress has differential effects on the cER and pnER, and that the loss of Slt2 affects only the function of the pnER.
The cER and the pnER remain interconnected during ER stress
The differential mobility of the same protein in the cER and pnER during ER stress was surprising because the two compartments are thought to be contiguous and interconnected; therefore, Kar2/BiP is expected to travel freely throughout the network. To examine if ER stress causes a disconnection between the pnER and cER, we performed fluorescence loss in photobleaching (FLIP) experiments. We found that after repeatedly photobleaching a small region of the cER, Kar2-sfGFP fluorescence in the pnER and a remote region of the cER decayed at a similar rate in DMSO- and Tm-treated cells, and eventually all fluorescence was lost (Figures 2A and 2B). These data demonstrate that the pnER and cER remain interconnected during ER stress and that Kar2-sfGFP mobility differences in the two compartments are not due to physical separation.
Figure 2. The ER remains connected and distribution of CPY*-mRFP, GFP-CFTR, and Ire1-GFP aggregates is similar between the cER and pnER in unstressed and ER stressed cells.

(A and B) Fluorescence loss in photobleaching (FLIP) analysis of Kar2-sfGFP-expressing WT cells incubated with DMSO or 1 μg/ml Tm for 30 min. A region in the cER was photobleached (black rectangle) and the loss of fluorescence in the cER (orange rectangle) or pnER (green rectangle) was monitored. Average fluorescence depletion curves normalized to the pre-bleach fluorescence intensity are shown. Graphs are the mean ± SD of 3 experiments, each examining ≥5 cells.
(C) WT cells expressing the Hmg1-GFP ER reporter were grown in synthetic media with 2% galactose for 2 hr (with DMSO or 1 μg/ml Tm) to induce CPY*-mRFP expression.
(D) WT cells expressing the ER reporter DsRed-HDEL were incubated with 100 μM copper sulfate for 2 hr (with DMSO or 1 μg/ml Tm) to induce GFP-CFTR expression.
(E) WT cells expressing the DsRed-HDEL ER reporter and Ire1-GFP were treated with DMSO or Tm (1 μg/ml) for 1 or 3 hr.
In all experiments, foci were quantified and shown by the ratio of foci in the pnER to that in cER per 50 μm2 surface area (SA) and are the mean ± SD of 3 experiments, each examining ≥100 cells. Scale bar is 2 μm.
ER stress-induced protein aggregates accumulate in both the cER and pnER
Two potential scenarios could explain why ER stress did not affect Kar2/BiP fluorescence recovery in the pnER: (1) the pnER could contain significantly fewer unfolded proteins than the cER, and (2) unfolded proteins could be generated in the pnER, but Kar2-sfGFP might not dissociate from Ire1 and thus be unable to bind unfolded proteins, as suggested by a recent study (Ishiwata-Kimata et al., 2013). If true, the latter scenario would suggest that pnER-localized and cER-localized Kar2-sfGFP acquire different properties during ER stress. To test the first possibility, we examined the formation of CPY*-mRFP or GFP-CFTR protein aggregates in each ER subdomain during ER stress (Kakoi et al., 2013; Fu and Sztul, 2003; Pina and Niwa, 2015). CPY*-mRFP aggregates activate both the UPR and ERSU pathways, whereas GFP-CFTR aggregates do not (Pina and Niwa, 2015). This difference allows us to observe the cER and pnER localization pattern of aggregates that do or do not induce ER stress. In both CPY*-mRFP- and CFP-CFTR-expressing cells, approximately twice as many foci were present in the pnER than in the cER. This ratio did not change in the presence of Tm, indicating that unfolded proteins are abundant in the pnER and that the ratio of protein aggregates in the pnER to the cER does not decrease under ER stress (Figures 2C and 2D). To test the second possibility, we monitored the distribution of Ire1 in the cER and pnER to examine UPR activation. During ER stress, activated Ire1 is released from Kar2/BiP and autophosphorylates, forming oligomers that can be detected as foci in cells expressing Ire1-GFP (Aragon et al., 2009; Kimata et al., 2007). Thus, the presence of Ire1 foci is a measure of Kar2/BiP–Ire1 dissociation. Using DsRed-HDEL as an ER reporter, we found that Ire1-GFP was distributed throughout the ER in unstressed cells, but discrete Ire1-GFP foci were evident in both the pnER and cER within 1 hr and persisted for at least 3 hr after Tm treatment (Figure 2E). Furthermore, Ire1-GFP foci were twice as abundant in the pnER as in the cER, as was observed for CYP* and CFTR aggregates. Taken together, these observations indicate that ER stress-induced protein aggregates are abundant in the pnER and that the difference in Kar2/BiP mobility in the two compartments during ER stress does not reflect a lack of unfolded proteins or a perceived lack of ER stress in the pnER.
