

Abstract
Cells possess adaptive biosynthetic systems to maintain cellular energy levels for survival under adverse environmental conditions. Autophagy is an evolutionarily conserved cellular catabolic process that breaks down and recycles cytosolic material including macromolecules and organelles through lysosomal degradation. This catabolic process, represented by macroautophagy, is induced by a variety of cellular stresses such as nutrient starvation, which causes a shortage of cellular energy for cells to maintain cellular homeostasis and essential biological activities. In contrast, upon nutrient availability, cells stimulate anabolic processes. The mechanistic/mammalian target rapamycin (mTOR), a serine/threonine protein kinase, is a key player in stimulating cellular anabolism in response to nutrients and growth factors, and plays a crucial role in suppressing autophagy activity. Growing evidence has suggested that autophagy activity is required for the maintenance and physiological functions of renal cells including proximal tubular cells and podocytes. In this section, we will discuss recent progresses in the regulation of autophagy by the mTOR signaling.
Keywords: Autophagy, rapamycin, mTOR, mTORC1, AMPK, renal proximal tubular cell, podocyte
Mechanistic/mammalian target of rapamycin (mTOR)
mTOR protein kinase stimulates many cellular anabolic processes and plays a key role in inhibiting the initiation of autophagy 1. mTOR is a phosphatidylinositide 3-kinase (PI3K)-related protein kinase conserved from yeast to mammal 2. mTOR forms at least two distinct functional complexes termed mTOR complex1 (mTORC1) and mTORC2 3-7. mTORC1 exists as a multi-protein complex containing mTOR, RAPTOR (Regulatory-associated protein of mTOR), PRAS40 (Proline-rich AKT substrate 40 kDa), MLST8 (Mammalian Lethal with SEC13 protein 8), and DEPTOR (DEP domain-containing protein 6) 8-11, while mTORC2 consists of mTOR, RICTOR (Rapamycin-insensitive companion of mTOR), SIN1 (Stress-activated map kinase-interacting protein1/MAPKAP1), PRR5 (Proline-rich protein 5/Protor-1), MLST8, and DEPTOR 4,5,12-16. The configuration of each mTORC is also conserved across species 3. Importantly, mTORC1 activity is sensitive to rapamcin, whereas mTORC2 activity is resistant.
mTOR possesses multiple domains including HEAT (Huntington, elongation factor-3, a subunit of protein phoshpatase-2A, TOR1) repeats, a FAT (FRAP, ATM, and TRRAP) domain, an FATC domain, a kinase domain, and an FRB (FKBP12-rapamycin-binding) domain 2. Rapamycin, a macrolide antibiotic originally purified from Streptomyces hygroscopicus, is an allosteric inhibitor of mTORC1 17. It interacts with the intracellular receptor FKBP12 to form a drugprotein complex. This formation is necessary to block mTORC1 phosphorylation of substrates such as S6 kinase (S6K). Recent structural studies provide insight into the differential sensitivity of mTORC1 and mTORC2 to rapamycin 18,19. The FRB domain of mTOR, which interacts with rapamycin, resides in close proximity to the active site of mTOR kinase. In mTORC1, the binding of rapamycin-FKBP12 complex to the FRB domain sterically hinders the kinase cleft of mTOR, thereby blocking the accessibility of substrates to the active site of mTOR kinase 19. In mTORC2, it is conceivable that rapamycin binding to FKBP12 prevents its interaction with the FRB domain of mTOR, likely due to steric hindrance of the FRB domain with a specific component of mTORC2 such as RICTOR or SIN1. However, at high, micromolar concentrations, rapamycin is also able to inhibit mTORC2 activity, when it binds to the FRB in the absence of FKBP12 20. It has also been demonstrated that rapamycin treatment reduces the integrity of mTORC1 where the rapamycin-FKBP12 complex destabilizes the interaction between mTOR and RAPTOR. Prolonged treatment of rapamycin however also decreases the integrity and activity of mTORC2 possibly by preventing RICTOR interaction with mTOR during de novo mTORC2 formation 21. These observations indicate that mTOR is unable to keep or form a multiprotein complex once the rapapmycin-FKBP12 complex binds to the FRB domain 19.
