

Abstract
Expression of fission yeast Pho1 acid phosphatase is repressed during growth in phosphate-rich medium. Repression is mediated by transcription of the prt locus upstream of pho1 to produce a long noncoding (lnc) prt RNA. Repression is also governed by RNA polymerase II CTD phosphorylation status, whereby inability to place a Ser7-PO4 mark (as in S7A) derepresses Pho1 expression, and inability to place a Thr4-PO4 mark (as in T4A) hyper-represses Pho1 in phosphate replete cells. Here we find that basal pho1 expression from the prt–pho1 locus is inversely correlated with the activity of the prt promoter, which resides in a 110-nucleotide DNA segment preceding the prt transcription start site. CTD mutations S7A and T4A had no effect on the activity of the prt promoter or the pho1 promoter, suggesting that S7A and T4A affect post-initiation events in prt lncRNA synthesis that make it less and more repressive of pho1, respectively. prt lncRNA contains clusters of DSR (determinant of selective removal) sequences recognized by the YTH-domain-containing protein Mmi1. Altering the nucleobase sequence of two DSR clusters in the prt lncRNA caused hyper-repression of pho1 in phosphate replete cells, concomitant with increased levels of the prt transcript. The isolated Mmi1 YTH domain binds to RNAs with single or tandem DSR elements, to the latter in a noncooperative fashion. We report the 1.75 Å crystal structure of the Mmi1 YTH domain and provide evidence that Mmi1 recognizes DSR RNA via a binding mode distinct from that of structurally homologous YTH proteins that recognize m6A-modified RNA.
Keywords: RNA recognition, YTH domain, phosphate homeostasis
INTRODUCTION
Phosphate homeostasis in the fission yeast Schizosaccharomyces pombe is governed by an intricate signaling network entailing positive and negative influences on the transcription of genes encoding proteins involved in extracellular phosphate mobilization and uptake, specifically a cell surface acid phosphatase Pho1, an inorganic phosphate transporter Pho84, and a glycerophosphate transporter Tgp1 (Carter-O'Connell et al. 2012). Expression of these genes is repressed during growth in phosphate-rich medium and induced during phosphate starvation. Induction of the phosphate-regulated genes is strictly dependent on transcription factor Pho7 (Henry et al. 2011; Carter-O'Connell et al. 2012). Repression of pho1 under phosphate replete conditions is itself an active process involving protein kinases Csk1 and Cdk9 and the phosphorylation of the carboxyl-terminal domain (CTD) of the Rpb1 subunit of RNA polymerase II (Pol2) (Carter-O'Connell et al. 2012; Schwer et al. 2014, 2015).
The S. pombe Pol2 CTD consists of tandemly repeated Y1S2P3T4S5P6S7 heptapeptides. Phosphorylation of the CTD Tyr1, Ser2, Thr4, Ser5, and Ser7 residues inscribes a dynamic “CTD code” that orchestrates transcription, cotranscriptional RNA processing, and chromatin modifications (Schwer and Shuman 2011; Schwer et al. 2012; Corden 2013; Eick and Geyer 2013; Doamekpor et al. 2014; Mbogning et al. 2015). Deletion of Csk1, missense mutations of the Cdk9 T-loop threonine, and alanine mutations of Pol2 CTD phospho-sites Ser5, Pro6, or Ser7 cause derepression of pho1 in phosphate replete cells (Carter-O'Connell et al. 2012; Schwer et al. 2014, 2015). In contrast, CTD mutations T4A and S7E, and inactivation of CTD phosphatase Ssu72, hyper-repress pho1 in phosphate replete cells (Schwer et al. 2014, 2015).
Recent studies have unveiled a role for long noncoding (lnc) RNAs in phosphate homeostasis. The lncRNA prt, initiating 1198 nucleotides (nt) upstream of the pho1 AUG start codon (Fig. 1), represses pho1 in cis during phosphate replete growth (Lee et al. 2013; Shah et al. 2014). Transcription of the pho1 mRNA initiates 51 nt upstream of the pho1 AUG start codon (Fig. 1). Unlike pho1, expression of prt is independent of Pho7 (Schwer et al. 2015). The cis and trans factors that control prt expression are presently uncharted. In the case of tgp1, the lncRNA nc-tgp1, initiating 1865 nt upstream of the tgp1 AUG start codon, represses tgp1 in cis during phosphate replete growth (Ard et al. 2014). prt and nc-tgp1 levels are restrained by the action of the nuclear exosome; they increase sharply when the Rrp6 nuclear exosome subunit is absent. The turnover of these lncRNAs by the exosome is aided by Mmi1 (Ard et al. 2014; Shah et al. 2014), a YTH domain-containing protein that recognizes determinants of select removal (DSRs) in the target RNA (Harigaya et al. 2006; Yamashita et al. 2012). YTH domain proteins from other eukaryal taxa are specific “readers” of the modified nucleoside N6-methyladenosine (m6A) in RNA (Li et al. 2014; Luo and Tong 2014; Xu et al. 2014, 2015), but it is not known whether or how inscription of an m6A mark might affect phosphate homeostasis.
FIGURE 1.
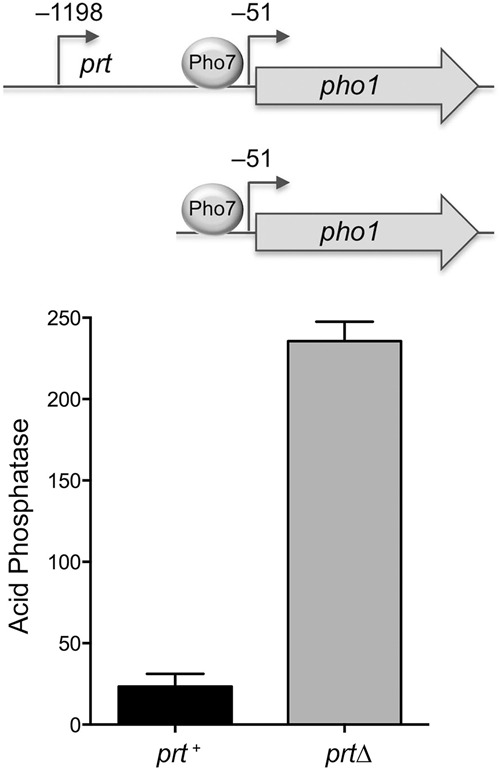
Plasmid reporter for dissection of prt control of pho1 expression. The plasmid-borne prt–pho1 cassette (prt+) is shown with the mapped transcription start sites of the prt lncRNA and pho1 mRNA (relative to the pho1 AUG start codon) indicated by arrows. Transcription factor Pho7 is required for pho1 transcription and binds to DNA upstream of the pho1 transcription start site. A prtΔ version of the reporter that contains only 283 nt of genomic DNA upstream of the pho1 transcription start site is shown. The prt+ and prtΔ plasmids (LEU2) were introduced into pho1Δ yeast cells. Acid phosphatase activity was assayed by conversion of p-nitrophenylphosphate to p-nitrophenol. The y-axis specifies the phosphatase activity (A410) normalized to input cells (A600). The error bars denote SEM.
Although there are divergent views on the role of heterochromatin in the lncRNA-mediated repression of fission yeast phosphate-responsive genes, the unifying model is that transcription of the upstream lncRNA in cis is what interferes with expression of the downstream genes encoding Pho1 or Tgp1 in phosphate replete cells (Ard et al. 2014). The signals that alter this dynamic during phosphate starvation, and the role of the Pol2 CTD in establishing or overriding the repressed state imposed by lncRNA synthesis and turnover, are presently ill-defined.
Here we interrogate influences on the repression of pho1 by prt. We address the following questions: (i) What comprises the prt and pho1 promoters? (ii) Does prt promoter strength correlate with pho1 repression? (iii) Do hyper-repressing (T4A) and derepressing (S7A) CTD alleles affect transcription driven by the prt and pho1 promoters? (iv) What are the DSR acting elements in the prt RNA and how does their mutation affect pho1 repression/derepression? (v) What determines DSR recognition by the Mmi1 YTH domain? We report the crystal structure of the Mmi1 YTH domain and provide evidence that Mmi1 recognizes DSR RNA via a binding mode distinct from that used by YTH m6A readers.
RESULTS
Plasmid-based reporter assay to study repression and derepression of pho1
Prior manipulations of the prt–pho1 chromosomal locus were performed by replacing prt or its putative promoter with a selectable marker gene ura4+ (Lee et al. 2013; Shah et al. 2014). To allow finer changes, we cloned the prt–pho1 cassette (spanning 633 nt upstream of the prt transcription initiation site to 647 nt downstream from the pho1 translation stop codon) into a fission yeast plasmid, which we introduced into a pho1Δ strain from which we had deleted the chromosomal pho1 gene plus 262 nt of 5′-flanking DNA. We constructed a prtΔ version of the plasmid that included the pho1 gene plus 283 nt of DNA upstream of the pho1 transcription start site (Fig. 1). Plasmid-bearing pho1Δ cells were grown to mid-log phase in liquid culture in phosphate replete medium. Acid phosphatase activity (a gauge of Pho1 enzyme level) was quantified by incubating suspensions of serial dilutions of the cells for 5 min with p-nitrophenylphosphate and assaying colorimetrically the formation of p-nitrophenol. Product formation in the linear response range, normalized to cell density, is plotted on the y-axis in Figure 1. The basal phosphatase activity of prt–pho1 cells was increased 10-fold by deletion of prt. (The pho1Δ strain itself had virtually undetectable phosphatase activity; not shown.) Thus, prt is critical for repression of Pho1 under phosphate replete conditions in the plasmid context.
prt transcription: What comprises a prt promoter?