ER–plasma membrane tethering proteins are not involved in the differential behavior of the cER and the pnER or in activation of the ERSU
Our results so far indicate that the differential effect of ER stress on the cER and pnER functional state, as reflected by Kar2-sfGFP mobility, was dependent on the ERSU (Slt2), independent of the UPR (Ire1), and was not caused by differences in unfolded proteins levels or a physical disconnection between the cER and pnER. We next hypothesized that ER stress might induce distinct structural changes in the two ER domains. Indeed, there are known differences in structural elements in the pnER and cER. For example, certain areas of the cER directly connect with the plasma membrane (PM) via tethering proteins such as lst2, Tcb1, Tcb2, Tcb3, Scs2, Scs22 (Manford et al., 2012), whereas the pnER does not form such contacts. Therefore, we examined Kar2-sfGFP mobility in a yeast strain lacking lst2, Tcb1, Tcb2, Tcb3, Scs2, Scs22, labeled Δtether. In the Δtether strain, the cER structure is severely altered such that it is no longer juxtaposed to the PM but is present in the middle of the cytoplasm (Figure S3A), as reported (Manford et al., 2012). However, we found that, like WT cells, Kar2-sfGFP mobility in the pnER of Δtether cells remained high compared with that in the cER (Figures 3A–3D; Figure S3B). Similar results were found using Δtether cells expressing Hmg1-GFP (Figures 3E–3H). Finally, stressed Δtether and WT cells blocked ER inheritance similarly (Figures 3I–3J). These findings indicate that the different cER and pnER functional responses cannot be explained by tethering protein-dependent structural differences, and that the tethering proteins are not required for the ERSU pathway.
Figure 3. Deletion of ER–plasma membrane tethering proteins has no effect on ER protein mobility or ER inheritance in response to ER stress.
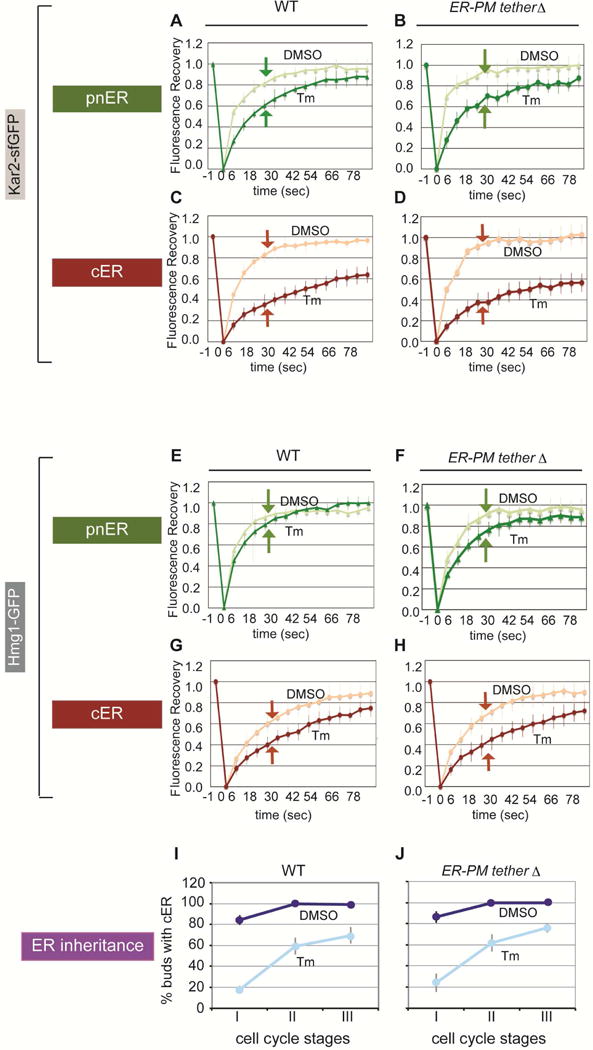
(A and C) Kar2-sfGFP-expressing WT cells treated with DMSO or 1 μg/ml Tm 3hr before FRAP analysis on the pnER (A) or cER (C).
(B and D) As described for A and C except Kar2-sfGFP FRAP analysis was performed on cells lacking six tethering genes (Δtether cells).
(E–H) As described for A–D except cells expressed Hmg1-GFP.
(I and J) Quantitation of cER inheritance in WT cells (I) and Δtether mutants (J) expressing Hmg1-GFP. Cells were treated with DMSO (dark blue) or 1 μg/ml Tm (pale blue).
For A–H, Graphs represent the mean ± SD of 3 experiments, each examining ≥7 cells. For I and J, graphs show the mean ± SD of 3 experiments, each with ≥200 cells counted. See also Figure S3.
