

Abstract
The peroxiredoxin (Prdx) family of antioxidant enzymes uses redox-active cysteines to reduce peroxides, lipid hydroperoxides, and peroxynitrites. Prdx1 is known to be important to protect red blood cells against reactive oxygen species and in tumor prevention. In this study, the role of Prdx1 in inflammation, thrombosis, and atherosclerosis was investigated. Using intravital microscopy, we showed that the number of leukocytes rolling per minute in unstimulated veins was increased by 2.5-fold in Prdx1−/− compared to Prdx1+/+ mice. In Prdx1−/− mice, 50% of leukocytes rolled at a velocity <10 μm/sec compared with 10% in Prdx1+/+ mice, suggesting that adhesion molecule density on the endothelium may have been increased by Prdx1 deficiency. Indeed, endothelial P-selectin, soluble P-selectin, and von Willebrand factor in plasma were increased in Prdx1−/− mice compared to Prdx1+/+ mice, indicating elevated Weibel–Palade body release. In contrast to this excessive endothelial activation, Prdx1−/− platelets showed no sign of hyperreactivity, and their aggregation both in vitro and in vivo was normal. We also examined the role of Prdx1 in the apoE−/− murine spontaneous model of atherosclerosis. Prdx1−/−/apoE−/− mice fed normal chow developed larger, more macrophage-rich aortic sinus lesions than Prdx1+/+/apoE−/− mice, despite similar amounts and size distributions of cholesterol in their plasma lipoproteins. Thus, Prdx1 protects against excessive endothelial activation and atherosclerosis, and the Prdx1−/− mice could serve as an animal model susceptible to chronic inflammation.
Keywords: inflammation, atherosclerosis, antioxidant enzymes, endothelium, peroxiredoxin
Reactive oxygen species (ROS) play an important role in many cellular metabolic and signaling processes. At low to moderate concentrations, ROS, such as nitric oxide (NO) and superoxide anion (O2−), are important because they can act as regulatory mediators in signaling processes in various cell types, including neurons and endothelial cells.1,2 For example, hydrogen peroxide (H2O2) can function as a second messenger and regulates inflammatory or growth-stimulatory signals by stimulating nuclear factor κB activation.3,4 At the same time, high concentrations of free radicals and radical-derived species are extremely hazardous for living organisms because they cause oxidative damage to many cellular components.5 There is growing evidence that an imbalance of redox homeostasis with elevated ROS/oxidative stress plays a role in various diseases such as cancer, diabetes, viral infection, chronic inflammation, and atherosclerosis.6,7 Atherosclerosis, for example, is thought to be a chronic inflammatory process in which oxidation may play an important role. Oxidized LDL has been proposed to contribute to the pathogenesis of atherosclerosis by causing endothelial dysfunction, increased monocyte adhesion (because of upregulation of adhesion molecules such as intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule [VCAM]-1, and E-selectin), foam cell formation, vascular inflammation, and cytotoxicity, resulting in formation and progression of atherosclerotic lesions.8,9
To protect against the toxic effects of ROS, cells and tissues have developed several enzymatic defense systems that regulate the concentration of these species inside and outside of the cells.10 Drugs used against cardiovascular diseases, such as statins, have been shown to exhibit antioxidant effects and simultaneously inhibit endothelial adhesion molecule expression.11 Angiotensin II receptor blockers, which are effective antiatherosclerosis agents, have been shown to suppress ROS in vascular smooth muscle cells.12,13 Furthermore, some naturally occurring antioxidants are proposed to be atheroprotective.14 One such antioxidant family is the peroxiredoxins (Prdxs).15
Prdxs are antioxidant proteins containing essential catalytic cysteine residues that use thioredoxin to scavenge hydrogen peroxide, lipid hydroperoxides, and peroxynitrite.15 Prdxs constitute a potent defense mechanism for maintaining redox balance under normal conditions and oxidative stress. The 6 mammalian Prdxs (Prdx1 to 6) are distributed at sites of ROS production, including the cytosol, mitochondria, and peroxisomes.15 Prdx1 is expressed in the cytosol of many types of cells and tissues and is induced in mouse peritoneal macrophages exposed to oxidative stresses.16 Prdx1-deficient fibroblasts in culture show decreased proliferation and increased sensitivity to oxidative DNA damage.17 Prdx1-deficient mice develop hemolytic anemia caused by an increase in erythrocyte ROS. Prdx1 has tumor-suppressor properties because Prdx1−/− mice develop an increased frequency of multiple malignant cancers as they age, presumably caused by accumulation of oxidative tissue damage.17
By scavenging H2O2,18 Prdx1 can suppress the nuclear factor κB pathway and the consequent inflammatory response,19 thus playing a crucial role in controlling the inflammation. The vascular endothelium controls inflammatory responses, and it has been shown that increased formation of ROS leads to endothelial dysfunction and promotes proinflammatory gene expression.20 Reactive oxygen intermediates induce secretion of endothelial Weibel–Palade bodies (WPBs) and consequently cause rapid endothelial activation.21 One manifestation of this activation is enhanced leukocyte rolling and attachment along the endothelium and, as a consequence of increased adhesive interactions, a reduction in the speed of rolling leukocytes. Prdx1 has been shown to be expressed in endothelial cells, and, interestingly, its expression is regulated by flow, suggesting that Prdx1 could serve as a mechanosensitive antioxidant.22 In addition, proteome and Western blot analysis revealed increased expression levels of Prdx1 in aortas from apoE−/− mice with advanced atherosclerosis.23 Therefore, the role of Prdx1 in endothelium and the effect of Prdx1 deficiency on inflammation in general, and atherosclerosis in particular, needs to be explored. We crossed Prdx1−/− mice onto apoE−/− mice, a model of spontaneous atherosclerosis, and studied the role of this antioxidant in atherosclerosis. Here, we show that Prdx1 prevents excessive endothelial activation and atherosclerotic lesion formation.