To address this question, we constructed a plasmid reporter in which the pho1 ORF was fused immediately downstream from a fragment of genomic DNA containing the prt transcription start site, a 20 nt 5′-UTR, and 633 nt of 5′ flanking DNA (Fig. 2A). Because this plasmid generated vigorous acid phosphatase activity when introduced into a pho1Δ strain (Fig. 2B), we surmised that the 633-nt segment embraces a prt promoter and potential regulatory elements. To demarcate cis-acting elements, we serially truncated the 5′ flanking DNA to positions −444, −110, −61 and −5 upstream of the prt transcription start site. The acid phosphatase activity of the plasmid-bearing pho1Δ cells (Fig. 2B) indicated that the 5′-flanking 110-nt segment sufficed for prt promoter-driven expression. Truncation to −61 reduced phosphatase activity to 22% of that driven by the longer flanking DNAs; further truncation to leaving only 5 nt upstream of the prt start site effaced phosphatase activity (Fig. 2B). Primer extension analysis of the pho1 transcript driven by the series of prt promoters revealed that the 5′ ends mapped to the expected prt initiation site and that transcript abundance, as gauged by the level of the primer extension product, correlated with acid phosphatase activity. To wit, prt-driven pho1 RNAs were similar for the −633, −444, and −110 reporters, reduced by truncation to −61, and eliminated by truncation to −5 (Fig. 2C). The DNA segment from −61 to −5 that is essential for prt transcription contains a candidate TATA box sequence 5′-TATATATA (Ahn et al. 2012).
FIGURE 2.
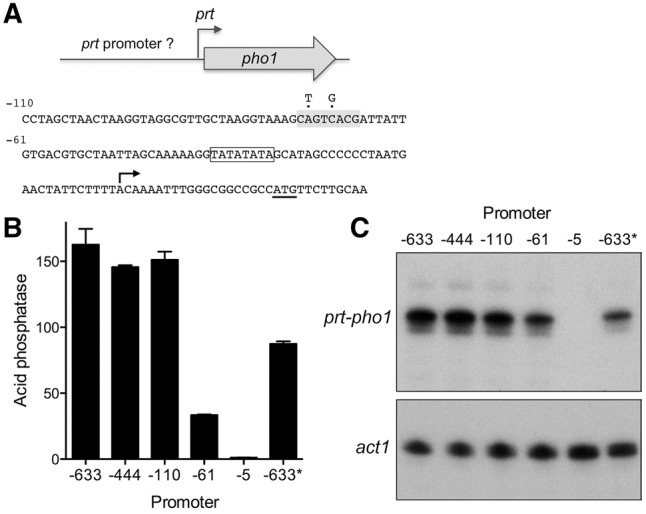
Delineation of the prt promoter. (A) Plasmid reporter of prt promoter activity. The pho1 ORF was fused immediately downstream from a fragment of genomic DNA containing the prt transcription start site, a 20-nt 5′-UTR, and 633 nt of 5′ flanking prt DNA (presumed to include the prt promoter). Serial truncation of the upstream margins of the 5′ flanking prt DNA were made at positions −444, −110, −61, and −5 relative to the prt transcription start site. The nucleotide sequence from position −110 to +32 is shown below the cartoon depiction of the reporter. The pho1 AUG start codon is underlined. The prt transcription start site is indicated by the arrow. A putative TATA element is outlined by a box. A putative HomolD element is shaded gray. Two nucleobase changes shown above the sequence were introduced into the HomolD element in the context of the −633 reporter (B) Acid phosphatase activity of pho1Δ cells bearing the indicated reporter plasmids (LEU2). The −633* reporter plasmid contains the two nucleobase changes in the HomolD element. (C) Primer extension analysis of the prt-driven pho1 transcript was performed with total RNA isolated from pho1Δ cells bearing the indicated reporter plasmids (top panel). Primer extension analysis of actin mRNA from the same samples was performed as a control (bottom panel).
The nucleotide sequence of the 110-nt DNA segment flanking the prt start site is shown in Figure 2A. The promoter deletion analysis suggested the presence of a positive transcriptional element in the interval between −61 and −110. Our inspection of this segment disclosed the sequence 5′-CAGTCACG (shaded gray in Fig. 2A) as a potential HomolD-box. The HomolD element CAGTCAC(A/G) functions as a core Pol2 promoter signal in fission yeast, especially in genes encoding ribosomal proteins (Witt et al. 1993, 1995; Gross and Käufer 1998). The HomolD sequence is a binding site for the essential fission yeast transcription factor Rrn7 (Rojas et al. 2011). To evaluate whether the HomolD box plays a role in prt promoter function, we introduced into the −633 reporter plasmid two nucleotide changes in the HomolD box that have been reported to inactivate the element (Rojas et al. 2011). The HomolD point mutations reduced acid phosphatase activity by half (Fig. 2B). The HomolD mutation also reduced the prt-driven pho1 transcript level, without affecting the site of transcription initiation from the prt promoter (Fig. 2C). These findings suggest a novel role for HomolD in lncRNA transcription.
pho1 transcription: demarcating a pho1 promoter
The plasmid reporter experiments in Figure 1 established that 283 nt of DNA 5′ of the pho1 transcription start site suffice to drive vigorous Pho1 expression. The −283 pho1 reporter plasmid generated no acid phosphatase activity in a pho7Δ strain (not shown). Because Pho1 expression is stringently dependent on transcription factor Pho7, the −283 flanking DNA segment must include the requisite Pho7 binding site. Genome-wide ChIP-seq analysis revealed a peak of Pho7 occupancy between positions −280 and −180 upstream of pho1 ATG start codon (Carter-O'Connell et al. 2012), which corresponds to the region from −229 to −121 upstream of the pho1 transcription start site. Here we further truncated the 5′ flanking DNA to positions −219, −189, −169, −113, and −58 from the pho1 transcription start site in a pho1 reporter plasmid and gauged the acid phosphatase activity of the plasmid-bearing pho1Δ cells. Pho1 activity declined progressively as the upstream DNA was serially deleted to positions −219, −189, and −169, whereby these constructs generated 76%, 31%, and 8% of the Pho1 activity of the −283 promoter plasmid, respectively (Fig. 3A). Further truncations to −113 and −58 yielded 4% and 2% Pho1 activity. Measurements of pho1 mRNA levels by RT-qPCR mirrored the incremental declines in Pho1 activity with promoter truncations (Fig. 3B). These results suggested that multiple Pho7 binding sites might populate the DNA segment between −283 and −169 from the transcription start site. The DNA element recognized by Pho7 is unknown, and prior attempts to identify a DNA binding motif based on genome-wide Pho7 ChIP-seq data were unsuccessful (Carter-O'Connell et al. 2012). Pho7 belongs to the zinc binuclear cluster family of fungal DNA-binding transcription regulators. Many of the zinc cluster proteins recognize CGG triplets, singly or as everted, inverted, or direct repeats (MacPherson et al. 2006). The −283 pho1 promoter DNA includes six CGG triplets. We found that deleting the GG dinucleotide of one of these triplets, within the sequence −158TATTCGGAAAT−148, abolished Pho1 expression (not shown). Finer delineation of the pho1 promoter will hinge on purification of active Pho7 protein and direct footprinting of its DNA contacts.
FIGURE 3.
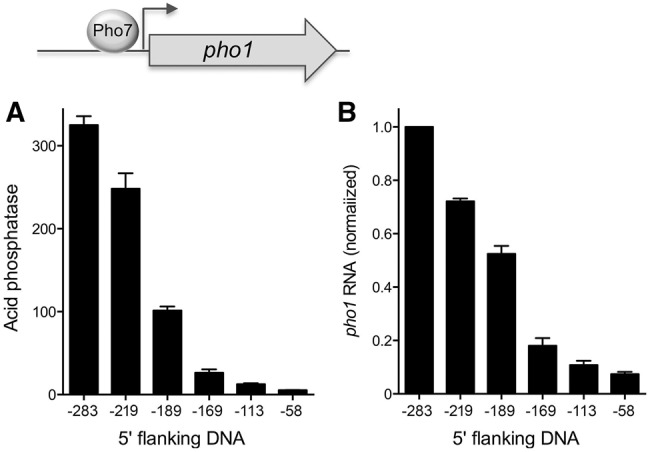
Demarcation of the pho1 promoter. Plasmid reporters of promoter activity contained 283, 219, 189, 169, 113, and 58 nt of genomic DNA upstream of the pho1 transcription start site. (A) Acid phosphatase activity of pho1Δ cells bearing the indicated pho1 reporter plasmids (LEU2). (B) RT-qPCR analysis of the pho1 transcript was performed with total RNA isolated from pho1Δ cells bearing the indicated reporter plasmids. The pho1 transcript levels are normalized to that of the −283 reporter (defined as 1.0). Error bars indicate SEM.
prt and pho1 promoter activities are not affected by Pol2 CTD mutations T4A and S7A
Repression of pho1 expression from the chromosomal locus in phosphate replete cells is affected by Pol2 CTD phosphorylation status, as gauged by the impact of CTD mutations that eliminate particular phosphorylation marks. For example, inability to place a Ser7-PO4 mark in S7A cells derepresses Pho1 expression and inability to place a Thr4-PO4 mark in T4A cells hyper-represses Pho1 under phosphate replete conditions. It is conceivable that these CTD effects on pho1 might simply reflect their impact on transcription of the upstream prt locus. To query whether the CTD mutations influence prt promoter-driven transcription, we introduced the prt −633 promoter-driven pho1 reporter plasmid into pho1Δ cells in which we replaced the rpb1 chromosomal locus with alleles rpb1-CTD-WT, -CTD-T4A, or -CTD-S7A (Schwer and Shuman 2011). We found that the three strains expressed similar levels of acid phosphatase (Fig. 4A) and pho1 mRNA (Fig. 4B) when grown in phosphate replete medium. We infer that hyper-repression of pho1 in T4A cells and derepression of pho1 in S7A cells under phosphate rich conditions is probably not caused by changes in the activity of the prt promoter.
FIGURE 4.