Loss of Reticulons/Yop1 differentially affects Kar2-sfGFP mobility in the cER and pnER
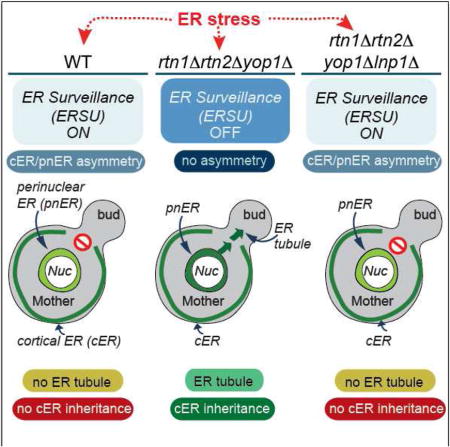
As described earlier, the ER network is composed of tubules and sheets. In both yeast and mammalian cells, ER sheets are found juxtaposed to the nucleus, whereas tubular ER is peripherally located away from the nucleus and close to the PM. The high membrane curvature of the ER is stabilized by two reticulon proteins, Rtn1 and Rtn2, and a reticulon-like protein, DP1/Yop1 (Stefano et al., 2014; Chiurchiu et al., 2014; Goyal and Blackstone, 2013; Hu et al., 2011; Friedman and Voeltz, 2011; English et al., 2009). To determine whether an intact cER tubular structure is necessary to establish ER stress-induced functional asymmetry between the cER and pnER, we examined Kar2-sfGFP mobility in cells lacking RTN1, RTN2, and YOP1. rtn1Δrtn2Δyop1Δ cells form extended sheets of cER, rather than the fine reticular structure seen in WT cells, which remain juxtaposed to the PM (Figure S3A) (Voeltz et al., 2006; De Craene et al., 2006; Hu et al., 2008; Shibata et al., 2008; Hu et al., 2009; West et al., 2011). In contrast to WT cells, we found that Kar2-sfGFP fluorescence recovery decreased to similar extents in the pnER and cER of Tm-treated rtn1Δrtn2Δyop1Δ cells, closely resembles the phenotype of slt2Δ cells (Figures 4A–C, 4F–H, and S3C). Kar2-sfGFP mobility decreased similarly in the cER of stressed WT and rtn1Δrtn2Δyop1Δ cells (Figures 4F, 4H, and S3C). Similar results were obtained with the Hmg1-GFP reporter in rtn1Δrtn2Δyop1Δ and slt2Δ cells (Figure S4A and S4B), indicating that the mobility of chaperone and non-chaperone pnER-resident proteins was affected. These data point to a central role for the reticulons and DP1/Yop1 in dictating how the function of the cER and pnER is affected by ER stress. Furthermore, the data indicate that ER stress has effects on ER function and inheritance that go beyond influencing the behavior of chaperones in the lumen and extend to altering the structure and/or composition of the ER.
Figure 4. The ER stress response and ER inheritance are defective in rtn1Δrtn2Δyop1Δ cells and this is rescued by deletion of Lnp1.
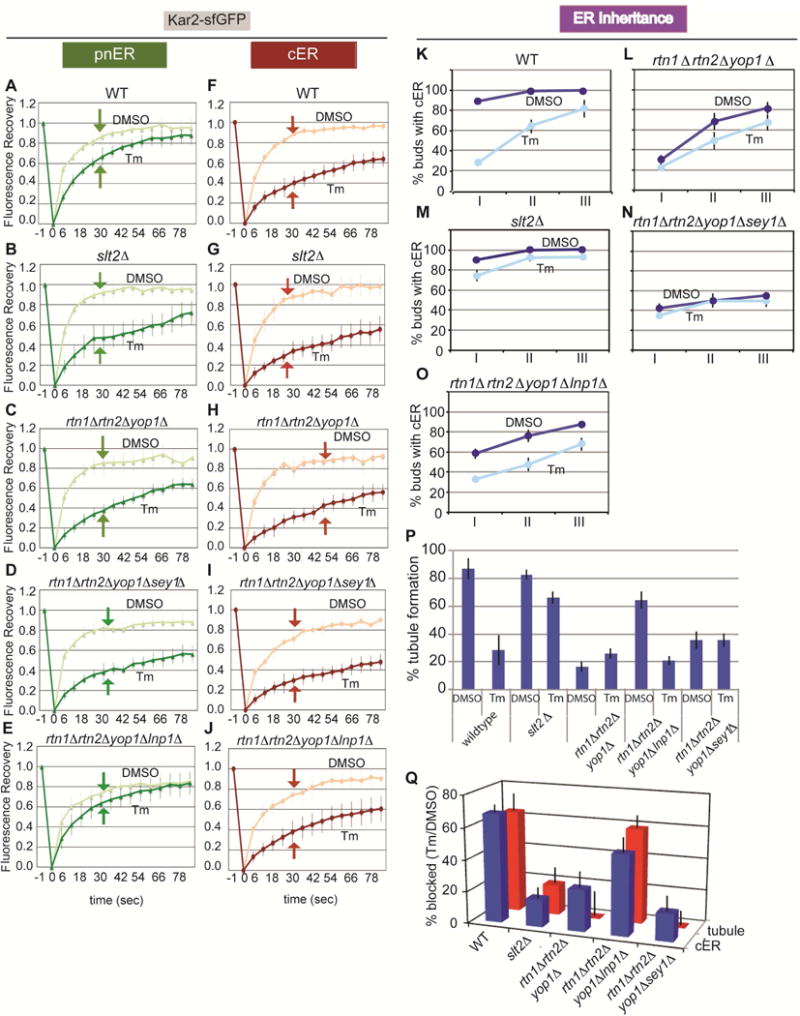
(A–J) FRAP analysis of the pnER and cER of Kar2-sfGFP-expressing WT (A and F), slt2Δ (B and G), rtn1Δrtn2Δyop1Δ (C and H), rtn1Δrtn2Δyop1Δsey1Δ (D and I), and rtn1Δrtn2Δyop1Δlnp1Δ cells (E and J). Graphs are the mean ± SD of 3 experiments, each examining ≥7 cells.