Materials and Methods
Animals
Mice used in this study were siblings obtained from crosses of Prdx1+/− mice on C57BL/6J/129Sv background. The mice used for intravital microscopy were 4 weeks old, both male and female, weighing 14 to 18 g. For atherosclerosis studies, Prdx1−/− mice were crossed to apoE−/− mice on C57BL/6J to eventually obtain Prdx1+/−/apoE−/− mice (they have genetic background of approximately 12.5% and 87.5% of 129J and C57Bl/6J, respectively). Prdx1+/−/apoE−/− mice were crossed to each other to obtain Prdx1+/+/apoE−/− and Prdx1−/−/apoE−/− siblings, which were used for atherosclerosis studies. Mice were fed a rodent chow diet until the age of 4 months. Animals were bred and housed on a 12-hour light/dark cycle at the Immune Disease Institute, and all experimental procedures were approved by its Animal Care and Use Committee.
Intravital Microscopy
Shear rates, leukocyte rolling per minute, and leukocyte-rolling velocity were done as previously described using phase-contrast intravital microscopy in unstimulated mesenteric veins with the shear rates 150 to 200 sec−1.24,25 To study leukocyte rolling in the histamine-stimulated veins, 200 μL of 1 mmol/L histamine per 15 g body weight of mouse was injected 15 minutes before intravital microscopy. To inhibit P-selectin specifically, we used aptamer ARC5690 (1 mg/kg, IV); scrambled aptamer ARC 5694 was used as control (both from Archemix). In addition, P-selectin was inhibited with a rat monoclonal anti–P-selectin immunoglobulin (Ig) (2 μg/g body weight, IV; BD Pharmingen). Control was purified rat Ig (BD Pharmingen). Images were visualized using a Zeiss inverted microscope connected to an SVHS Panasonic AG-6720A video recorder (Matsushita Electric, Kadoma City, Japan) using a charge-coupled device video camera (Hamamatsu Photonics Systems, Hamamatsu City, Japan). The analysis was done by an investigator blinded to genotype.
In Vivo Detection of P-selectin and VCAM-1 on Endothelial Surface
Yellow green (excitation/emission, 505 nm/515 nm) and red (excitation/emission, 580 nm/605 nm) carboxylate-modified microspheres (1 μm) were covalently coupled to anti–P-selectin monoclonal antibody (BD Pharmingen) or anti–VCAM-1 antibody (Santa Cruz Biotechnology) and respective control Ig as previously described.26 Mesenteric veins were observed by fluorescent intravital microscopy.
Detection of Adhesion Molecules in Plasma
For the quantitative determination of mouse soluble P-selectin, E-selectin, L-selectin, VCAM-1, ICAM-1, and von Willebrand factor (VWF) in plasma, we used the quantitative sandwich enzyme immunoassay (ELISA, R&D Systems). For VWF, the number of units in each sample was determined based on the absorbance value (A450) of normal pooled plasma obtained from 10 wild-type mice. We defined normal pooled plasma as containing 1 U of VWF antigen per milliliter of plasma.26
Platelet Preparation
Platelet collection and preparation were done as described.27 Platelets were fluorescently labeled with calcein acetoxymethyl ester 2.5 μg/mL (Invitrogen) for 10 minutes at room temperature and used for intravital microscopy.
Thrombosis Model
A previously described model was used.27 In brief, mice were anesthetized, and fluorescent platelets were infused through the retroorbital plexus of the eye. An incision was made through the abdominal wall to expose the mesentery, and arterioles 100 to 150 μm in diameter were studied. The shear rate was calculated as described.27 Arterioles were visualized using the same microscope set up as for leukocyte rolling. Whatman paper saturated with FeCl3 (10%) solution was applied topically for 5 minutes, and the vessel was monitored for 40 minutes after injury or until occlusion. One arteriole was chosen per mouse.
Platelet Aggregation
To determine platelet aggregation, light transmission was measured using platelet-rich plasma adjusted to a platelet concentration of 3×108 platelets/mL with modified Tyrode’s buffer containing 1 mmol/L CaCl2. The agonists ADP, collagen, convulxin, and protease-activated receptor (PAR)4 were added and transmission was recorded over 14 minutes on a Chrono-Log 4-channel optical aggregation system (Chrono-Log).
Analysis of Aortic Root Atherosclerotic Lesions
Mice were perfused with ice cold PBS embedded in Tissue-Tec OCT compound, frozen at −<80°C, and sectioned (10 μm). Sections with aortic sinus lesions were stained with oil red O and counter-stained with hematoxylin. For each animal, 5 sections, 80 μm apart, were analyzed without knowledge of the genotype to determine atherosclerotic lesion size. Macrophage infiltration was visualized with rat antimouse MOMA-2 antibody (Serotec), followed by Alexa 488 goat anti-rat Ig and nuclear staining with Hoechst (Molecular Probes). MOMA-2 expression was quantified using Axiovision computer-assisted imaging software (Zeiss) and expressed as an area of positive immunofluorescent staining.