Specific components that interact with mTOR kinase determine substrate specificity for mTORC1 and mTORC2. For example, RAPTOR, an essential scaffolding component of mTORC1 recruits mTORC1 substrates including S6Ks, eIF4E-binding proteins, and ATG1 2,22,23, while RICTOR or SIN1 may recruit the mTORC2 substrates Akt, PKC, and SGK1 for phosphorylation 4,24-28. Through substrate phosphorylation, mTORC1 stimulates a wide array of cellular anabolic processes including protein and lipid synthesis, and mitochondria biogenesis, whereas it inhibits catabolic processes such as autophagy (see later section). In contrast, the biological roles of mTORC2 are relatively unknown. However, by activating SGK1 and Akt, and stabilizing conventional PKCs, mTORC2 is likely to play major roles in the regulation of cell survival and cytoskeletal reorganization that are the known functions of these AGC kinases 29,30.
Regulation of mTOR signaling: Growth factor-mediated mTORC1 activation
mTORC1 activity is regulated by multiple extra- and intracellular cues including growth factors, oxidative stress, and nutrients such as glucose and amino acids (Figure 1). Among these cellular cues, both growth factor and amino acid inputs are indispensable for the full activation of mTORC1. The most proximal molecule that elicits a key role in activating mTORC1 activity is the small GTPase Rheb (Ras homolog enriched in brain). Rheb is a Ras-related GTPase originally identified as a gene rapidly induced in brain neurons by synaptic activity 31. Genetic studies in both Drosophila and mice have shown that Rheb functions as an essential activator of mTORC1 32,33. Biochemical studies have demonstrated that active Rheb directly associates with mTOR and potently stimulates its kinase activity in vitro 8,34,35. Loss of Rheb function eliminates mTORC1 activity and blunts any effects of stimuli including growth factors and amino acid. However, the molecular mechanism by which active Rheb stimulates mTORC1 kinase activity in vitro remains unclear. Since active Rheb specifically stimulates mTORC1 but not mTORC2, the direct target of Rheb is likely to be a specific component of mTORC1. Active Rheb may change the conformation of mTORC1 and open the kinase cleft of mTOR to increase the accessibility of substrates to the active site of mTOR kinase 19. Consistent with this idea, amino acid stimulation, oxidative stress, or active Rheb overexpression weakens the association between mTOR and Raptor 6,36.
Figure 1. Signal transduction in the regulation mTORC1 activation and autophagy inhibition.
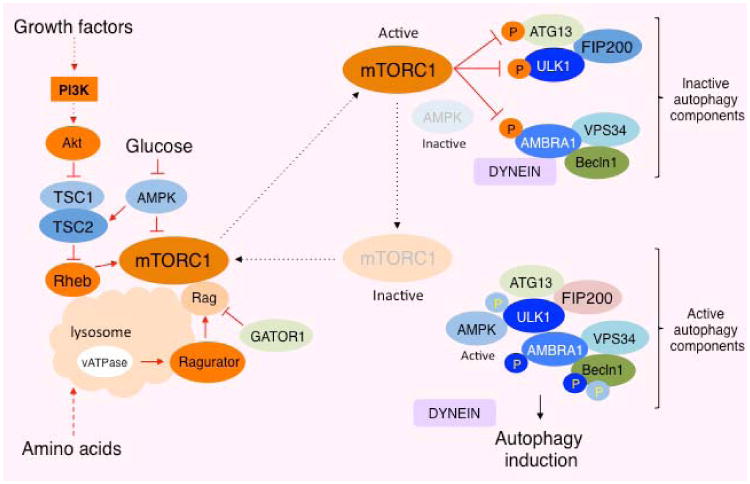
Two small GTPases, Rheb and Rags cooperatively stimulate mTORC1 on the lysosomal membrane in response to growth factor and amino acid, respectively. Active mTORC1 phosphorylates multiple components in the ULK1 (ATG1) complex and inhibits its function, whereas AMPK and ULK1 phosphorylation of the components in the complex stimulates its function to induce autophagy.