(K–O) Quantification of cER inheritance in WT (K), rtn1Δrtn2Δyop1Δ (L), slt2Δ (M), rtn1Δrtn2Δyop1Δsey1Δ (N), and rtn1Δrtn2Δyop1Δlnp1Δ cells (O) expressing Hmg1-GFP. Cells were treated with DMSO (dark blue) or Tm (pale blue) and cER inheritance was scored as described in Figure 3I–J. Graphs show the mean ± SD of 3 experiments, each with ≥200 cells counted.
(P) Quantitation of early ER tubule formation in WT, slt2Δ, rtn1Δrtn2Δyop1Δ, rtn1Δrtn2Δyop1Δlnp1Δ, and rtn1Δrtn2Δyop1Δsey1Δ cells expressing Hmg1-GFP. Cells were treated with DMSO or Tm for 3 hr. Quantitations are the mean ± SD of 3 experiments, each analyzing ≥40 cells.
(Q) Correlation of the block in initial tubule formation [100 – 100 × (% tubule formation with Tm)/(% tubule formation with DMSO)] (Figure 4K–4O) and cER inheritance [100 – 100 × (% cER inheritance with Tm)/(% cER inheritance with DMSO)] (Figure 4P) for WT, slt2Δ, rtn1Δrtn2Δyop1Δ, rtn1Δrtn2Δyop1Δlnp1Δ, and rtn1Δrtn2Δyop1Δsey1Δ cells expressing Hmg1-GFP. The data represent the mean ± SD of 3 experiments. See also Figures S3 and S4.
rtn1Δrtn2Δyop1Δ cells are defective in the formation of tubular ER from the pnER
Since we observed a similar reduction in Kar2/BiP mobility in the pnER of stressed slt2Δ cells and rtn1Δrtn2Δyop1Δ cells (Figures 4B, 4C, 4G, and 4H), we asked whether rtn1Δrtn2Δyop1Δ cells had also lost the ability to prevent transmission of a dysfunctional ER to the daughter cells. Compared to WT cells, ER inheritance in rtn1Δrtn2Δyop1Δ cells was reduced even under normal growth conditions, indicating the importance of reticulons for normal ER inheritance. However, ER stress had no further effect on ER inheritance in these cells (Figures 4K and 4L); a phenotype also observed in stressed slt2Δ cells (Figure 4M). These observations revealed an intriguing correlation between Kar2/BiP mobility in the pnER and the ability to block cER inheritance in response to ER stress.
To probe this further, we examined the association between the function of the pnER and its ability to form ER tubules. For this, we quantified the appearance of tubular ER from the pnER in WT, slt2Δ, and rtn1Δrtn2Δyop1Δ cells. While more than 80% of unstressed WT cells showed evidence of early tubular ER formation, ER stress reduced this to ~30%, consistent with the block in ER inheritance under these conditions (Figures 4K and 4P, Figure S3E). In contrast to WT cells, a high percentage of slt2Δ cells formed pnER tubules in stressed and unstressed cells. Moreover, ER inheritance was also unaffected by ER stress in slt2Δ cells (Figures 4M and 4P). Similarly in rtn1Δrtn2Δyop1Δ cells, although tubular ER formation and ER inheritance were much lower than in slt2Δ or WT cells under normal growth conditions, ER stress did not further decrease ER inheritance or tubule formation (Figures 4L, 4P, and S3E). Although the emergence of tubular ER and ER inheritance were quantitatively different in rtn1Δrtn2Δyop1Δ and slt2Δ cells, ER stress had no effect on either function in both mutant strains. Collectively, these findings suggest that the functional status of the pnER plays a pivotal role in determining the formation of initial tubular ER from the pnER and that the ability to block ER inheritance in response to ER stress depends on the function of the reticulons and Yop1.
Loss of Lunapark 1 restores Kar2/BiP mobility in the pnER and the ability to activate the ERSU pathway during ER stress
Our results thus far suggest two intriguing hypotheses: (1) the pnER functional state regulates the initial ER tubule formation for ER inheritance and (2) ER stress induces the different functional states of the pnER and cER. The formation and maintenance of ER sheet-like (pnER) and reticular (cER) structures are regulated by a balance between the activity of the reticulons/Yop1 and two other structural proteins: Lunapark 1 (Lnp1) and the dynamin-like GTPase Sey1/Atlastin. Lnp1 and Sey1 are reported to play roles in forming the proper ER network by interacting with Rtn1, Rtn2, and Yop1 (Chen et al., 2012; Hu et al., 2009). Lnp1 and Sey1 both localize to three-way junctions where they act antagonistically (Anwar et al., 2012; Chen et al., 2012; Chen et al., 2015). In yeast cells, simultaneous loss of Sey1 and reticulons/Yop1 results in less branched tubular ER, while deletion of Lnp1 alone results in densely reticulated ER in yeast cells and formation of more sheet-like ER in mammalian cells (Chen et al., 2012; Shemesh et al., 2014; Chen et al., 2015; Hu et al., 2009). The localization of Sey1 and Lnp1 is also interdependent, in that Sey1 accumulates throughout the cER when Lnp1 is absent and Lnp1 localizes throughout the cER and pnER when Sey1 is absent (Chen et al., 2012). Thus, the defects observed in the reticulon/Yop1 mutants could result from a loss of balance in the relative functions of Lnp1 and Sey1 in tubule and three-way junction formation.