Plasma Lipids and Lipoprotein Measurements
Blood samples were collected to heparinized tubes by retroorbital venous plexus puncture after a 4-hour fast. Plasma was separated by centrifugation and analyzed. Total cholesterol and total phospholipid levels were determined by using enzymatic colorimetric assays (Wako Biochemicals). Lipoproteins were isolated from plasma by fast protein liquid chromatography as previously described.28
Statistical Analysis
The values are presented as mean±SEM. Differences between means were determined by using the Student’s 2-tailed t test or ANOVA, if more than 2 groups were compared. Probability values of 0.05 or less were regarded as statistically significant.
Results
Peroxiredoxin1 Deficiency Results in Increased Systemic Leukocyte Rolling and Decreased Leukocyte-Rolling Velocity
To study the effect of Prdx1 deficiency on endothelial activation, we visualized basal leukocyte–endothelium interactions (numbers of rolling leukocytes per minute and rolling speed) in real time by intravital microscopy. A previous study established that Prdx1 deficiency in mice does not affect the total leukocyte counts in murine peripheral blood.17 Using unstimulated veins exhibiting similar shear rates, we observed a 2.5-fold increase in the number of rolling leukocytes/min in Prdx1−/− mice (98±11) compared to Prdx1+/+ mice (39±6, P<0.0001; Figure 1A and 1B). Furthermore, leukocyte-rolling velocities were significantly lower in Prdx1−/− mice (≈50% of leukocytes exhibiting a velocity of <10 μm/sec) than in Prdx1+/+ mice (≈10% leukocytes with a velocity <10 μm/sec, P<0.001; Figure 1C). The abnormally high number of slowly rolling leukocytes in veins of Prdx1−/− mice suggests that their endothelium is preactivated.
Figure 1.
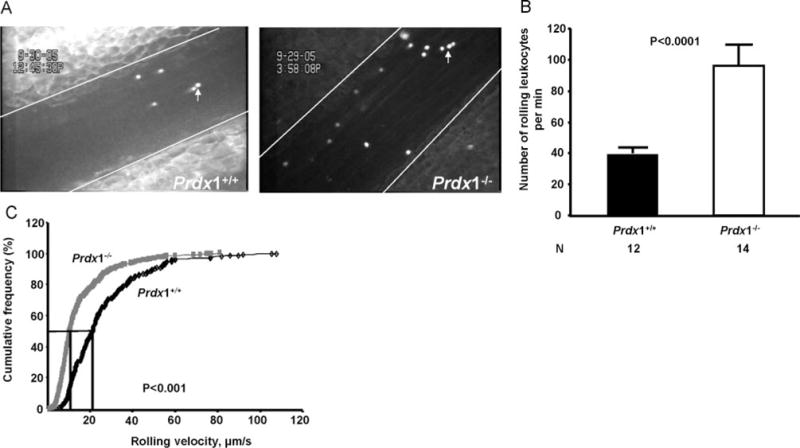
Increased leukocyte rolling and decreased leukocyte-rolling velocity in Prdx1−/− mice. A, Representative images of the analyzed mesenteric veins. Lines delineate the vessel and arrows point to the leukocytes stained with rhodamine 6G. Shear rates for Prdx1+/+ (161±9 sec−1) and Prdx1−/− (175±9 sec−1) mice were not significantly different (P>0.05). B, In the unstimulated veins, the number of rolling leukocytes was calculated by counting rolling leukocytes passing a line perpendicular to the vessel axis during a 1-minute time period. The number of leukocytes rolling per minute was significantly increased in Prdx1−/− when compared to Prdx1+/+ mice. C, Leukocyte-rolling velocity in veins from Prdx1+/+ (black line) and Prdx1−/− (gray line) mice. Velocity curves were constructed by sampling the velocities of 255 rolling leukocytes in Prdx1+/+ mouse (n=7) and 466 rolling leukocytes in Prdx1−/− mouse (n=11) veins. In Prdx1−/− mice, leukocytes roll slowly compared to Prdx1+/+ mice (P<0.001). Error bars represent means±SEM.
Increased WPB Release in Prdx1−/− Mice
The observed increase in number of rolling leukocytes along with the decrease in leukocyte-rolling velocity in Prdx1−/− mice compared to Prdx1+/+ suggests upregulation of adhesion molecules such as P-selectin and E-selectin on the endothelium. To evaluate which selectin is responsible for the increased leukocyte rolling in Prdx1−/− mice, we inhibited P-selectin specifically using a new inhibitor, aptamer ARC5690, developed by Archemix. Blocking of P-selectin with aptamer ARC5690 in Prdx1−/− mice completely abolished leukocyte rolling, whereas control scrambled aptamer ARC5694 did not (Figure 2A). These results were confirmed with a blocking monoclonal antibody to P-selectin (Figure 2B and Movies 1 and 2 in the online data supplement, available http://circres.ahajournals.org). Together, these results demonstrate that the increased leukocyte rolling in Prdx1−/− mice is attributable to P-selectin rather than E-selectin expression. To confirm an increase in endothelial P-selectin expression, we measured expression of P-selectin on the veins in Prdx1+/+ and Prdx1−/− mice by simultaneous IV infusion of fluorescent microspheres coupled to either anti–P-selectin Ig (yellow-green) or control Ig (red) in the same mouse (Figure 3A). Control Ig-conjugated microspheres did not bind significantly to the endothelium in either genotype (not shown). There were significantly more anti–P-selectin Ig-conjugated microspheres bound to the vessel wall in the veins of Prdx1−/− mice than in Prdx1+/+ mice (Figure 3B), confirming that there is abnormal activation of the endothelium along with increased WPB secretion in Prdx1−/− mice.