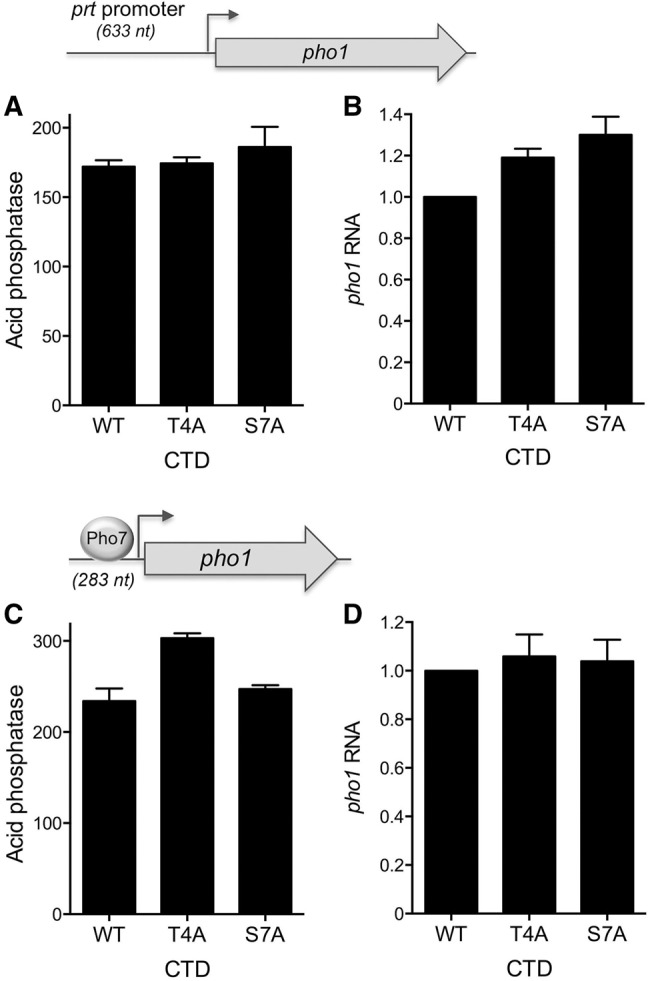
Effect of rpb1-CTD T4A and S7A mutations on prt and pho1 promoter activities. S. pombe pho1Δ rpb1-CTD-WT, rpb1-CTD-T4A, and rpb1-CTD-S7A strains were transformed with reporter plasmids (LEU2) in which pho1 expression was driven by the prt promoter (top panels A and B) or the pho1 promoter (bottom panels C and D). Acid phosphatase activity is shown in A and C. RT-qPCR analysis of the pho1 transcript is shown in B and D. The pho1 transcript levels in T4A and S7A cells are normalized to those of rpb1-CTD-WT cells (defined as 1.0). Error bars indicate SEM.
An alternative explanation of the hyper-repression of pho1 in T4A cells and the derepression in S7A cells is that the T4A and S7A mutations, respectively, lower and raise the intrinsic activity of the pho1 promoter, conceivably affecting the response of Pol2 to the key transcription factor Pho7. To evaluate this idea, we introduced the pho1 −283 promoter-driven pho1 reporter plasmid into pho1Δ CTD-WT, CTD-T4A, and CTD-S7A cells and gauged their acid phosphatase activity in phosphate replete medium (Fig. 4C). There was no difference in Pho1 expression in WT versus S7A cells. Pho1 activity in T4A cells was slightly higher than WT. pho1 mRNA levels were similar in WT, T4A, and S7A cells (Fig. 4D). These results signify that the effects of T4A and S7A on the isolated pho1 promoter do not mimic their effects on pho1 expression in the context of the prt–pho1 locus. Rather, the results suggest that CTD mutations influence the response of pho1 transcription to upstream prt transcription.
prt promoter mutations derepress pho1
The inverse relationship between prt promoter strength and expression of the downstream pho1 gene was affirmed by introducing the prt promoter truncations into the tandem prt–pho1 cassette of the reporter plasmid, which revealed that deletion from −110 to −61 elicited an acute increase in acid phosphatase activity in phosphate replete cells (Fig. 5A). The contribution of the HomolD element CAGTCACG was gauged by replacing it with the mutated version CTGTGACG in the context of the −633 promoter of the tandem prt–pho1 cassette. This mutation resulted in a 50% increase in acid phosphatase activity associated with a 50% decrement in prt transcript level (Fig. 5B). The putative TATA box was apparently critical for prt promoter function and prt-mediated pho1 repression, as surmised from the effects of replacing the TATATATA sequence with GCGGCCGC (Fig. 5B). This maneuver resulted in a fourfold increase in acid phosphatase activity associated with a 16-fold decrease in prt transcript level. Combining the HomolD and TATA mutations had an additive effect in further raising the acid phosphatase activity and lowering the prt RNA level (Fig. 5B).
FIGURE 5.

prt promoter deletion derepresses pho1. (A) The indicated prt promoter truncations were introduced into the tandem prt–pho1 cassette depicted in the cartoon. The graph shows the acid phosphatase activity of pho1Δ cells bearing the indicated reporter plasmids (kanMX). (B) prt promoters with the indicated mutations (mut) of the HomolD and TATA elements were introduced into the −633 prt promoter of the tandem prt–pho1 cassette. The left panel shows the acid phosphatase activity of pho1Δ cells bearing reporter plasmids with wild-type (WT) or mutated prt promoter elements as specified. The right panel shows RT-qPCR analysis of the prt transcript levels; values are normalized to that of cells with the wild-type prt promoter (defined as 1.0). Error bars indicate SEM.
prt lncRNA contains multiple potential DSR sequences
The prt lncRNA is targeted for decay by the nuclear exosome, which limits its steady-state level in wild-type cells. prt was initially identified in rrp6Δ cells (which lack a catalytic subunit of the nuclear exosome) as an RNA transcribed from the ∼1.6-kb region between the pho84 and pho1 genes and extending through the pho1 open reading frame (Lee et al. 2013; Shah et al. 2014). The prt transcript, initiating 1198 nt upstream of the pho1 start codon, was also detected in wild-type rrp6+ cells by primer extension analysis (Schwer et al. 2014). The level of the prt transcript, as gauged by RT-qPCR, was 50-fold higher in phosphate replete rrp6Δ cells versus rrp6+ cells (Schwer et al. 2015), which accords with the hyper-repression of Pho1 acid phosphate activity in phosphate replete rrp6Δ cells (Shah et al. 2014; Schwer et al. 2015). Shah et al. (2014) noted that the prt RNA contains a cluster of three DSR consensus sequences (5′-UUAAAC) in the region from +399 to +438 (underlined in Fig. 6A as DSRx3-2). Tandem DSRs promote the selective degradation of meiotic mRNAs expressed during vegetative growth of fission yeast, via their recognition by the YTH-domain protein Mmi1 (Harigaya et al. 2006; Yamashita et al. 2012), which interacts with the nuclear exosome and with the Ccr4-Not complex (Cotobal et al. 2015; Ukleja et al. 2016). During meiosis, Mmi1 is sequestered by a DSR-containing lncRNA transcribed at the sme2 locus (Harigaya et al. 2006; Shichino et al. 2014), allowing stabilization of DSR-containing meiotic mRNAs. Our inspection of the prt RNA sequence revealed six additional potential DSR elements upstream of the DSRx3-2 cluster noted previously. These are 5′-UUAAAU hexanucleotides that differ from the consensus DSR by a C-to-U change at position 6. Moreover, three of these 5′-UUAAAU elements form a tandem cluster in the prt region from +89 to +108 (underlined in Fig. 6A as DSRx3-1). It was shown previously that certain DSR variant hexanucleotide motifs can augment the function of the consensus DSR element to promote RNA elimination (Yamashita et al. 2012). However, 5′-UUAAAU was not among the variant motifs tested.
FIGURE 6.

Effect of mutating the DSR clusters in the prt lncRNA on pho1 expression. (A) Cartoon of the prt and pho1 genes, highlighting the presence of two clusters of DSR elements (boxes 1 and 2) within the 5′ segment of the prt transcription unit. The nucleotide sequence of the 5′ segment of prt lncRNA is shown below the cartoon, with the transcription start site indicated by the arrow. DSR consensus hexamers UUAAAC and variant hexamers UUAAAU are shaded gray. Clustered DSR triple-repeats DSRx3-1 (box 1) and DSRx3-2 (box 2) are underlined. Mutant 1 and mutant 2 versions of the prt–pho1 reporter cassette had DNA mutations that resulted in the indicated changes in the sequence of the prt lncRNA. (B, left panel) Acid phosphatase activity of pho1Δ cells bearing the indicated prt–pho1 reporter plasmids (kanMX). (Right panel) RT-qPCR analysis of the prt transcript was performed with total RNA isolated from pho1Δ cells bearing the DSR WT or DSR mut1 + mut2 reporter plasmids. The prt transcript levels are normalized to that of the wild-type DSR reporter (defined as 1.0). (C) Effect of DSR mutations on the phosphate starvation response. Fission yeast pho1Δ cells bearing the indicated prt–pho1 reporter plasmids were grown in YES medium to A600 of 0.5–0.7. The cells were harvested, washed in water and, after withdrawing an aliquot to measure phosphatase activity (time 0), were transferred to PMG medium (+ kanamycin) lacking exogenous phosphate. Pho1 activity was assayed after incubation for the times specified. Error bars indicate SEM.
Mutations of the prt UUAAAC and UUAAAU clusters hyper-repress pho1
To evaluate the role of the DSR clusters in prt-promoted pho1 repression, we introduced compound mutations (mut1 and mut2) into each of the hexanucleotide motifs comprising the DSRx3-1 and DSRx3-2 sequences of a prt–pho1 reporter plasmid (Fig. 6A). Mut2 of the consensus DSRs in the DSRx3-2 cluster reduced acid phosphatase activity in phosphate replete cells to 18% of the wild-type prt control (Fig. 6B). Mut1 of the variant DSRs in the DSRx3-1 cluster lowered acid phosphatase activity to 44% of wild-type prt. Combining the mut1 and mut2 DSR cluster mutations had an additive effect, reducing Pho1 activity to 9% of the wild-type prt level (Fig. 6B). The hyper-repression of Pho1 expression by the mut1 + mut2 DSR mutant correlated with fivefold increased levels of the prt transcript in phosphate replete mut1 + mut2 cells (Fig. 6B).