To test this, we asked whether perturbation of the Lnp1/Sey1 balance in rtn1Δrtn2Δyop1Δ cells could restore a WT response to ER stress; that is, the differential cER/pnER functional responses, inhibition of initial tubular ER formation, and reduction in ER inheritance. FRAP analysis showed that SEY1 deletion had no effect on Kar2sfGFP mobility in the cER and pnER in rtn1Δrtn2Δyop1Δ cells (Figures 4D and 4I), or on cER inheritance (Figure 4N), or tubule formation (Figure 4P) even in the absence of ER stress. Although the ER was present in the bud of a small percentage of rtn1Δrtn2Δyop1Δsey1Δ cells, it failed to spread throughout the bud cortex. In addition, Tm treatment did not further decrease ER inheritance or tubule formation, similar to what we see for unstressed rtn1Δrtn2Δyop1Δsey1 cells. In contrast, we found that deletion of LNP1 in rtn1Δrtn2Δyop1Δ cells rescued the WT phenotype with respect to Kar2-sfGFP mobility in the pnER (Figures 4E, 4J, and S3D), tubular ER formation from the pnER (Figures 4P and S3E), and the ER inheritance block upon ER stress (Figure 4O). To determine whether there is a direct relationship between initial tubule formation and cER inheritance, we graphed the block in initial tubule formation (Figure 4Q). The plot shows that both the initial ER tubule formation and cER inheritance are effectively blocked by ER stress in WT and rtn1Δrtn2Δyop1Δlnp1Δ cells. In contrast, only small reductions in these events are seen in stressed slt2Δ, rtn1Δrtn2Δyop1Δ, and rtn1Δrtn2Δyop1Δsey1Δ cells. Thus, tubule formation and cER inheritance are closely linked and are regulated by a precise balance in the function of ER-shaping proteins. Interestingly, we found that ~20% of rtn1Δrtn2Δyop1 and rtn1Δrtn2Δyop1Δsey1Δ cells displayed a block in cER inheritance but not in initial tubule formation. These observations suggest the presence of an additional regulatory determinant(s) that contributes to the ER stress-induced cER inheritance block even when initial ER tubule formation proceeds. Taken together, the data presented here point to a critical role for the functional status of the pnER in the regulation of ER inheritance.
Discussion
We previously identified the ERSU pathway as a cell cycle regulatory checkpoint that prevents ER inheritance and halts cytokinesis during ER stress (Babour et al., 2010; Bicknell et al., 2007). In this study, we probed the structure/function relationships underlying the ER stress-induced block in cER inheritance and show that the initial emergence of tubular ER from the pnER is a key regulatory event. We found that the functional asymmetry in the stress response of the cER and pnER correlated with a block in initial ER tubule formation. Thus, ER stress diminished the mobility of two ER-resident proteins, the chaperone Kar2/BiP and the transmembrane protein Hmg1, in the cER but not in the pnER, despite a similar level of unfolded proteins and free exchange of proteins between the two ER subdomains. Furthermore, genetic manipulations that eliminated functional asymmetry with respect to Kar2 and Hmg1, such as deletion of SLT2 or RTN1, RTN2, and YOP1, also eliminated the ability of ER stress to modulate the initial ER tubule formation and subsequent ER inheritance. Collectively, these data highlight a role for the pnER structural and functional response to ER stress in regulating the ER inheritance block.
Surprisingly, we found that ER stress had little effect on Kar2-sfGFP mobility in the pnER and this lack of effect may enable the initial tubule formation block. The slower Kar2 mobility during ER stress could largely be attributed to its interaction with unfolded proteins (Lajoie et al., 2012). However, the kar2-1 and Hmg1 FRAP results support the conclusion that other aspects of ER function contribute to the reduced chaperone mobility and, by inference, to ERSU pathway activation. Further support for this idea comes from our previous result that UPR deficient cells (e.g., ire1Δ cells) can still activate the ERSU pathway in response to ER stress (Babour et al., 2010). Our data also suggest that the ER architecture can affect the mobility of ER components, since loss of RTN1, RTN2, and YOP1 eliminates the asymmetric behavior mobility of Kar2 and Hmg1 in the cER and pnER. If shown to be true, such an observation would suggest that the cell might continuously monitor the balance between sheet-like and tubular ER structures to establish the fate of the ER during the cell cycle (Figure S4C). In this regard, Slt2 activation is required to establish functional asymmetry between the cER and the pnER and to inhibit tubular ER emergence in response to ER stress, possibly by directly or indirectly acting on Rtn1/2, Yop1, Sey1, or Lnp1 (Figure S4C and S4D). Interestingly, Slt2 activity has been reported to increase transiently in the M phase of the cell cycle (Li et al., 2010), and this increase might provide the initial signal for the rapid establishment of functional asymmetry. Under conditions of ER stress, perhaps Slt2 remains activated to ensure that ER inheritance is blocked until its function recovers sufficiently for the cell to re-engage the cell cycle. When that occurs, Slt2 activity would decrease, allowing ER tubule formation and ER inheritance to proceed.