The activity of Rheb, and mTORC1, is inhibited by the tuberous sclerosis complex gene products, TSC1 (hamartin) and TSC2 (tuberin), which form a GTPase activating protein (GAP) complex 37-42. Mutations in TSC1 or TSC2 are associated with the disease tuberous sclerosis complex (TSC), characterized by the formation of hamartomas in multiple organs. TSC1 stabilizes TSC2, which possesses a GAP domain in its carboxyl terminus 43,44. Multiple growth-related kinases such as AKT, ERK (Extracellular signal-regulated kinase), and RSK (p90 ribosomal protein S6 kinase) phosphorylate and inhibit TSC2 function, thereby activating the Rheb-mTORC1 pathway 45-48. Consistently, in TSC1 or TSC2 deficient cells, mTORC1 is constitutively activated and no longer sensitive to the inhibitory effects of growth factor deprivation. Accordingly, autophagy activity is largely diminished in TSC null cells 49. Although both biochemical and genetic studies demonstrate that Rheb is a critical activator of mTORC1 in response to growth factor and amino acid stimulation, amino acid starvation still inhibits mTORC1 activity even in TSC null cells 50, where Rheb is constitutively active, suggesting that the mechanism underlying amino acid-induced mTORC1 is parallel but dominant to Rheb-mediated mTORC1 activation.
Regulation of mTOR signaling: Amino acid-mediated mTORC1 activation
How amino acids, especially leucine, play critical roles in activating mTORC1 remains a longstanding question in the mTOR field. A series of recent studies have elucidated the molecular mechanisms by which amino acids enhance the mTORC1 pathway in coordination with growth factor signaling. Two independent studies identified that Rag (Ras-related GTP-binding protein), another Ras-related GTPase, mediates amino acid-induced mTORC1 activation 51,52 (Figure1). The mammalian Rag subfamily of GTPase consists of Rag A, B, C, and D 53. RagA and RagB are homologous to yeast Gtr1p, while RagC and RagD are homologous to yeast Gtr2p 54. The mammalian RagA or RagB forms a heterodimer with RagC or RagD. This formation can also be seen in the yeast Gtr1p/Gtr2p complex. A unique feature of this conserved hetrodimeric Rag complex is that RagA or B (RagA/B) is the GTP form whereas RagC/D is the GDP form in the active complex. The Rag heterodimer is expressed on the lysosomal membrane, and upon amino acid stimulation, GTP-bound RagA/B interacts with mTORC1 through Raptor 51. Indeed, immunofluorescence studies revealed that mTORC1 translocates to the LAMP2/Rab7-positive endosome (late endosome/lysosome) in response to amino acid stimulation 51 (Figure 1). Importantly, ectopic expression of GDP-bound RagA/B prevents translocation of mTORC1 to the lysosomal membrane and its activity, whereas GTP-bound active RagA/B renders mTORC1 resistant to amino acid deprivation 51,52. Accordingly, mTORC1 constitutively localizes to the lysosomal membrane in cells expressing GTP-bound RagA/B even under amino acid starvation conditions. These data indicate that Rags play a critical role in recruiting mTORC1 to the lysosome where mTORC1 can be activated by Rheb 51. This spatial regulation of mTORC1 by Rag and Rheb explains how the signals from amino acids and growth factors are integrated to fully activate the mTORC1 pathway (Figure 1).