We gauged the effect of the DSR mutations on the responsiveness of Pho1 expression to phosphate starvation, as reflected in the induction of acid phosphatase activity as a function of time after transfer to medium lacking phosphate (Fig. 6C). Wild-type DSR cells evinced a steady increase in acid phosphatase activity from 2 to 5 h post-starvation. Whereas DSR-mut1 and DSR-mut2 cells maintained lower basal levels of phosphatase activity than WT for up to 2 h post-starvation, both strains responded to starvation with steady increases in Pho1 activity between 2 and 5 h post-starvation. Combining the mut1 and mut2 DSR mutations delayed the onset of acid phosphatase production, such that the starvation response was evident between 3 and 5 h (Fig. 6C).
Effect of DSR mutations on prt and pho1 expression in rpb1-CTD-WT and -S7A cells
We examined the effects of the DSR mut1 + mut2 mutation in rpb1-CTD-WT versus rpb1-CTD-S7A cells under phosphate replete conditions. The DSR mut1 + mut2 change elicited a 12-fold decrement in acid phosphatase activity in CTD-WT cells (Fig. 7A), associated with a sixfold increase in the level of the prt transcript (Fig. 7B). CTD-S7A derepressed the Pho1 acid phosphatase, with little effect on prt transcript level. The salient finding was that DSR mut1 + mut2 resulted in a sevenfold reduction in Pho1 activity in CTD-S7A cells (Fig. 7A) while increasing prt transcript level by 10-fold relative to CTD-S7A cells bearing the prt–pho1 reporter with wild-type DSRs (Fig. 7B). Thus, the hyper-repressive effect of DSR erasure on Pho1 expression, mediated via increasing prt lncRNA, is preserved in S7A cells, notwithstanding that the baseline Pho1 activity is higher absent the Ser7 CTD mark.
FIGURE 7.
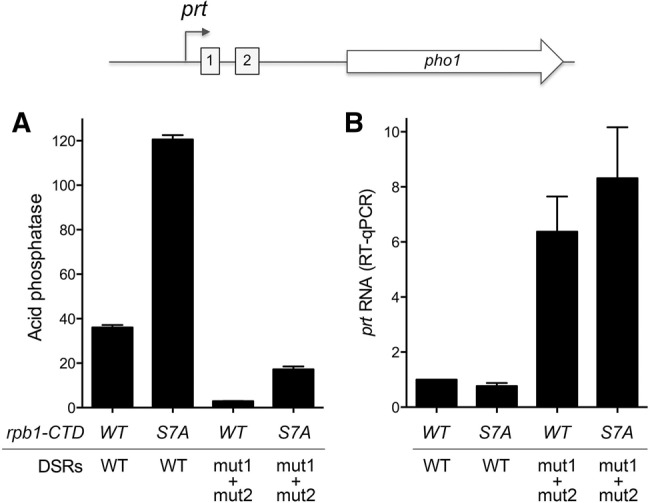
Effect of DSR mutation on prt and pho1 expression in rpb1-CTD-WT and -S7A cells. pho1Δ rpb1-CTD-WT and pho1Δ rpb1-CTD-S7A strains were transformed with prt–pho1 reporter plasmids (kanMX) with wild-type or mut1 + mut2 DSRs. (A) Acid phosphatase activity. (B) RT-qPCR analysis of the prt transcript. The prt transcript levels are normalized to that of rpb1-CTD-WT cells with the wild-type DSR reporter (defined as 1.0). Error bars indicate SEM.
DSR RNA binding by the Mmi1 YTH domain
Schizosaccharomyces pombe Mmi1 is a 488-amino acid polypeptide; it shares 275 positions of side chain identity/similarity with the homologous Mmi1 protein of S. japonicum, with the greatest degree of conservation spanning their C-terminal putative YTH domains (Fig. 8B). YTH domain proteins have attracted great attention recently as “readers” of the N6-methyladenine (m6A) mark in eukaryal mRNA (Li et al. 2014; Luo and Tong 2014; Xu et al. 2014, 2015). m6A-reading YTH proteins recognize the RNA sequence 5′-GG(m6A)C, which bears no resemblance to the fission yeast DSR sequence that attracts Mmi1. Initial experiments that demonstrated RNA binding by Mmi1 were performed by electrophoretic mobility shift assay (EMSA) using a GST-Mmi1 fusion protein and RNA ligands containing tandem copies of the consensus UUAAAC sequence (Yamashita et al. 2012; Shichino et al. 2014). It was found that whereas a full-length GST-Mmi1 fusion bound RNA, a truncated version GST-Mmi1(1–292) lacking the C-terminal YTH domain did not. Because GST-fusion forces homodimerization of the Mmi1 protein, and because the RNA probes were shifted into the polyacrylamide gel wells when bound by GST-Mmi1, this approach does not provide accurate information about RNA–protein complex stoichiometry, or potential cooperativity of binding. Our aim here was to interrogate the RNA binding properties of the Mmi1 YTH domain, absent any tag that affects its quaternary structure.
FIGURE 8.

Purification of recombinant Mmi1 YTH domain. An alignment of the amino acid sequences of S. pombe (Spo) and S. japonicum (Sja) Mmi1 proteins is shown in panel B. Positions of side-chain identity/similarity are indicated by dots above the alignment. Gaps in the alignment are denoted by dashes. Three versions of the Mmi1 C-terminal YTH domain, extending from the Ser301, Ser311, or Ser319 sites (demarcated by arrows in panel B) to the C-terminus were produced in E. coli and purified as described in Materials and Methods. Aliquots (5 µg) of the three purified Mmi1 YTH domains were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown in panel A. The N-terminus of each YTH domain is indicated above the lane. The positions and sizes (kDa) of marker polypeptides are indicated on the left.
We produced three recombinant versions of the Mmi1 C-terminal YTH domain, initiating from the sites demarcated by arrows in Figure 8B, in E. coli as His10Smt3 fusions. The recombinant Mmi1-YTH proteins were isolated from soluble bacterial extracts by nickel-affinity chromatography. The His10Smt3 tags were removed with the Smt3-specific protease Ulp1 and the native Mmi1-YTH proteins were separated from the tag by a second round of nickel-affinity chromatography and purified further by gel filtration, during which they eluted as monomers. The Mmi1 YTH domains were pure and displayed the expected incremental size changes when analyzed by SDS-PAGE (Fig. 8A).
EMSA analysis of the binding of Mmi1(301–488) to two 5′ 32P-labeled RNA probes is shown in Figure 9A. The 14-mer RNA in the left panel corresponds to the second consensus DSR element of the prt DSRx3-2 cluster; the 24-mer RNA in the right panel contains the tandem first and second consensus DSR sequences of DSRx3-2, which are separated by an 8-nt U-rich spacer. Mixture of 100 nM labeled 14-mer single-DSR RNA with Mmi1(301–488) resulted in the formation of a single discrete RNA•protein complex that migrated into the polyacrylamide gel; the abundance of the complex increased with Mmi1(301–488) concentration until virtually all of the input RNA was shifted (Fig. 9A, left panel). The two shorter versions of the YTH domain, Mmi1(311–488) and Mmi1(319–488), also formed single discrete RNA•protein complexes on the 14-mer single-DSR RNA (Fig. 9B). We conclude that the C-terminal YTH domain suffices for Mmi1 recognition of a single DSR.
FIGURE 9.
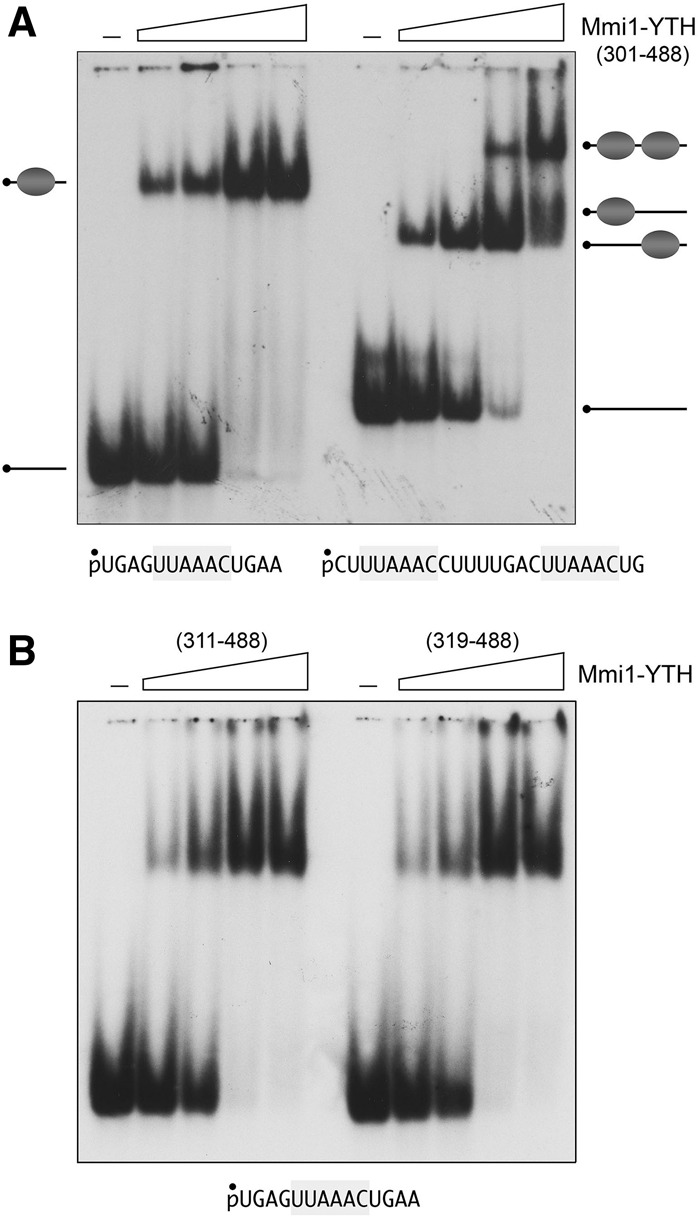
DSR RNA binding by the Mmi1 YTH domain. EMSA was performed as described in Materials and Methods. (A) The RNA binding reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer or 24-mer RNAs (shown at bottom, with one or two DSR hexamers, highlighted in gray), and increasing concentrations of Mmi1(301–488) protein (125, 250, 500, or 1000 nM, proceeding from left to right in each titration series). (B) RNA binding reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer DSR RNA and 125, 250, 500, or 1000 nM Mmi1(311–488) or Mmi1(319–488). Mmi1-YTH was omitted from the control reactions in lanes indicated by (–). The free RNAs and RNA•protein complexes were resolved by native PAGE and visualized by autoradiography of the dried gels.