One key question remaining is how architectural differences in the pnER and cER affect their ability to respond to ER stress. Our results with rtn1Δrtn2Δyop1Δ, rtn1Δrtn2Δyop1Δsey1Δ, and rtn1Δrtn2Δyop1Δlnp1Δ cells point to the importance of a balance between reticular and sheet-like structures. RTNs and Yop1 localize preferentially to the cER to maintain the ER tubular structure (Stefano et al., 2014; Chiurchiu et al., 2014; Goyal and Blackstone, 2013; Hu et al., 2011; Friedman and Voeltz, 2011; English et al., 2009). Lnp1 and Sey1 act at three-way junctions in the peripheral ER and at junctions between the reticular and pnER (Chen et al., 2012) and function together to balance the formation of the reticular network. Although the functional relationships between Lnp1 and Sey1 are incompletely understood, it is clear that deletion of either protein alone results in contrasting ER morphology phenotypes, suggesting that they have opposing functions in three-way junction formation and stabilization (Chen et al., 2012). We propose that one function of these ER-shaping proteins is to generate the initial pnER tubule required for ER inheritance during the normal cell cycle. Our results show that rtn1Δrtn2Δyop1Δ cells are less able to form the initial pnER tubule, even under normal growth conditions, but this can be restored, at least in part, by LNP1 deletion. Furthermore, during ER stress, a specific balance between the structural functions of Reticulons/Yop1, Sey1 and Lnp1 is required to establish the block in ER tubule formation and ER inheritance, as demonstrated by the ability of LNP1 deletion, but not of SEY1 deletion, in rtn1Δrtn2Δyop1Δ cells to restore the block in both functions. Although reticulons are enriched in the cER, our study suggests that they also accumulate at specific locations within the sheet-rich structure of the pnER in order to support ER tubule formation, which must occur in a spatially and temporally regulated fashion. Recent studies have shown that loss of Reticulons/Yop1 causes defects in nuclear pore complex formation and spindle pole body formation (Dawson et al., 2009; Casey et al., 2015; Shemesh et al., 2014; Casey et al., 2012), suggesting that at least some Reticulons/Yop1 are present in close proximity to the nuclear membrane and/or the pnER.
The observed effects of Reticulon/Yop1 deficiency may have important ramifications for understanding the contribution of these proteins to human disease. Recently, it was reported that RTN1 is overexpressed in humans with diseased kidneys, and its expression inversely correlated with renal function. Moreover, overexpression of the RTN1A isoform caused ER stress in isolated kidney cells, and knockdown of RTN1A attenuates ER stress and renal fibrosis in mice (Fan et al., 2015). In addition, a subset of human hereditary spastic paraplegias is caused by mutations in RTN-2 and Atlastin-1, which also regulates ER morphology (Goyal and Blackstone, 2013; Hu et al., 2009; Chang et al., 2013). Interestingly, Reticulon-4a regulates the ER chaperone protein disulfide isomerase and protects against neurodegeneration in a mouse model of amyotrophic lateral sclerosis (Yang and Strittmatter, 2007). We do not yet have a clear understanding of the molecular mechanisms by which changes in Reticulons/Yop1 contribute to such diseases. However, these examples do highlight the intimate relationship between altered RTN function and the capacity of the cell to cope with ER stress, and raise the possibility that some disease phenotypes might be due to defects in RTN/Yop1-dependent coordination of ER architecture and function.
Experimental Procedures
Yeast strains, plasmids, and primers used are listed in Supplemental Experimental Material.
FRAP and FLIP assays
Cells were grown in filter-sterilized 0.5× YPD (0.5% yeast extract, 1% peptone, and 2% dextrose) and treated with dimethyl sulfoxide (DMSO) or tunicamycin (Tm 1 μ/ml) for 30 min or 3 hr at 30°C. Cells were transf erred to 1.6% agarose pads made with 0.5× YPD ± 1 μg/ml Tm and maintained at 30°C for the duration of the experiment. Photobleaching was achieved with one 0.2-sec pulse from a 488-nm argon laser set to 50% power and images were acquired immediately before and at 6-sec intervals after photobleaching. The data represent the mean ± SD of 3 experiments, each examining at least 7 cells. For FLIP experiments, photobleaching was achieved with one 0.4-sec pulse from a 488-nm argon laser set to 50% power, after which two images were captured at 10-sec intervals before the next round of photobleaching for the duration of the experiment. Average fluorescence recovery curves were obtained by averaging the fluorescence recovery values, after normalized to neighboring non-photobleached cells to account for fluorescence loss during image acquisition, as described previously (Fleming et al., 2010). The data represent the mean ± SD of 3 experiments, each examining at least 5 cells.