Figure 2.
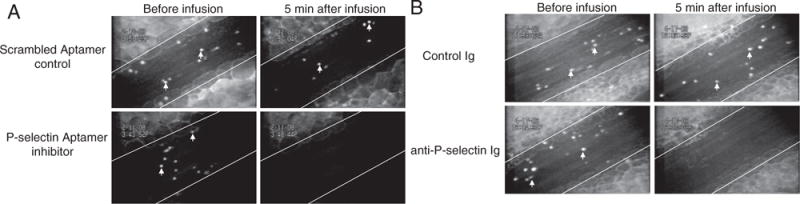
Increased leukocyte rolling in Prdx1−/− mice is P-selectin–dependent. Baseline leukocyte rolling in mesenteric veins (left images) of Prdx1−/− mice. A, Aptamer ARC 5690 infusion (P-selectin inhibitor) (bottom right) completely inhibited leukocyte rolling, whereas scrambled aptamer ARC 5694 (control) did not (top right). B, Control Ig infusion did not have any effect on leukocyte rolling (top right), whereas a blocking antibody to P-selectin completely abolished leukocyte rolling (bottom right). Endogenous leukocytes (white arrows) were labeled with rhodamine 6G. White lines delineate the mesenteric veins. Two to 3 veins from 3 to 4 mice in each group were evaluated.
Figure 3.
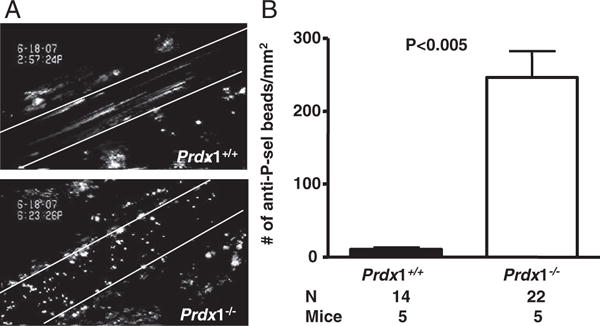
Increased endothelial P-selectin in Prdx1−/− mice. Fluorescent microspheres (1 μm) coupled to anti–P-selectin antibody were infused through retroorbital venous plexus before intravital microscopy, and their binding to veins of Prdx1+/+ and Prdx1−/− mice was analyzed. A, Representative images of the veins are shown. Lines delineate the blood vessel. B, Quantification of anti–P-selectin–conjugated microspheres bound to endothelium.
To further examine the effects of Prdx1 deficiency on endothelial activation, we measured plasma levels of soluble adhesion molecules that are markers for endothelial activation. P-selectin and VWF are 2 major proteins stored in WPBs. We found statistically significantly greater levels of circulating soluble P-selectin and VWF in Prdx1−/− than in Prdx1+/+ mice (Figure 4A and 4B). There was no significant difference in the levels of soluble E-selectin in Prdx1−/− compared to Prdx1+/+ mice (not shown). In terms of endothelial adhesion molecules upregulated in atherosclerosis, we found a small increase in the levels of soluble VCAM-1 (Figure 4C), but not soluble ICAM-1 (not shown). We could not document an increase in endothelial VCAM-1 expression. We infused fluorescent microspheres coupled to anti–VCAM-1 antibody, and their binding to Prdx1−/− veins was visualized and quantified. Control Ig was infused simultaneously to the same mouse. There was no difference in the number of microspheres bound to veins of Prdx1+/+ mice (25±8/mm2) compared to Prdx1−/− mice (23±5/mm2, P>0.05; supplemental Figure I).
Figure 4.
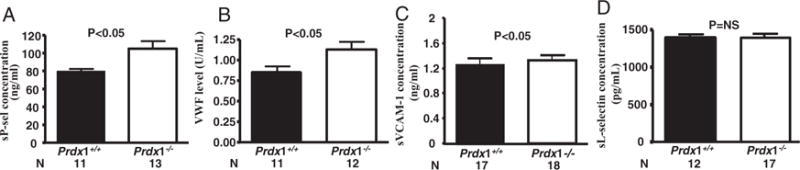
Prdx1 deficiency results in an increase in adhesion molecules. Plasma was collected from Prdx1+/+ and Prdx1−/− mice. ELISA was performed to determine levels of circulating soluble P-selectin (A), VWF (pooled plasma from 10 wild-type mice was defined as 1 U/mL) (B), soluble VCAM-1 (sVCAM-1) (C), and soluble L-selectin (sL-selectin) (D).
Activated platelets were recently shown to induce WPB release and leukocytes rolling in vivo.26 Therefore, we measured classical markers of platelet activation such as P-selectin expression and activated αIIbβ3 levels by flow cytometry. No differences were found between Prdx1−/− and Prdx1+/+ platelets (not shown). Next, we measured markers of leukocyte activation by measuring, soluble L-selectin (Figure 4D), L-selectin shedding, and αMβ2 (CD18) expression on leukocytes. Again, expression levels were similar between Prdx1−/− and Prdx1+/+ mice (not shown). Together, these results suggest that Prdx1 deficiency does not lead to basal platelet and leukocyte activation but does promote secretion of WPBs as a consequence of endothelial activation.