When presented with the 24-mer dual-DSR RNA, Mmi1(301–488) formed a single discrete RNA•protein complex at lower protein concentrations that was converted progressively to a more slowly migrating complex as Mmi1(301–488) concentration was increased (Fig. 9A, right panel). The evolution of the RNA•YTH and RNA•(YTH)2 complexes as a function of protein concentration is indicative of sequential (noncooperative) mode of Mmi1-YTH binding to the DSR elements of the 24-mer RNA. [If binding of two YTH domains had been highly cooperative, we would have expected to see accumulation of the RNA•(YTH)2 complex at protein concentrations at which there was a significant fraction of residual unbound RNA.] The EMSA format does not distinguish whether Mmi1-YTH binds randomly to one or the other of the two tandem DSRs in the 24-mer, or if there is a preferred order of binding. For convenience, we depict a random process in Figure 9A, in which the RNA•YTH shifted species is a mixture of RNAs with the YTH domain bound to the first and second DSRs.
Specificity of RNA binding
The YTH domain Mmi1(319–488) failed to bind to a 5′ 32P-labeled 14-mer DNA of identical nucleobase sequence (with T instead of U) to that of the single-DSR RNA (Fig. 10A). Mmi1-YTH also failed to bind to a 14-mer RNA in which the consensus DSR hexanucleotide was mutated to CCGGGU, i.e., changing each nucleobase without altering the pyrimidine/purine content of the motif (Fig. 10B). In contrast, Mmi1-YTH bound equally well to a 14-mer RNA containing a consensus UUAAAC motif or the variant UUAAAU motif (Fig. 10C). This result hints that the contribution of the prt variant DSRx3-1 cluster to the modulation of Pho1 activity and prt level in phosphate replete cells could reflect a direct role in attracting Mmi1.
FIGURE 10.
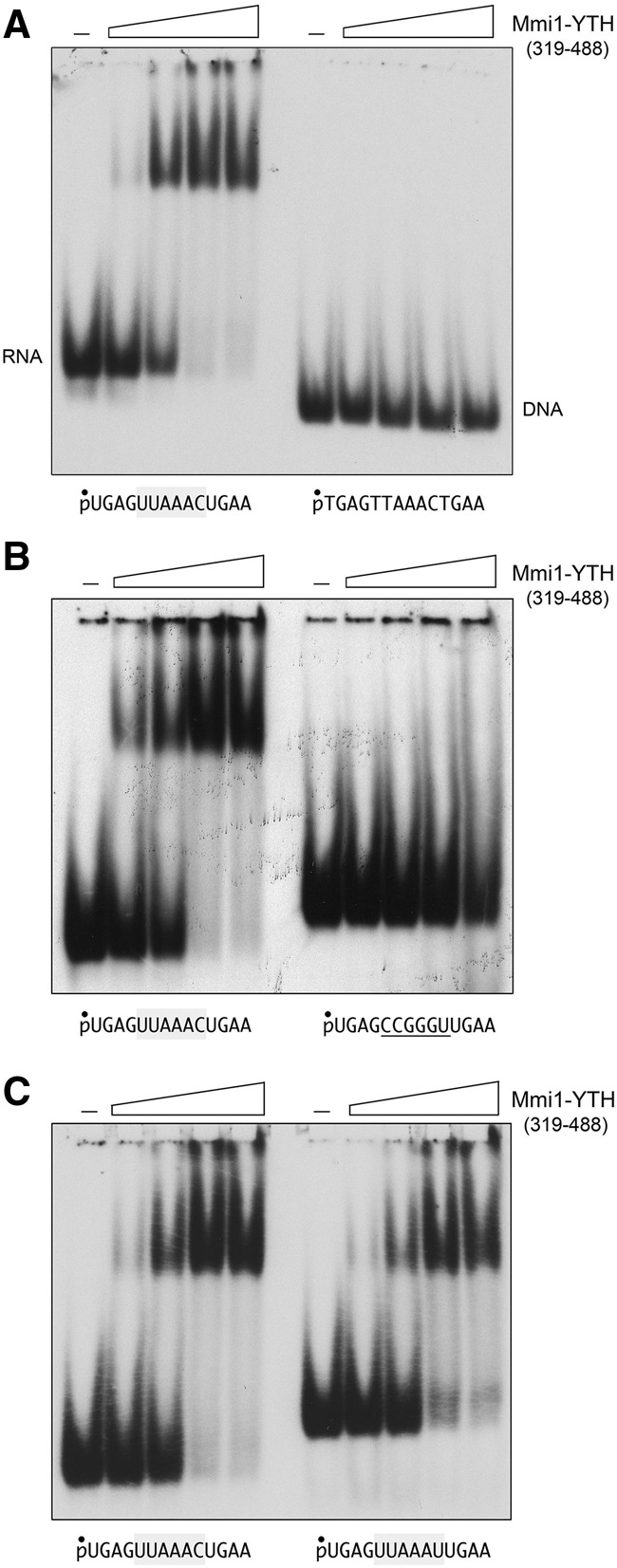
Specificity of RNA binding by the Mmi1 YTH domain. (A) RNA versus DNA recognition. The nucleic acid binding reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer DSR RNA or 14-mer DSR DNA (shown at bottom) and 125, 250, 500, or 1000 nM Mmi1(319–488). Mmi1-YTH was omitted from the control reactions in lanes indicated by (–). (B) DSR RNA hexanucleotide recognition. The RNA binding reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer RNA with a consensus DSR hexamer UUAAAC (left panel) or 14-mer RNA with CCGGGU in lieu of the consensus DSR (right panel) and 125, 250, 500, or 1000 nM Mmi1(319–488). (C) Variant DSR recognition. RNA binding reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer RNA with a consensus DSR hexamer UUAAAC (left panel) or 14-mer RNA with a variant UUAAAU hexamer (right panel) and 125, 250, 500, or 1000 nM Mmi1(319–488). Mmi1(319–488) was omitted from the control reactions in lanes indicated by (–). The free nucleic acids and nucleic acid•protein complexes were resolved by native PAGE and visualized by autoradiography.
Structure of the Mmi1 YTH domain
Crystals of native and SeMet-substituted Mmi1(311–488) (grown as described in Materials and Methods) were in space group P212121 and diffracted X-rays to 1.75 Å and 2.05 Å resolution, respectively. SAD phases were used to solve the structure of the native Mmi1(311–488) protein, which was refined to R/Rfree of 18.7/20.5 at 1.75 Å resolution (Supplemental Table S1). The asymmetric unit contained four protomers. Analysis of the protomer interfaces in PISA (Krissinel and Henrick 2007) calculated Complex Formation Significance scores of 0.000, signifying that protomer contacts may be solely a result of crystal packing, i.e., Mmi1(311–488) crystallized as a monomer. The tertiary structure of Mmi1(311–488) is shown in stereo view in Figure 11A. The secondary structure elements are aligned over the Mmi1(311–488) amino acid sequence in Figure 11B. Mmi1(311–488) comprises a central five-strand β barrel with topology β1↑•β3↑β4↓•β5↑•β2↓. The barrel is flanked by four α helices (Fig. 11A). A DALI search (Holm et al. 2008) of the Mmi1(311–488) structure against the protein database identified several known YTH domain proteins as top hits, with Z scores of 13.1–15.4 and rmsds of 2.1–3.2 Å at 126–132 Cα positions. A structure-based alignment of the primary and secondary structures of Mmi1 YTH domain and human YTHDF2 (pdb 4RDN) (Li et al. 2014) highlights 47 positions of side chain identity/similarity (denoted by • in Fig. 11B). One salient difference between Mmi1(311–488) and YTHDF2 resides in the segment between the β5 strands and the C-terminal α helices. Whereas this region embraces α helix 3 in Mmi1(311–488), it comprises two 310 helices and three short β strands in YTHDF2 (Fig. 11B).
FIGURE 11.
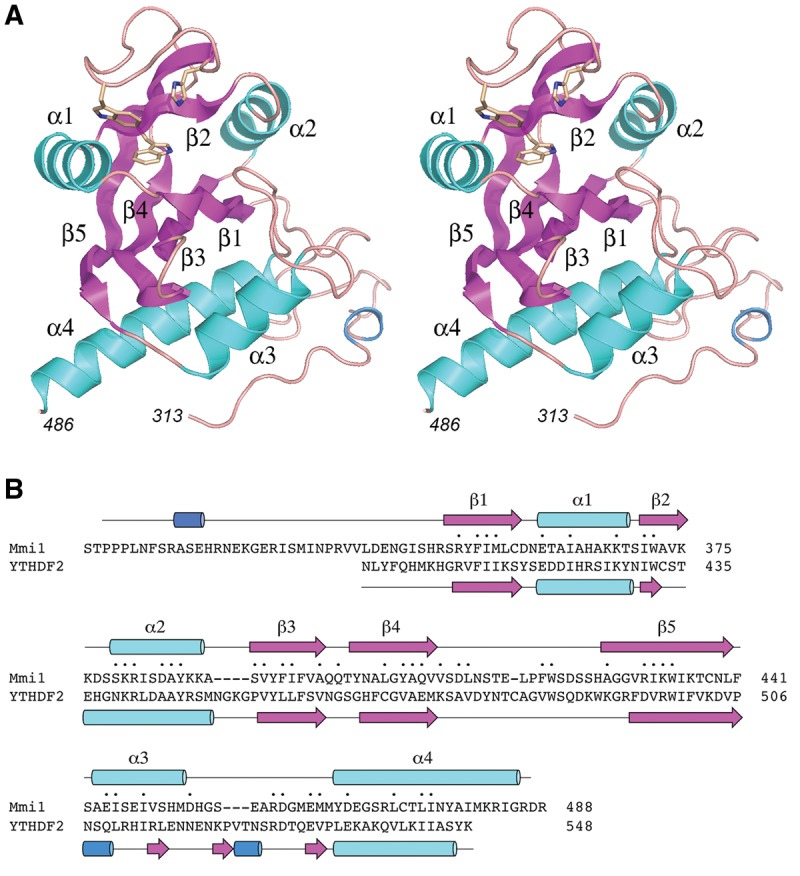
Structure of the Mmi1 YTH domain. (A) Stereo view of the tertiary structure of Mmi1(311–488), depicted as a ribbon model composed of five β strands (magenta), four α helices (cyan), and one 310 helix (blue). N- and C-terminal amino acids are numbered in italics. The three amino acids corresponding to the aromatic cage that binds to m6A in other YTH proteins are shown as stick models. (B) The Mmi1 and YTHDF2 primary structures are aligned according to DALI. Gaps in the alignment are indicated by dashes. Positions of amino acid side chain identity/similarity are indicated by (•) above the sequence. The secondary structure elements of Mmi1 and YTHDF2 (colored as in panel A) are shown above and below the respective amino acid sequences.