ER inheritance and ER aggregate assays
ER inheritance and ER aggregate formation was imaged and analyzed as described previously (Pina and Niwa, 2015; Babour et al., 2010). The graphs represent the mean ± SD of 3 experiments, each analyzing ≥200 cells for cER inheritance or ≥100 cells for aggregate formation. See Supplemental Experimental Procedures for details.
Ire1-GFP foci formation assay
MNY2704 cells were grown to mid-log phase in YPD and treated with DMSO or 1 μg/ml Tm. Cells were imaged after 1 hr or 3 hr of treatment. The graphs represent the mean ± SD of 3 experiments, each analyzing ≥100 cells.
Supplementary Material
Highlights.
The cortical ER and peri-nuclear (pn) ER respond to ER stress asymmetrically
Reduced asymmetry of the ER stress response blocks initial tubular ER formation
Initial tubule formation from the pnER is a key determinant of ER inheritance
Reticulons/Yop1 govern the asymmetric ER stress response and ER inheritance block
Acknowledgments
We thank Drs. Susan Ferro-Novick, for the rtn1Δrtn2Δyop1Δlnp1Δ and rtn1Δrtn2Δyop1Δsey1Δ strains, Scott Emr for Δtether strain and William A. Prinz for the rtn1Δrtn2Δyop1Δ strain, Jeff Brodsky for kar2-1 strain and Erik Snapp for Kar2-sfGFP plasmid. This work was supported by grants from the NIH (R01GM087415) and American Cancer Society (118765-RSG-10-027-01-CSM) to M.N., and NIH (RO1GM57045) to K.P. and F. J. P. was supported by an NIH 5T32AI007469-20UCSD/LIAI allergy postdoctoral training grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, M.N. and F.J.P.; Methodology, M.N., K.P., T.F.; Investigation, F.J.P.; Writing, M.N. and F.J.P.; Funding Acquisition, M.N. and K.P.; Resources, M.N. and K.P.; Supervision, M.N., K.P. and T.F.
References
- Abbas T, Keaton MA, Dutta A. Genomic Instability in Cancer. Cold Spring Harbor perspectives in biology. 2013;5 doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar K, Klemm RW, Condon A, Severin KN, Zhang M, Ghirlando R, Hu J, Rapoport TA, Prinz WA. The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. The Journal of Cell Biology. 2012;197:209–217. doi: 10.1083/jcb.201111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon T, Van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–40. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babour A, Bicknell AA, Tourtellotte J, Niwa M. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell. 2010;142:256–69. doi: 10.1016/j.cell.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell AA, Babour A, Federovitch CM, Niwa M. A novel role in cytokinesis reveals a housekeeping function for the unfolded protein response. The Journal of Cell Biology. 2007;177:1017–27. doi: 10.1083/jcb.200702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey AK, Chen S, Novick P, Ferro-Novick S, Wente SR. Nuclear pore complex integrity requires Lnp1, a regulator of cortical endoplasmic reticulum. Molecular biology of the cell. 2015;26:2833–44. doi: 10.1091/mbc.E15-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey AK, Dawson TR, Chen J, Friederichs JM, Jaspersen SL, Wente SR. Integrity and function of the Saccharomyces cerevisiae spindle pole body depends on connections between the membrane proteins Ndc1, Rtn1, and Yop1. Genetics. 2012;192:441–55. doi: 10.1534/genetics.112.141465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Lee S, Blackstone C. Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14954–9. doi: 10.1073/pnas.1307391110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Desai T, Mcnew JA, Gerard P, Novick PJ, Ferro-Novick S. Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:418–23. doi: 10.1073/pnas.1423026112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Novick P, Ferro-Novick S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nature cell biology. 2012;14:707–16. doi: 10.1038/ncb2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurchiu V, Maccarrone M, Orlacchio A. The Role of Reticulons in Neurodegenerative Diseases. NeuroMolecular Medicine. 2014;16:3–15. doi: 10.1007/s12017-013-8271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TR, Lazarus MD, Hetzer MW, Wente SR. ER membrane‚Äìbending proteins are necessary for de novo nuclear pore formation. The Journal of Cell Biology. 2009;184:659–675. doi: 10.1083/jcb.200806174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene JO, Coleman J, Estrada De Martin P, Pypaert M, Anderson S, Yates JR, 3rd, Ferro-Novick S, Novick P. Rtn1p is involved in structuring the cortical endoplasmic reticulum. Molecular biology of the cell. 2006;17:3009–20. doi: 10.1091/mbc.E06-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martin PE, Novick P, Ferro-Novick S. The organization, structure, and inheritance of the ER in higher and lower eukaryotes. Biochemistry and Cell Biology. 2005;83:752–761. doi: 10.1139/o05-159. [DOI] [PubMed] [Google Scholar]