Hemolytic Anemia Is Not the Contributing Factor for Increased Leukocyte Rolling in Prdx1−/− Mice
Prdx1−/− mice develop severe hemolytic anemia, starting at 9 months of age.17 In our study, we used younger mice aged 1 to 4 months. To rule out the possibility that free hemoglobin released from RBC lysis activated the endothelium in Prdx1−/− mice, we determined blood cell count, hemoglobin, hematocrit, and reticulocyte numbers in Prdx1+/+, Prdx1−/−, and Prdx1−/−/apoE−/− mice. No differences were found between groups (Table 1). Together, these data show that hemolytic anemia was not the contributing factor for increased leukocyte rolling in Prdx1−/− mice.
Table 1.
Blood Parameters of Prdx1+/+, Prdx1−/−, and Prdx1−/−/apoE−/− Mice
| Parameter | Prdx1+/+ | Prdx1−/− | Prdx1−/−/apoE−/− |
|---|---|---|---|
| Erythrocytes (×106/μL) | 6±0.2 | 7±0.1 | 7±0.1 |
| Hematocrit | 48±1 | 48±1 | 48±1 |
| Hemoglobin (g/dL) | 13±1 | 13±1 | 13±1 |
| Reticulocytes (%) | 3±0.3 | 3±0.7 | 2±0.4 |
| White blood cells (×103/μL) | 7±1 | 6±1 | 6±1 |
| Platelets (×103/μL) | 956±139 | 1000±116 | 1027±81 |
Peripheral blood was obtained from Prdx1+/+, Prdx1−/−, and Prdx1−/−/apoE−/− mice (n=6–9), and blood parameters were measured using a Beckman Coulter counter. Peripheral blood smears stained with new methylene blue were used to quantify the amount of reticulocytes. For all parameters tested, there were no significant differences between the groups. Values represent the means±SEM (P>0.05, ANOVA).
Prdx1−/− Deficiency Does Not Affect Exocytosis of WPBs Under Stimulated Conditions
Next, we asked whether there is an increased exocytosis of WPBs in stimulated conditions. Veins were stimulated with histamine, a secretagogue of WPBs, and resulting leukocyte rolling was observed 15 minutes later. In Prdx1−/− mice, the number of rolling leukocytes was insignificantly higher (154±22) than in Prdx1+/+ mice (130±20) (n=9 veins from 4 mice, each genotype, P>0.05). Thus, deficiency of Prdx1 does not result in increased exocytosis of WPBs when challenged with an agonist such as histamine.
Arterial Thrombosis Is Not Affected in Prdx1−/− Mice
Vascular oxidative stress can be associated with elevated thrombosis and may affect the function of platelets by suppressing NO.29 Therefore, we evaluated whether Prdx1 deficiency in mice would affect platelet function both in vitro and in vivo. In vitro aggregation initiated by adding agonists (ADP, collagen, convulxin, and PAR4) to platelet-rich plasma was monitored by light transmission. No significant differences were found in the response of Prdx1−/− and Prdx1+/+ platelets (Figure 5A). Next, we evaluated in vivo FeCl3-induced thrombus formation in mesenteric arterioles of Prdx1−/− and Prdx1+/+ mice. The mean occlusion times (Prdx1+/+, 12.9±0.7 minutes; Prdx1−/−, 11.3±0.8 minutes; P>0.05; Figure 5B) were similar in both groups. Taken together, these results suggest that Prdx1 deficiency does not appear to modulate platelet aggregation and thrombus formation.
Figure 5.
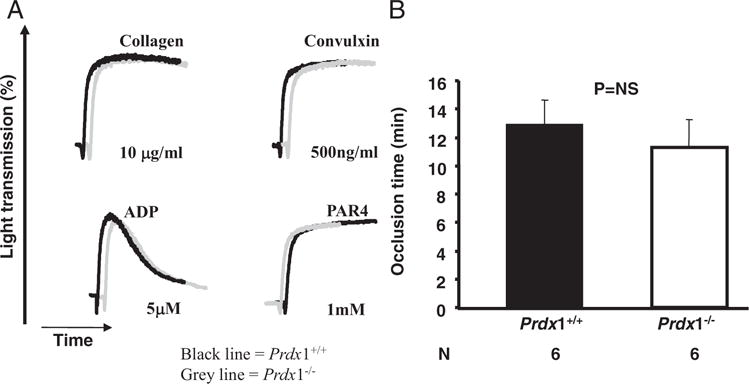
Prdx1 deficiency does not affect platelet function. Platelets were prepared from Prdx1+/+ and Prdx1−/− mice as described in Materials and Methods. A, Platelet aggregation was initiated in platelet-rich plasma of Prdx1+/+ (black line) and Prdx1−/− (gray line) using the following agonists: collagen, convulxin, ADP, and PAR4. Results are representative of 3 experiments. B, Time to occlusion in FeCl3 arterial injury model was similar in Prdx1−/− compared to Prdx1+/+mice.