Another distinctive structural feature of the Mmi1 YTH domain is its N-terminal peptide segment (aa 313–347), which has no obvious counterpart in m6A-reading YTH proteins, but is well conserved in the S. pombe and S. japonicum Mmi1 proteins. Although this N peptide has no secondary structure other than a short 310 helix (colored blue in Fig. 11), it is well-ordered in the Mmi1(311–488) crystal structure by virtue of an extensive network of side chain–side chain and side chain–main chain hydrogen bonds and salt bridges, within the N-peptide itself and between the N-peptide and the α3 and α4 helices (Fig. 12). The inherent ordered structure of the N peptide was underscored by crystallizing the shorter YTH domain Mmi1(319–488) in a different lattice (space group P3121) than Mmi1(311–488). The structure of Mmi1(319–488), refined to R/Rfree of 20.5/25.1 at 1.95 Å resolution (Supplemental Table S1), was virtually identical to that of the longer version (Supplemental Fig. S1). Subtraction of the 311STPPPLNF318 peptide had no apparent effect on the structure of the residual N peptide from amino acids 319 to 347.
FIGURE 12.
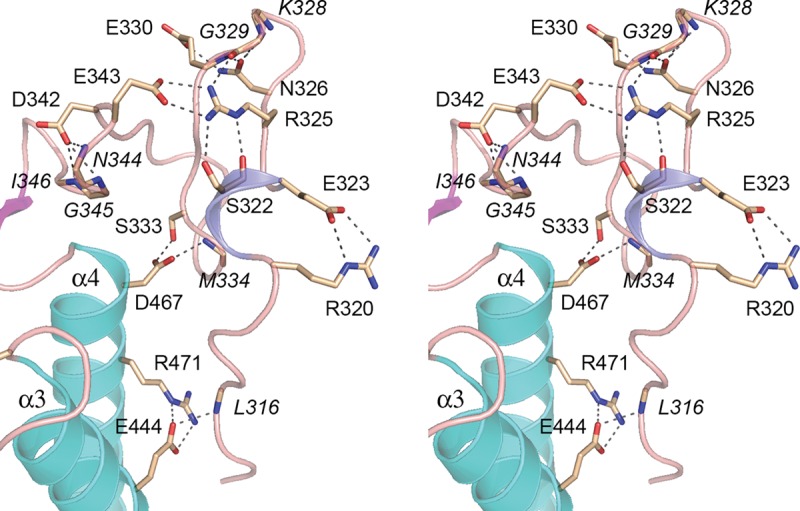
Distinctive N-terminal peptide of the Mmi1 YTH domain. Detailed stereo view of the N-terminal segment, highlighting an extensive network of side chain–side chain and side chain–main chain hydrogen bonds and salt bridges (depicted as dashed lines), within the N-peptide itself and between the N-peptide and the α3 and α4 helices. Interacting amino acid side chains are labeled in plain font. Main chain interactors are labeled in italics.
Is the aromatic cage relevant to DSR recognition by Mmi1?
The three aromatic residues that form the m6A cage in YTHDF2 (Trp432, Trp486, and Trp491) are conserved in the Mmi1(311–488) primary structure as Trp372, Trp421, and His426 (Fig. 11B). These three side chains, depicted as stick models in Figure 11A, are located in the β barrel, between the α1 and α2 helices of Mmi1(311–488). Figure 13A shows a close-up stereo view of the aromatic cage of YTHDF2 bound to an m6A mononucleotide (Li et al. 2014), superimposed on the equivalent region of Mmi1(311–488). In YTHDF2, the planar m6A base is sandwiched by Trp432 and Trp491, while the N6-methyl group projects into a three-sided box formed by the pseudoparallel Trp432 and Trp491 indoles and the orthogonal Trp486 side chain. The adenine N–CH3 makes van der Waals contacts with Trp432 and Trp486 (yellow dashed lines in Fig. 13A). Whereas Mmi1 Trp372 superimposes perfectly on YTHDF2 Trp432, the Mmi1 Trp421 side chain is rotated (compared with YTHDF2 Trp486) so that it effectively obstructs a putative binding pocket for m6A in Mmi1. (Indeed, this orientation of Trp421 would clash with an unmodified adenosine.) Although we cannot rule out the prospect that Trp421 reorients when Mmi1 binds to RNA, the fact that DSR recognition by Mimi-YTH clearly does not require N6-methylation of any of the three adenines in the DSR motif, raises the question of whether Mmi1 relies on the aromatic cage for RNA binding.
FIGURE 13.
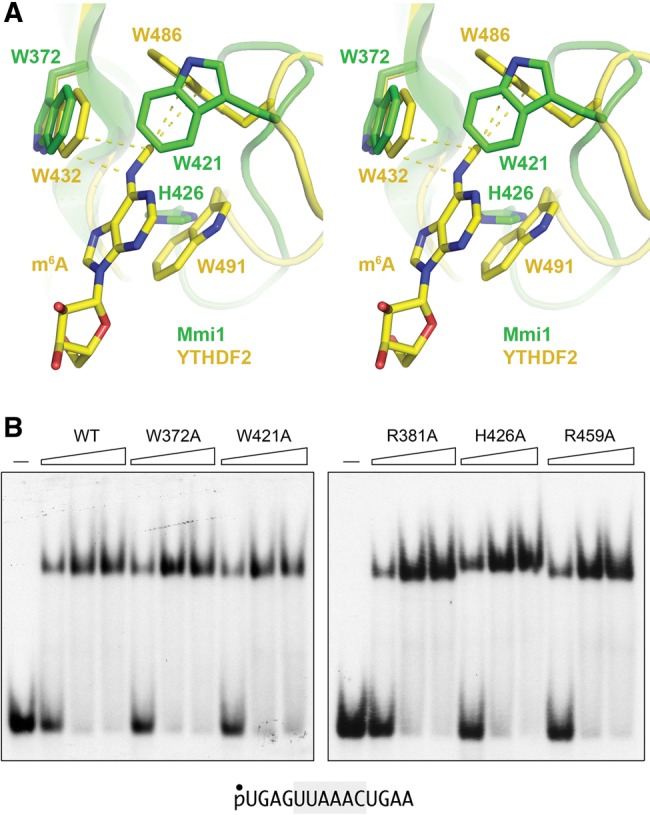
Mutagenesis of amino acids that contact m6A RNA in other YTH proteins. (A) Stereo view of the superimposed YTH domains of Mmi1 (green) and YTHDF with m6A nucleoside bound (yellow), highlighting the three aromatic amino acids (Trp432, Trp486, Trp491) that form a cage around the m6A nucleobase and make van der Waals contacts to the N6-methyl atoms (indicated by dashed lines). The corresponding Mmi1 amino acids are Trp372, Trp421, and His426. (B) DSR RNA binding by Mmi1 YTH mutants. Reaction mixtures contained 100 nM 5′ 32P-labeled 14-mer DSR RNA and 250, 500, or 1000 nM wild-type Mmi1(319–488) or the indicated alanine mutants. Mmi1(319–488) was omitted from the control reactions in lanes indicated by (–). The free RNA and RNA•protein complexes were resolved by native PAGE and visualized by autoradiography.
To probe this issue, we mutated the three Mmi1 aromatic cage residues to alanine. We also mutated two conserved arginines—Arg381 and Arg459—that correspond to arginines that contact the 5′-base and 3′-phosphate flanking m6A in the YTH•RNA complex solved by Luo and Tong (2014). We purified the recombinant W372A, R381A, W421A, H426A, and R459A mutant YTH domains in parallel with the wild-type YTH domain (Supplemental Fig. S2) and tested them by EMSA for binding to the 14-mer DSR-containing RNA (Fig. 13B). The instructive findings were that none of these mutations affected binding to the DSR RNA. We surmise that Mmi1 recognizes DSR RNA via a binding mode distinct from YTH m6A readers.
DISCUSSION
The present study sheds new light on fission yeast phosphate homeostasis, especially the role of the prt lncRNA in dampening the expression of the downstream pho1 gene under phosphate replete conditions. We find that basal pho1 expression from the prt–pho1 locus is inversely correlated with the activity of the prt promoter, which resides in a 110-nt DNA segment preceding the prt transcription start site. Within the prt promoter, there are two elements that dictate its strength: (i) a TATA box sequence (TATATATA) that is essential for prt transcription; and (ii) an upstream segment from −110 to −62 that augments prt transcription. The positive contributions of upstream segment stem, at least in part, from the presence of a HomolD sequence (CAGTCACG). Manipulations of the prt promoter in the context of the prt–pho1 locus that either delete the upstream DNA segment or mutate the TATA box cause derepression of pho1 in phosphate replete cells.