- English AR, Zurek N, Voeltz GK. Peripheral ER structure and function. Current Opinion in Cell Biology. 2009;21:596–602. doi: 10.1016/j.ceb.2009.04.004. doi: http://dx.doi.org/10.1016/j.ceb.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Xiao W, Li Z, Li X, Chuang PY, Jim B, Zhang W, Wei C, Wang N, Jia W, Xiong H, Lee K, He JC. RTN1 mediates progression of kidney disease by inducing ER stress. Nature communications. 2015;6:7841. doi: 10.1038/ncomms8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher KL, Davis D, Wu M, Boldogh I, Pon LA. Endoplasmic reticulum dynamics, inheritance, and cytoskeletal interactions in budding yeast. Molecular biology of the cell. 2002;13:854–65. doi: 10.1091/mbc.01-04-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming TC, Shin JY, Lee SH, Becker E, Huang KC, Bustamante C, Pogliano K. Dynamic SpoIIIE assembly mediates septal membrane fission during Bacillus subtilis sporulation. Genes & development. 2010;24:1160–72. doi: 10.1101/gad.1925210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends in cell biology. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. doi: http://dx.doi.org/10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Sztul E. Traffic-independent function of the Sar1p/COPII machinery in proteasomal sorting of the cystic fibrosis transmembrane conductance regulator. The Journal of Cell Biology. 2003;160:157–63. doi: 10.1083/jcb.200210086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal U, Blackstone C. Untangling the web: Mechanisms underlying ER network formation. Biochimica et biophysica acta. 2013;1833:2492–2498. doi: 10.1016/j.bbamcr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Koning A, Wright R, Rine J. In vivo examination of membrane protein localization and degradation with green fluorescent protein. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:828–33. doi: 10.1073/pnas.93.2.828. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Prinz WA, Rapoport TA. Weaving the Web of ER Tubules. Cell. 2011;147:1226–1231. doi: 10.1016/j.cell.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport TA, Prinz WA. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–50. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–61. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiwata-Kimata Y, Promlek T, Kohno K, Kimata Y. BiP-bound and nonclustered mode of Ire1 evokes a weak but sustained unfolded protein response. Genes to cells: devoted to molecular & cellular mechanisms. 2013;18:288–301. doi: 10.1111/gtc.12035. [DOI] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Molecular biology of the cell. 2003;14:3437–48. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakoi S, Yorimitsu T, Sato K. COPII machinery cooperates with ER-localized Hsp40 to sequester misfolded membrane proteins into ER-associated compartments. Molecular biology of the cell. 2013;24:633–42. doi: 10.1091/mbc.E12-08-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. The Journal of Cell Biology. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CW, Aronson DE, Snapp EL. BiP Availability Distinguishes States of Homeostasis and Stress in the Endoplasmic Reticulum of Living Cells. Mol Biol Cell. 2010;21:1909–1921. doi: 10.1091/mbc.E09-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Moir RD, Willis IM, Snapp EL. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Molecular biology of the cell. 2012;23:955–64. doi: 10.1091/mbc.E11-12-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The Spindle Assembly Checkpoint. Current Biology. 2012;22:R966–R980. doi: 10.1016/j.cub.2012.10.006. doi: http://dx.doi.org/10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Li X, Du Y, Siegel S, Ferro-Novick S, Novick P. Activation of the mitogen-activated protein kinase, Slt2p, at bud tips blocks a late stage of endoplasmic reticulum inheritance in Saccharomyces cerevisiae. Molecular biology of the cell. 2010;21:1772–82. doi: 10.1091/mbc.E09-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Developmental cell. 2012;23:1129–40. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Mcmaster CR. Lipid metabolism and vesicle trafficking: more than just greasing the transport machinery. Biochem Cell Biol. 2001;79:681–692. doi: 10.1139/o01-139. [DOI] [PubMed] [Google Scholar]
- Pina FJ, Niwa M. The ER Stress Surveillance (ERSU) pathway regulates daughter cell ER protein aggregate inheritance. eLife. 2015;4 doi: 10.7554/eLife.06970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Russell P. Signaling Pathways that Regulate Cell Division. Cold Spring Harbor perspectives in biology. 2012;4:a005942. doi: 10.1101/cshperspect.a005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews. Molecular cell biology. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends in cell biology. 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Shemesh T, Klemm RW, Romano FB, Wang S, Vaughan J, Zhuang X, Tukachinsky H, Kozlov MM, Rapoport TA. A model for the generation and interconversion of ER morphologies. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E5243–51. doi: 10.1073/pnas.1419997111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. The Journal of biological chemistry. 2008;283:18892–904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Sharma A, Lippincott-Schwartz J, Hegde RS. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proceedings of the National Academy of Sciences. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano G, Hawes C, Brandizzi F. ER ‚Äì the key to the highway. Current Opinion in Plant Biology. 2014;22:30–38. doi: 10.1016/j.pbi.2014.09.001. doi: http://dx.doi.org/10.1016/j.pbi.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–86. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang S, Romano FB, Field CM, Mitchison TJ, Rapoport TA. Multiple mechanisms determine ER network morphology during the cell cycle in Xenopus egg extracts. The Journal of Cell Biology. 2013;203:801–14. doi: 10.1083/jcb.201308001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. The Journal of Cell Biology. 2011;193:333–46. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YS, Strittmatter SM. The reticulons: a family of proteins with diverse functions. Genome biology. 2007;8:234–234. doi: 10.1186/gb-2007-8-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasutis KM, Kozminski KG. Cell cycle checkpoint regulators reach a zillion. Cell cycle. 2013;12:1501–9. doi: 10.4161/cc.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.