Unlike other small GTPases, the Rag family of GTPases lack lipid modification motifs such as those for farnesylation or myristoylation, even though they localize on the lysosomal membrane. Using proteomics approaches, Sabatini and colleagues identified a Rag heterodimer-associated complex termed as “Ragulator” that consists of at least five distinct proteins including MP1 (MAPK scaffolding protein 1), p14, p18 (MAKSP1), ROBLD3 (Roadblock domain-containing protein 3), and c11orf59. Three (MP1/p14/p18) of these five proteins were known to be associated with the lysosomal membrane to regulate endosome/lysosome organization 55. Disruption of Ragulator inhibits amino acid-induced mTORC1 activation and causes mislocalization of Rags, indicating that Ragulator plays an important role for lysosomal localization of Rags. Further analysis demonstrated that the Ragulator possesses guanidine exchange factor (GEF)-like activity for both RagA and RagB 56. These results indicate that Ragulator plays key roles in not only localization but also activation of Rags, thereby stimulating mTORC1 activity on the lysosomal membrane (Figure 1). Furthermore, the activity of vATPases required for lysosomal acidification plays an important role in activating Ragulator to stimulate Rag GTPases in response to amino acid availability 57. Recent studies also revealed that two protein complexes, termed “GATOR1 (GAP activity toward Rags) and GATOR2”, regulate Rag activity in response to cellular amino acid availability 58. GATOR1 consists of at least three proteins including NPRL2 (Nitrogen permease regulator 2-like protein), NPRL3, and DEPDC5 (DEP domain-containing protein 5), of which NPRL2 and NPRL3 have been demonstrated to inhibit mTORC1 activity in response to amino acid starvation in yeast 59. In addition, a recent study by Sabatini and colleagues identified GATOR1 as specifically possessing GAP activity for RagA and RagB 58. Intriguingly, GATOR2, which consists of 5 distinct WD40 repeat-containing proteins, associates with and inhibits GATOR1 to suppress RagA/B activity. However, the precise molecular mechanisms by which the GATOR complexes sense amino acids and which component of GATOR1 has GAP activity remain unclear. Overall, these series of studies have clarified the pathway and signals from amino acid sufficiency to mTORC1 activation. Unexpectedly, these studies also revealed that the activity of lysosomes plays critical roles for mTORC1 activation, which are also paradoxically important for cellular autophagy.
Mechanism of mTORC1-dependent autophagy inhibition
Although the activity of lysosomes is essential for both mTORC1 activation and autophagy, mTORC1 has been long recognized as an essential negative regulator for autophagy induction. Autophagy is an evolutionarily conserved process that recycles macromolecules and organelles through lysosome-mediated degradation to generate the source of cellular energy during nutritional stress 60. Upon activation of autophagy, unnecessary cellular components are encapsulated in a double-membrane vesicle structure (autophagosomes), which targeted to lysosomes (autolysosome). Fusion of the outer autophagosomal membrane with the lysosome releases the cargo-containing inner membrane to the lumen of the lysosome for further breakdown and recycling, thereby providing a nutrient source to maintain vital cellular activities 61.
TORC1 in S. cerevisiae (budding yeast) negatively regulates autophagy. Rapamycin treatment is sufficient to induce autophagy even in the presence of nutrients, providing key evidence that TORC1 elicits an essential negative role in suppressing autophagy. Previous genetic and biochemical studies demonstrated that TORC1 suppressed the function of ATG1, an autophagy-initiating kinase 62,63. The budding yeast atg1 mutant is defective in autophagy induction even under nutrient starvation or rapamycin treatment conditions, indicating that ATG1 acts downstream of TORC1 to induce autophagy. ATG1 forms a complex with other autophagy proteins such as ATG13 and ATG17. The integrity of the ATG1-ATG13-ATG17 complex is important for ATG1 kinase activity, and rapamycin treatment or nutrient starvation enhances the integrity of this complex 62. It has been postulated that TORC1 enhances the phosphorylation of ATG13 on multiple residues to weaken the integrity of the ATG1 complex and repress autophagy induction 63,64. Interestingly, the molecular mechanism underlying TORC1-mediated autophagy inhibition through the post-translational modifications of the ATG1 complex seems to be conserved albeit more complicated in higher eukaryotes 65. The mammalian ATG1 orthologs, Unc-51-like kinase 1 (ULK1) and ULK2, also play important roles in autophagy induction