Earlier findings implicated Ser7-PO4 as a component letter of a Pol2 CTD code “word” needed to repress pho1 under phosphate replete conditions (Schwer et al. 2015). We show here that the CTD-S7A mutation resulted in derepression of pho1 in the context of the plasmid-based prt–pho1 locus in a pho1Δ strain, i.e., recapitulating the S7A effect reported previously for the native chromosomal locus. The CTD-T4A mutation has the opposite effect of hyper-repressing pho1 in phosphate replete cells, whether at the chromosomal locus or the plasmid prt–pho1 locus (Schwer et al. 2015; and data not shown). Key findings here are that S7A and T4A had no effect on the activity of the prt promoter or the pho1 promoter. These results prompt us to conjecture that S7A either: (i) desensitizes Pol2 to the effects of upstream prt transcription; or (ii) affects post-initiation events in prt lncRNA synthesis that render it less repressive. In the same vein, we envision that T4A might either make Pol2 more susceptible to upstream prt transcription or have post-initiation effects on prt lncRNA synthesis that make it more repressive at pho1.
A simplified model of the status of the prt–pho1 locus in the pho1 repressed state (in phosphate replete cells) is depicted in Figure 14A, whereby the prt promoter is active in driving synthesis of the prt lncRNA. We envision that Pol2 elongation across the pho1 promoter is the crucial event in dampening pho1 expression, by antagonizing the binding of the Pho7 transcriptional activator to the pho1 promoter. This might entail direct displacement of Pho7 by collision with the Pol2 elongation complex traveling down the prt gene. [Alternative explanations are that Pol2 transcription through prt: (i) lays down an RNA:DNA hybrid over the pho1 promoter that interferes with Pho7 binding; or (ii) causes a change in supercoiling topology of the pho1 promoter that affects Pho7 binding or its activation of pho1 transcription.]
FIGURE 14.
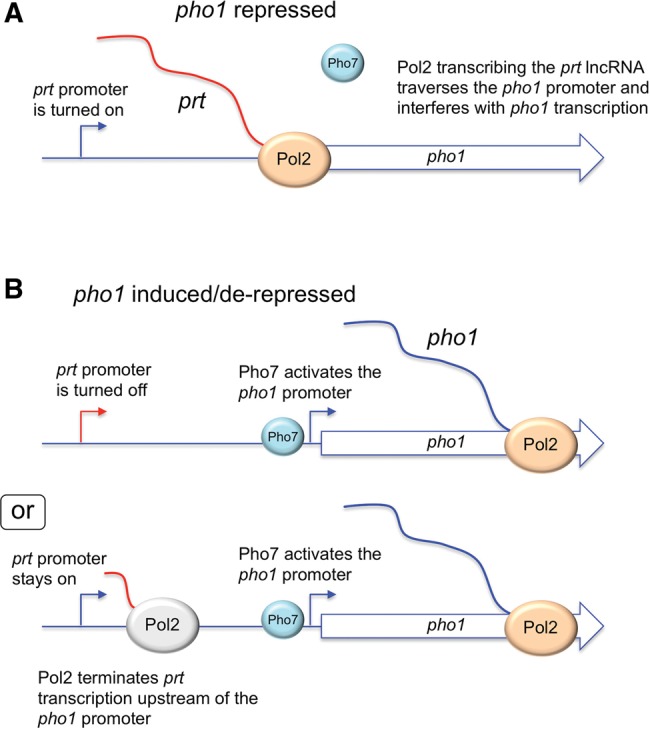
Models for the pho1 repressed and induced/derepressed states of prt–pho1. See Discussion text.
Two plausible models of the prt–pho1 locus in pho1 activated/derepressed states are shown in Figure 14B. In one scenario, the activation of pho1 transcription is achieved simply by shutting off the prt promoter, which would eliminate transcriptional interference with Pho7 at the downstream pho1 gene (by any of the mechanisms discussed above). Indeed, lncRNA promoter shut-off has been suggested to underlie the inductive response to phosphate starvation (Ard et al. 2014). In a second scenario, derepression of pho1 can occur even when the prt promoter remains on, via precocious termination of prt transcription by Pol2 before it reaches the pho1 promoter. The termination control model is attractive to explain pho1 derepression by the CTD S7A mutation, which does not silence the prt promoter, by positing that S7A polymerase is more susceptible to early termination of prt lncRNA synthesis. (Reciprocally, a model whereby T4A polymerase is less prone to terminate during prt RNA synthesis would explain the observed hyper-repression of pho1 in phosphate replete T4A cells.) Of course, it is possible that both mechanisms are operative in achieving phosphate homeostasis.
We provide here direct evidence that cis-acting elements of the prt lncRNA can influence its repressive effect on pho1. Specifically, we altered the nucleobase sequence of two DSR clusters that contain bona fide recognition elements for the Mmi1 YTH domain and found that these mutations caused hyper-repression of pho1 in phosphate replete cells, concomitant with increased steady-state levels of the prt transcript. We speculate that the DSRs contribute positively to termination of prt transcription, such that mutations of the DSRs (that preclude their interaction with Mmi1-YTH) lead to a lower probability of Pol2 terminating prt synthesis early and thus a greater probability of traversing the pho1 promoter from upstream to establish a pho1 repressed state. Mmi1 binding to the DSRs of nascent prt RNA would recruit the nuclear exosome, which can in turn promote Pol2 termination (Lemay et al. 2014).
Finally, we have investigated the RNA binding properties of the Mmi1 YTH domain and determined its atomic structure. Mmi1-YTH specifically recognizes DSR RNA (not DNA), binds to tandem DSR elements found in the prt DSR cluster in a noncooperative mode, and does not distinguish in vitro between the originally defined consensus DSR hexanucleotide UUAAAC and the UUAAAU variant present in the prt RNA. Mmi1-YTH does not require m6A modification to bind its DSR target, notwithstanding that many of the amino acids that comprise the m6A RNA binding site of other YTH proteins are conserved in Mmi1. Our demonstration that mutation of five of these conserved amino acids had no effect on Mmi1-YTH binding to DSR RNA prompted us to conclude that Mmi1 YTH has a distinctive RNA binding site. While we were preparing this work for publication, Wang et al. (2016) reported the crystal structure of Mmi1(322–488) in complex with a 10-mer RNA containing a consensus DSR hexamer (pdb 5EIM), which revealed that the DSR RNA binds on the face of the protein opposite the “aromatic cage” and is indeed a novel binding mode vis à vis the m6A reader clade of YTH proteins.
MATERIALS AND METHODS
Pho1 reporter plasmids
We constructed plasmid pLEU2-(prt–pho1) by inserting a 3866-bp prt–pho1 gene cassette between the SphI and SacI sites in pREP41x, thereby eliminating the nmt1 promoter and terminator sequences. The prt–pho1 cassette, comprising the pho1 ORF plus 1831 and 647 bp of upstream and downstream flanking genomic DNA, was assembled from DNA segments amplified by PCR using genomic DNA as a template and oligonucleotides that introduced restriction sites for cloning. A NotI site was introduced immediately upstream of the pho1 ATG start codon and an NheI site was placed at the 5′ end of the cassette. The prt gene was deleted to derive plasmid pLEU2-(pho1), which includes the pho1 ORF and 283 nt of DNA 5′ of the pho1 transcription start site that spans the pho1 promoter. Serially truncated pho1 promoters were generated by PCR amplification using oligonucleotides that introduced Nhe1 and Not1 sites at their 5′ and 3′ ends; the PCR products were restricted and inserted into pLEU2-(pho1) in lieu of the original 283-nt pho1 promoter. In plasmid pLEU2-(prtprom–pho1) the expression of pho1 is driven by a 633-nt DNA segment 5′ of the prt transcription start site that contains the prt promoter. This prt promoter was then exchanged for serially truncated or mutated versions of the prt promoter. Plasmid pKAN-(prt–pho1) was derived by replacing the LEU2 gene of pLEU2-(prt–pho1) with a kanMX gene that confers resistance to G418. PCR mutagenesis was used to introduce prt genes with truncated or mutated promoters or mutated DSRs in the prt lncRNA into pKAN-(prt–pho1). The inserts of all plasmids were sequenced to exclude unwanted mutations.
Plasmid-based reporter assays
We generated a pho1Δ strain, in which the chromosomal locus spanning the pho1 ORF plus 262 nt of 5′-flanking DNA was replaced by a hygMX cassette. LEU2 or kanMX reporter plasmids were transfected into pho1Δ cells. LEU2 transformants were selected on PMG (pombe glutamate) agar medium lacking leucine; kanMX transformants were selected on YES (yeast extract with supplements) agar medium containing 150 µg/mL G418. Single colonies of transformants (≥20) were pooled and grown at 30°C in plasmid-selective liquid medium. Aliquots of exponentially growing cultures were harvested, washed, and suspended in water to attain A600 of 1.25. To quantify acid phosphatase activity, reaction mixtures (200 µL) containing 100 mM sodium acetate (pH 4.2), 10 mM p-nitrophenylphosphate, and cells (ranging from 0.005 to 0.1 A600 units) were incubated for 5 min at 30°C. The reactions were quenched by adding 1 mL of 1 M sodium carbonate; the cells were removed by centrifugation, and the absorbance of the supernatant at 410 nm was measured. Acid phosphatase activity is expressed as the ratio of A410 (p-nitrophenol production) to A600 (cells). Each datum in the bar graphs is the average (±SEM) of three phosphatase assays using cells from three independent cultures.
To test the responsiveness of Pho1 expression to phosphate starvation, cells transformed with pKAN-(prt–pho1) were grown in YES + G418 liquid medium, aliquots of exponentially growing cultures were harvested, washed in water, and adjusted to A600 of ∼0.3 in PMG + G418 medium without phosphate. After incubation for various times at 30°C, cells were harvested, washed, and assayed for acid phosphatase activity as described above.