Prdx1 Deficiency Accelerates Atherosclerosis in apoE−/− Mice
To examine the potential pathological consequences of the increased endothelial activation found in Prdx1−/− mice on a chronic inflammatory disease, we determined the effects of Prdx1 deficiency on atherosclerosis in apoE−/− mice fed standard chow diet. Because lipoproteins play a critical role in atherogenesis, we characterized plasma lipoproteins in Prdx1+/+/apoE−/− and Prdx1−/−/apoE−/− mice in addition to measuring the extent of aortic root atherosclerosis. Levels of total cholesterol and total phospholipids, as well as body weights, were not significantly different in these mice (Table 2). Furthermore, there were no differences in the lipoprotein size distributions of the plasma lipoproteins (intermediate-density lipoprotein/LDL, HDL, and very-low-density lipoprotein), as assessed by size exclusion chromatography (not shown).
Table 2.
Weights, Plasma Total Cholesterol, and Total Phospholipid Levels in Chow Diet–Fed Prdx1+/+/apoE−/− and Prdx 1−/−/apoE−/− Mice
| Genotype (Sample Size) |
Weight (g) |
Total Cholesterol (mg/dL) |
Total Phospholipids (mg/dL) |
|---|---|---|---|
| Prdx1+/+/apoE−/− (n=5) | 27±1 | 435±50 | 428±24 |
| Prdx1−/−/apoE−/− (n=6) | 28±1 | 407±30 | 433±32 |
Values are represented as means±SEM. P>0.05 for all values using unpaired Student’s t test.
We examined aortic root atherosclerotic lesions in the mice at 16 weeks of age, both by quantifying the areas of neutral lipid accumulation in cross-sections of the aortic sinus stained with oil red O (Figure 6A and 6B) and areas of monocyte/macrophage accumulation by MOMA-2 antibody staining in the atherosclerotic plaque (Figure 6C and 6D). Because female apoE−/− mice usually develop larger lesions than corresponding males,30,31 we compared the extent of atherosclerosis for the different sexes, as well as for the combined group of males and females. Compared to Prdx1+/+/apoE−/− controls, there was a 1.9-fold increase in total aortic sinus lesion area in Prdx1−/−/apoE−/− females (1.0±0.3×105 μm2 versus 1.8±0.4, P<0.005) and a 1.5-fold increase in males (0.5±0.2×105 μm2 versus 0.8±0.2, P<0.05). Immunofluorescent staining showed a significant increase in MOMA-2–stained lesion area in Prdx1−/−/apoE−/− (0.5±0.09×105 μm2) compared to Prdx1+/+/apoE−/− mice (0.2±0.03×105 μm2, P<0.01). Thus, Prdx1 deficiency leads to increased monocyte/macrophage recruitment and enhanced fatty streak formation in apoE−/− mice.
Figure 6.
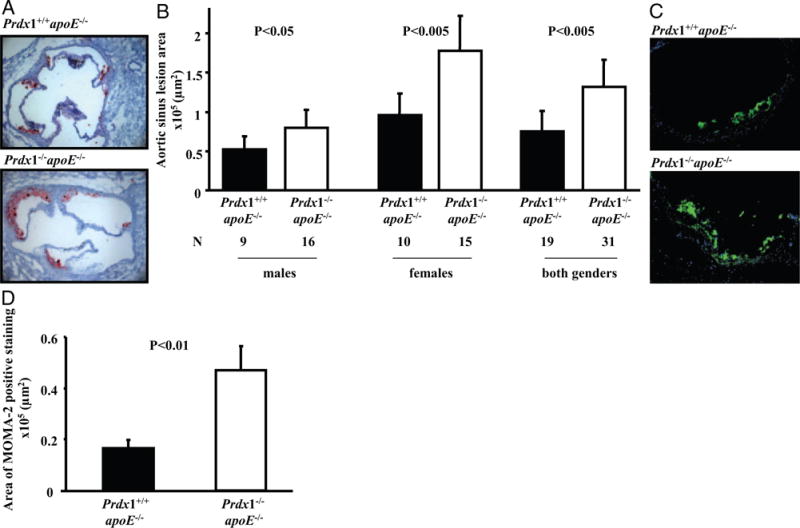
Atherosclerosis is accelerated in Prdx1−/−/apoE−/− mice. Animals were fed a normal chow diet and evaluated for aortic sinus lesions at 16 weeks of age. A, Atherosclerotic lesions were measured by lipid deposition detected with oil red O staining, which is represented here in red within aortic sinus. Representative images showing plaque (red) in female Prdx1−/−/apoE−/− and Prdx1+/+/apoE−/− mice. B, Quantitative analysis of aortic sinus lesion areas in male (left), female (middle), and combined male and female (right) Prdx1−/−/apoE−/− and Prdx1+/+/apoE−/− mice. C, Representative images of immunofluorescent staining with MOMA-2 in aortic root atherosclerotic lesions of females. D, Quantitative image analysis of MOMA-2 immunofluorescent staining in the aortic sinus atherosclerotic plaques (combined male and female). MOMA-2 (green) and HOECHST (blue) (N=15 to 21).
Discussion
In the present study, using Prdx1-deficient mice, we show that the antioxidant enzyme Prdx1 suppresses basal levels of endothelial activation and, thus, inflammation. Prdx1 deficiency caused increased numbers of rolling leukocytes with decreased velocity in unstimulated veins. This was apparently a consequence of abnormally high endothelial activation (increased WPB exocytosis and an accompanying increase in the exposure of adhesion molecules, eg, P-selectin and VWF). The observed endothelial activation attributable to Prdx1 deficiency was not caused by RBC hemolysis because there was no difference in hemoglobin content between the young (1 to 4 months) Prdx1−/− and Prdx1+/+ mice used in our studies (Table 1). Hydrogen peroxide, which is inactivated by Prdx1,32 has previously been shown to induce WPB exocytosis.33 Thus, it seems likely that the loss of ROS inactivation by Prdx1 in Prdx1−/− mice and consequent increased oxidative stress may play a major role in the abnormally high endothelial activation in these mice. Activated platelets were shown to increase leukocyte rolling in vivo.26 Circulating platelets and leukocytes were not overtly activated in Prdx1−/− mice, suggesting that oxidative stress in endothelium is most likely the prominent factor in endothelial activation.