in mammalian cells 66,67. ULK1 is phosphorylated and activated by 5′-AMP-activated protein kinase (AMPK), an essential energy sensor, in response to metabolic stress 23,68. In contrast, ULK1 is phosphorylated and inactivated by mTORC1 in response to nutrient availability (Figure 1). ULK1 is stably bound to AMPK, and this interaction is suppressed by the mTORC1-dependent ULK1 phosphorylation, indicating that mTORC1 disrupts the process of ULK1 activation by AMPK under nutrient-rich conditions 23. Consistently, the interaction between ULK1 and AMPK, and the phosphorylation of ULK1 by AMPK are enhanced by rapamycin treatment. These results indicate that mTORC1 phosphorylation of ULK1 maintains ULK1 in an inactive state. Although ATG13 can also be subjected to mTORC1-dependent phosphorylation in mammalian cells as it is in yeast, it remains unclear the physiological roles of ATG13 phosphorylation in the ULK1 complex because the interaction between ULK1 and ATG13 is maintained even under nutrient-rich conditions 69. In addition to the above mechanisms, recent studies also revealed that mTORC1 directly phosphorylates AMBRA1 (activating molecule in Beclin-1-regulated autophagy) 70, a component of the VPS34-Beclin1 (ATG6) complex 71, which recruits downstream effectors to the site where nucleation of autophagosomes occurs (Figure 1). AMPBRA1 induces autophagosome nucleation by promoting Beclin1 interaction with the lipid kinase VPS34 72. AMBRA1 plays a key role in stabilizing ULK1 and activating ULK1 kinase activity by facilitating ULK1 dimerization 70. Interestingly, mTORC1 directly phosphorylates AMBRA1 and inhibits its function in activating ULK1 under nutrient-rich conditions. Taken together, mTORC1 phosphorylates multiple autophagy proteins leading to the blockade of ULK1 functions and inhibiting the induction phase of autophagy.
Role of the mTORC1-autophagy pathway in kidney cells
A series of studies demonstrated that induction of autophagy plays an important role in protecting renal tubular cells (especially proximal tubular cells) from many stresses including ischemia 73-75. Renal tubular cells show the highest level of mTORC1 activity in renal tissues as determined by rapamycin-sensitive S6 phosphorylation, suggesting that basal autophagy activity is presumably low under normal physiological conditions. A recent study also demonstrated that autophagy in renal tubular cells is stimulated by proteinuria, and the autophagy induction in tubular cells plays an important role in protecting cells from proteinuria-induced apoptosis 76. Intriguingly, excess calorie uptake, such as in high fat diets, enhances mTORC1 activity and suppresses autophagy induction in the renal tubular cells, resulting in higher susceptibility of tubular injury to proteinuria. The role of autophagy has also been studied in glomerular podocytes 77-79. Glomerular podocytes display higher autophagy activity compared to other glomerular cells 79. Lack of autophagy in podocytes causes slowly progressive podocyte loss and glomerulosclerosis in aged mice, indicating that basal autophagy activity plays an important role in maintaining healthy podocytes in older mice. Intriguingly, podocytes also exhibit higher mTORC1 activity compared to other glomerular cells 80-82. These observations suggest that podocytes create a unique environment where both mTORC1 and autophagy mutually and exclusively function in a single cell. The mechanism by which autophagy activity is maintained in podocytes may be due to their specific cellular shape and the structure of their organelles, which are coupled to provide fundamental podocyte functions. Recent studies demonstrate that podocytes possess large Golgi apparatuses and develop lysosomes at the trans-side of the Golgi, where a large amount of cellular mTORC1 is sequestered on the lysosomal surface in perinuclear regions 83. Furthermore, podocytes have long foot processes that provide a large surface area for filtration. This unique structure and organellar position may provide a gradient of mTORC1 expression within a podocyte, and allow the cells to activate both autophagy and mTORC1 in different areas. Such a system may have beneficial roles in generating sufficient secretory proteins with a constant energy supply derived from autophagy. Consistently, phosphorylated S6, a substrate of mTORC1 localized to active polysomes, is predominantly expressed in the perinuclear region where active mTORC1 stimulates translation in podocytes 80,82. It will be important to explore the physiological functions of this coordinated spatial regulation of mTORC1 and autophagy in podocytes and address the questions of whether disruption of this system causes podocyte and glomerular dysfunction.