Interestingly, Prdx1 deficiency primarily enhanced the regulated secretion pathway in endothelial cells. This likely promoted the excessive release of several proinflammatory components of WPBs,34 such as VWF25 and angiopoietin-2,35 in addition to P-selectin. In contrast, Prdx1 deficiency did not appreciably affect transcriptional regulation of receptors such as ICAM-1 and VCAM-1.
Atherosclerosis is considered a chronic inflammatory disease in which endothelial activation results in monocyte adhesion, as well as transmigration and uptake of lipoprotein cholesterol, resulting in macrophage foam cell formation and eventual progression of the initial fatty streak into advanced, occlusive lesions. A variety of studies suggest that disturbed blood flow and inflammatory mediators, with subsequent generation of ROS such as superoxide anions resulting from oxidative bursts by macrophages, may significantly contribute to the pathogenesis of atherosclerosis.36–38 Such superoxide anions can be detoxified by reduction to water and oxygen by enzymes such as superoxide dismutase, catalases, Prdxs 1 to 6, and other peroxidases.15 Indeed, Prdx1 has been shown to be expressed and specifically induced in endothelium in vitro by laminar shear stress22 and to protect red blood cells from ROS.17
In this study, we have shown that Prdx1 suppresses atherosclerotic lesion formation in apoE−/− mice when fed a standard chow diet. Atherosclerosis is a complex disease, and it is possible that the antioxidant activities of Prdx1 protect mice against atherosclerosis in multiple ways. Our findings suggest that the antiatherosclerotic activity of Prdx1 is likely attributable, at least in part, to its antiinflammatory activity (reduces basal endothelial cell activation and directly or indirectly lowers monocyte recruitment into lesions). By suppressing the build up of intracellular H2O2, Prdx1 probably influences signaling pathways in both endothelial cells and macrophages that suppress an inflammatory phenotype and thus would be expected to be antiatherogenic. H2O2 is a second messenger for various cytokines and growth factors (such as tumor necrosis factor, interferon-γ, interleukin-1) that are considered proatherogenic.39–42 Others have shown that macrophage Prdx1 synthesis is induced by exposure to oxidized LDL,43 which has been proposed to participate in atherogenesis.44 Oxidized LDL has been shown to promote adhesion and migration of monocytes to injured or activated vessel walls, where they differentiate into macrophages that are converted into lipid-laden foam cells, resulting in formation of fatty streaks.45 ROS were reported to serve as a second messenger for oxidized LDL, which induces the activation of nuclear factor κB in endothelial cells.3,42 Future studies will be required to determine whether mechanisms independent of endothelial activation are involved in Prdx1-mediated atheroprotection.
In conclusion, our study indicates that Prdx1 is an important downregulator of inflammation. Its activity is particularly important in prevention of endothelial activation. We propose that mice lacking Prdx1 could become a useful animal model to study oxidative stress at the vessel wall and chronic inflammation.
Supplementary Material
Acknowledgments
We thank Lesley Cowan for assistance in preparing the manuscript; Dr Robert Schaub from Archemix for providing the P-selectin inhibitor, aptamer ARC 5690, and control aptamer ARC 5694; Dr Petr Jarolim, MD, PhD, and Patricia Fitzpatrick from Brigham and Women’s Hospital for help with hematocrit measurement.
Sources of Funding
This work was supported by the NIH: National Heart, Lung, and Blood Institute grants R37 HL041002 (to D.D.W) and PO1 HL066105 (to D.D.W and M.K.) and National Cancer Institute grant CA077691 (R.A.V.).
Footnotes
Disclosures
None.
References
- 1.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 3.Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 4.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 5.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 6.Heistad DD. Oxidative stress and vascular disease: 2005 Duff lecture. Arterioscler Thromb Vasc Biol. 2006;26:689–695. doi: 10.1161/01.ATV.0000203525.62147.28. [DOI] [PubMed] [Google Scholar]
- 7.Ottaviano FG, Handy DE, Loscalzo J. Redox regulation in the extracellular environment. Circ J. 2008;72:1–16. doi: 10.1253/circj.72.1. [DOI] [PubMed] [Google Scholar]
- 8.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 9.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 10.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des. 2004;10:1611–1626. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 11.Tousoulis D, Antoniades C, Vassiliadou C, Toutouza M, Pitsavos C, Tentolouris C, Trikas A, Stefanadis C. Effects of combined administration of low dose atorvastatin and vitamin E on inflammatory markers and endothelial function in patients with heart failure. Eur J Heart Fail. 2005;7:1126–1132. doi: 10.1016/j.ejheart.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, Bohm M, Meinertz T, Munzel T. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 13.Yan C, Kim D, Aizawa T, Berk BC. Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler Thromb Vasc Biol. 2003;23:26–36. doi: 10.1161/01.atv.0000046231.17365.9d. [DOI] [PubMed] [Google Scholar]
- 14.Keaney JF., Jr Atherosclerosis: from lesion formation to plaque activation and endothelial dysfunction. Mol Aspects Med. 2000;21:99–166. doi: 10.1016/s0098-2997(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 15.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 16.Ishii T, Yamada M, Sato H, Matsue M, Taketani S, Nakayama K, Sugita Y, Bannai S. Cloning and characterization of a 23-kDa stress-induced mouse peritoneal macrophage protein. J Biol Chem. 1993;268:18633–18636. [PubMed] [Google Scholar]
- 17.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 18.Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med. 2005;11:571–578. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Alom-Ruiz SP, Anilkumar N, Shah AM. Reactive oxygen species and endothelial activation. Antioxid Redox Signal. 2008;10:1089–1100. doi: 10.1089/ars.2007.2007. [DOI] [PubMed] [Google Scholar]
- 21.Vischer UM, Jornot L, Wollheim CB, Theler JM. Reactive oxygen intermediates induce regulated secretion of von Willebrand factor from cultured human vascular endothelial cells. Blood. 1995;85:3164–3172. [PubMed] [Google Scholar]
- 22.Mowbray AL, Kang DH, Rhee SG, Kang SW, Jo H. Laminar shear stress up-regulates peroxiredoxins (PRX) in endothelial cells: PRX 1 as a mechanosensitive antioxidant. J Biol Chem. 2008;283:1622–1627. doi: 10.1074/jbc.M707985200. [DOI] [PubMed] [Google Scholar]
- 23.Mayr M, Chung YL, Mayr U, Yin X, Ly L, Troy H, Fredericks S, Hu Y, Griffiths JR, Xu Q. Proteomic and metabolomic analyses of atherosclerotic vessels from apolipoprotein E-deficient mice reveal alterations in inflammation, oxidative stress, and energy metabolism. Arterioscler Thromb Vasc Biol. 2005;25:2135–2142. doi: 10.1161/01.ATV.0000183928.25844.f6. [DOI] [PubMed] [Google Scholar]
- 24.Smith ML, Smith MJ, Lawrence MB, Ley K. Viscosity-independent velocity of neutrophils rolling on p-selectin in vitro or in vivo. Microcirculation. 2002;9:523–536. doi: 10.1038/sj.mn.7800165. [DOI] [PubMed] [Google Scholar]
- 25.Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13 - a new link between thrombosis and inflammation. J Exp Med. doi: 10.1084/jem.20080130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106:2334–2339. doi: 10.1182/blood-2005-04-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauhan AK, Motto DG, Lamb CB, Bergmeier W, Dockal M, Plaimauer B, Scheiflinger F, Ginsburg D, Wagner DD. Systemic antithrombotic effects of ADAMTS13. J Exp Med. 2006;203:767–776. doi: 10.1084/jem.20051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubos E, Handy DE, Loscalzo J. Role of oxidative stress and nitric oxide in atherothrombosis. Front Biosci. 2008;13:5323–5344. doi: 10.2741/3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and HDL-lipid levels in male, female, and testosterone-treated female mice from strains C57BL/6, BALB/c, and C3H. Atherosclerosis. 1987;64:215–221. doi: 10.1016/0021-9150(87)90249-8. [DOI] [PubMed] [Google Scholar]
- 31.Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis. 1999;145:301–308. doi: 10.1016/s0021-9150(99)00081-7. [DOI] [PubMed] [Google Scholar]
- 32.Kang SW, Baines IC, Rhee SG. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J Biol Chem. 1998;273:6303–6311. doi: 10.1074/jbc.273.11.6303. [DOI] [PubMed] [Google Scholar]
- 33.Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. 2005;15:302–308. doi: 10.1016/j.tcm.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271–5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004;103:4150–4156. doi: 10.1182/blood-2003-10-3685. [DOI] [PubMed] [Google Scholar]
- 36.Libby P, Plutzky J. Inflammation in diabetes mellitus: role of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma agonists. Am J Cardiol. 2007;99:27B–40B. doi: 10.1016/j.amjcard.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Cardena G, Gimbrone MA., Jr Biomechanical modulation of endothelial phenotype: implications for health and disease. Handb Exp Pharmacol. 2006:79–95. doi: 10.1007/3-540-36028-x_3. [DOI] [PubMed] [Google Scholar]
- 38.Fischer B, von Knethen A, Brune B. Dualism of oxidized lipoproteins in provoking and attenuating the oxidative burst in macrophages: role of peroxisome proliferator-activated receptor-gamma. J Immunol. 2002;168:2828–2834. doi: 10.4049/jimmunol.168.6.2828. [DOI] [PubMed] [Google Scholar]
- 39.Rus HG, Niculescu F, Vlaicu R. Tumor necrosis factor-alpha in human arterial wall with atherosclerosis. Atherosclerosis. 1991;89:247–254. doi: 10.1016/0021-9150(91)90066-c. [DOI] [PubMed] [Google Scholar]
- 40.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 41.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 42.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conway JP, Kinter M. Dual role of peroxiredoxin I in macrophage-derived foam cells. J Biol Chem. 2006;281:27991–28001. doi: 10.1074/jbc.M605026200. [DOI] [PubMed] [Google Scholar]
- 44.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 45.Gleissner CA, Leitinger N, Ley K. Effects of native and modified low-density lipoproteins on monocyte recruitment in atherosclerosis. Hypertension. 2007;50:276–283. doi: 10.1161/HYPERTENSIONAHA.107.089854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.