In summary, a series of studies have proposed that autophagy plays important roles in keeping renal cells healthy by protecting them from metabolic stress. Given that mTORC1 is a potent suppressor for autophagy, any inappropriate mTORC1 activation should elicit deleterious effects on renal cell function. Thus, future studies clarifying the signals and mechanisms underlying dys-regulation of mTORC1 activity in renal cells promises to shed further light into the interplay between mTORC1 activity and autophagy in renal cell function.
Acknowledgments
Financial Support: K.I. is supported by the NIH (grant DK083491).
Footnotes
Conflict of Interest Statement: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Molecular cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current biology : CB. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 5.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 6.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 7.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 8.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 10.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 11.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 13.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current biology : CB. 2006;16:1865–70. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes & development. 2006;20:2820–32. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, et al. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem. 2007;282:25604–12. doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 16.Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PloS one. 2007;2:e1217. doi: 10.1371/journal.pone.0001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annual review of immunology. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 18.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–42. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–23. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, et al. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer research. 2008;68:2934–43. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 21.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 22.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular biology of the cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 25.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO journal. 2008;27:1919–31. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. The EMBO journal. 2010;29:3939–51. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) The Biochemical journal. 2008;416:375–85. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 28.Yan L, Mieulet V, Lamb RF. mTORC2 is the hydrophobic motif kinase for SGK1. The Biochemical journal. 2008;416:e19–21. doi: 10.1042/BJ20082202. [DOI] [PubMed] [Google Scholar]
- 29.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su B, Jacinto E. Mammalian TOR signaling to the AGC kinases. Critical reviews in biochemistry and molecular biology. 2011;46:527–47. doi: 10.3109/10409238.2011.618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, et al. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem. 1994;269:16333–9. [PubMed] [Google Scholar]
- 32.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–71. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 33.Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–65. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 34.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Current biology : CB. 2005;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem. 2009;284:12783–91. doi: 10.1074/jbc.M809207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, et al. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem. 2011;286:32651–60. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Human molecular genetics. 2002;11:525–34. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 39.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–6. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 40.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & development. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 42.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Molecular cell. 2003;11:1457–66. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 43.Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Annals of human genetics. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 44.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 45.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 46.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Molecular cell. 2002;10:151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 47.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 48.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13489–94. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou X, Ikenoue T, Chen X, Li L, Inoki K, Guan KL. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8923–8. doi: 10.1073/pnas.0903621106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–27. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 51.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–45. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem. 2001;276:7246–57. doi: 10.1074/jbc.M004389200. [DOI] [PubMed] [Google Scholar]
- 54.Hirose E, Nakashima N, Sekiguchi T, Nishimoto T. RagA is a functional homologue of S. cerevisiae Gtr1p involved in the Ran/Gsp1-GTPase pathway. Journal of cell science. 1998;111(Pt 1):11–21. doi: 10.1242/jcs.111.1.11. [DOI] [PubMed] [Google Scholar]
- 55.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–6. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neklesa TK, Davis RW. A genomewide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS genetics. 2009;5:e1000515. doi: 10.1371/journal.pgen.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 61.Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 62.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tormediated induction of autophagy via an Apg1 protein kinase complex. The Journal of cell biology. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TORmediated autophagy regulation. Molecular biology of the cell. 2009;20:2004–14. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Molecular and cellular biology. 2010;30:1049–58. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy. 2009;5:758–65. doi: 10.4161/auto.8709. [DOI] [PubMed] [Google Scholar]
- 66.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 67.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of cell biology. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–16. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 71.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. The Biochemical journal. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 72.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. The Journal of cell biology. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. The American journal of pathology. 2010;176:1181–92. doi: 10.2353/ajpath.2010.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–37. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 75.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–13. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamahara K, Kume S, Koya D, Tanaka Y, Morita Y, Chin-Kanasaki M, et al. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2012111080. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cina DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012;23:412–20. doi: 10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, et al. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol. 2011;22:2193–202. doi: 10.1681/ASN.2011020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. The Journal of clinical investigation. 2010;120:1084–96. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, et al. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23:1351–63. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. The Journal of clinical investigation. 2011;121:2181–96. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. The Journal of clinical investigation. 2011;121:2197–209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]