RNA analyses
Total RNA was extracted via the hot phenol method from 20 A600 units of yeast cells that had been grown at 30°C in PMG-leu medium (for selection of LEU2 plasmids), or YES + G418 (for selection of kanMX plasmids). For RT-qPCR, the RNAs were treated with DNase I, extracted serially with phenol:chloroform and chloroform, and then precipitated with ethanol. The RNAs were resuspended in 10 mM Tris–HCl (pH 6.8), 1 mM EDTA and adjusted to a concentration 600 ng/µL. Reverse transcription (RT) and gene-specific quantitative PCR (qPCR) were performed as previously described (Schwer et al. 2014). Primer extension assays were programmed by 5 µg of total yeast RNA as templates for M-MuLV reverse transcriptase-catalyzed elongation of a 5′ 32P-labeled oligodeoxynucleotide primers complementary to nucleotides +33 to +51 of the pho1 ORF as previously described (Schwer et al. 2014).
Purification of recombinant Mmi1-YTH proteins
DNA fragments encoding Mmi1(301–488), Mmi1(311–488), and Mmi1(319–488) were amplified from S. pombe cDNA by PCR with primers that introduced a BamHI site at the start codon and a HindIII site immediately after the stop codon. The PCR products were digested with BamHI and HindIII and inserted between the BamHI and HindIII sites of pET28b-His10Smt3 to generate expression plasmids encoding the Mmi1-YTH polypeptides fused to N-terminal His10Smt3 tags. Missense mutations W372A, R381A, W421A, H426A, and R459A were introduced into the Mmi1-(319–488) open reading frame by PCR with mutagenic primers. The inserts in each plasmid were sequenced to verify the fusion junctions to the tag and to ensure that no unwanted coding changes were introduced during amplification and cloning.
The Mmi1-YTH expression plasmids were transfected into E. coli BL21(DE3) cells. Cultures (2 L) amplified from single transformants were grown at 37°C in Terrific Broth containing 50 µg/mL kanamycin until the A600 reached 0.8. The cultures were chilled on ice for 1 h and adjusted to 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and then incubated for 20 h at 18°C with constant shaking. All subsequent steps of purification were performed at 4°C. Cells were harvested by centrifugation and resuspended in 35 mL buffer A (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 20 mM imidazole, 10% glycerol). The cells were lysed by sonication and the insoluble material was removed by centrifugation at 18,000 rpm for 45 min. Supernatants were mixed for 1 h with 4 mL of Ni-NTA resin (QIAGEN) that had been equilibrated with buffer A. The resin was recovered by centrifugation and resuspended in 50 mL buffer A. The washed resin was centrifuged again, resuspended in 50 mL buffer A, and then poured into columns. The bound material was step-eluted with buffer A containing 300 mM imidazole. The polypeptide compositions of the flow-through and eluate fractions were monitored by SDS-PAGE. The 300 mM imidazole eluate fractions containing Mmi1-YTH protein were supplemented with Smt3-specific protease Ulp1 and then dialyzed overnight against 2 L buffer B (50 mM Tris–HCl, pH 7.5, 200 mM NaCl, 10% glycerol). The dialysates were mixed for 1 h with 2 mL of Ni-NTA resin that had been equilibrated with buffer B. Tag-free Mmi1-YTH proteins were recovered in the flow-through fractions. Mmi1-YTH proteins were then subjected to gel filtration through a Superdex-200 column (GE Healthcare) equilibrated in 50 mM Tris–HCl, pH 7.5, 200 mM NaCl, 10% glycerol. Peak fractions were pooled, concentrated by centrifugal ultrafiltration to 9.2 mg/mL [Mmi1(301–488)], 20.1 mg/mL [Mmi1(311–488)], 25.5 mg/mL [Mmi1(319–488)], 30.1 mg/mL [W372A], 35.6 mg/mL [R381A], 41.3 mg/mL [W421A], 36.5 mg/mL [H426A] and 34.4 mg/mL [R459A], and then stored at −80°C. Protein concentrations were determined from the A280 measured with a Nanodrop spectrophotometer (Thermo Scientific), applying extinction coefficients calculated using Protparam.
Preparation of SeMet-Mmi1(311–488)
The pET28-His10Smt3-Mmi1(311–488) expression plasmid was transformed into E. coli B834(DE3) cells. A single transformant was inoculated into 10 mL of LB medium containing 50 µg/mL kanamycin and incubated for 8 h at 37°C. The bacteria were harvested by centrifugation and then resuspended in 200 mL of complete LeMaster medium containing 50 µg/mL kanamycin and 50 µg/mL SeMet (L-[+]-enantiomer; ACROS Organics). After overnight incubation, the culture volume was increased to 8 L with fresh LeMaster medium (+kanamycin +SeMet), and growth was continued at 37°C with constant shaking until the A600 reached 0.6. The culture was placed on ice for 1 h, adjusted to 0.5 mM isopropyl-β-D-thiogalactoside, and then incubated for 18 h at 17°C with continuous shaking. Cells were harvested by centrifugation, and the pellet was stored at −80°C. The SeMet-Mmi1(311–488) protein was purified via the same procedure described above for native Mmi1(311–488), except that the final storage buffer contained 5 mM DTT.
Nucleic acid binding assays
RNA and DNA oligonucleotides were purchased from Dharmacon and ThermoFisher Scientific, respectively. Oligonucleotides were 5′ 32P-labeled by reaction with T4 polynucleotide kinase and [γ32P]ATP, then gel-purified. Nucleic acid binding reaction mixtures (10 µL) containing 50 mM Tris–HCl (pH 7.5), 1 mM DTT, 10% glycerol, 100 nM 5′ 32P-labeled RNA or DNA oligonucleotide as specified, and Mmi1-YTH protein as specified were incubated at 4°C for 10–20 min. The mixtures were analyzed by electrophoresis at 4°C for 2.5 h at 110 V through a 15-cm native 6% polyacrylamide gel containing 22.5 mM Tris–borate.
Crystallization
Crystals of native and SeMet-substituted Mmi1(311–488) were grown by the sitting drop and hanging drop vapor diffusion method, respectively, at 22°C. Aliquots (1 µL) of 0.5 mM protein were mixed with 1 µL of reservoir buffer containing 0.1 M lithium sulfate, 0.1 M Tris (pH 8.5), 25% PEG4000. Crystals grew to their full size within 5 d. Crystals were harvested, cryoprotected by transfer to a solution containing 0.1 M lithium sulfate, 0.1 M Tris (pH 8.5), 25% PEG4000, 16% xylitol and then flash-frozen in liquid nitrogen.
Crystals of native Mmi1(319–488) were grown by the sitting drop vapor diffusion method at 22°C. A mixture of 0.5 mM Mmi1(319–488) and 1 mM 14-mer RNA (5′-UGAGUUAAACUGAA-3′) was incubated for 20 min at 22°C. Aliquots (1 µL) were then mixed with 1 µL of reservoir buffer containing 0.1 M lithium chloride, 0.1 M HEPES (pH 7.3), 25% PEG6000. Hexagonal-shaped crystals grew to their full size within 5 d. Crystals were harvested, cryoprotected by transfer to a solution containing 0.1 M lithium chloride, 0.1 M Hepes (pH 7.3), 25% PEG6000, 25% xylitol and then flash-frozen in liquid nitrogen.
Structure determination
Diffraction data were collected from single crystals at APS beamlines 24ID-C for SeMet-Mmi1(311–488) (to 2.05 Å resolution) and 24ID-E for Mmi1(311–488) and Mmi1(319–488) (to 1.75 Å and 1.95 Å resolution, respectively). Indexing and merging of the diffraction data were performed in HKL2000. The structure of Mmi1(311–488) was solved using single-wavelength anomalous dispersion (SAD) data from a SeMet-Mmi1(311-488) crystal. Location of the heavy atom sites corresponding to SeMet residues, phasing, density modification, and initial model building were accomplished in Phenix (Adams et al. 2002). The structure of Mmi1(319–488) was solved by molecular replacement in Phenix using the structure of Mmi1(311–488) as a search model. Both crystal forms contained four polypeptide chains in the asymmetric unit. Although an RNA oligonucleotide was present in the crystallization mixture for Mmi1(319–488), no electron density corresponding to RNA was observed. Diffraction data collection and refinement statistics are summarized in Supplemental Table S1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grant GM52470 (to S.S. and B.S.). Diffraction data were collected at the Northeastern Collaborative Access Team beamlines, which are funded by National Institutes of Health grant P41 GM103403, at the Advanced Photon Source operated by Argonne National Laboratory under DOE Contract DE-AC02-06CH11357. The Pilatus 6M detector at beam line 24-ID-C is funded by NIH-ORIP HEI grant S10-RR029205.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056515.116.
REFERENCES
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58: 1948–1954. [DOI] [PubMed] [Google Scholar]
- Ahn S, Spatt D, Winston F. 2012. The Schizosaccharomyces pombe inv1+ regulatory region is unusually large and contains redundant cis-acting elements that function in a SAGA- and Swi/Snf-dependent fashion. Eukaryot Cell 11: 1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard R, Tong P, Allshire RC. 2014. Long non-coding RNA-mediated transcriptional interference of a permease gene confers drug tolerance in fission yeast. Nat Commun 5: 5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O'Connell I, Peel MT, Wykoff DD, O'Shea EK. 2012. Genome-wide characterization of the phosphate starvation response in Schizosaccharomyces pombe. BMC Genomics 13: 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL. 2013. RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotobal C, Rodríguez-López M, Duncan C, Hasan A, Yamashita A, Yamamoto M, Bähler J, Mata J. 2015. Role of Ccr4-Not complex in heterochromatin formation at meiotic genes and subtelomeres in fission yeast. Epigenetics Chromatin 8: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD. 2014. How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes. Genes Dev 28: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D, Geyer M. 2013. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113: 8456–8490. [DOI] [PubMed] [Google Scholar]
- Gross T, Käufer NF. 1998. Cytoplasmic ribosomal protein genes of the fission yeast Schizosaccharomyces pombe display a unique promoter type: a suggestion for nomenclature of cytoplasmic ribosomal proteins in databases. Nucleic Acids Res 26: 3319–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y, Tanaka H, Yamanaka S, Tanaka K, Watanabe Y, Tsutsumi C, Chikashige Y, Hiraoka Y, Yamashita A, Yamamoto M. 2006. Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 442: 45–50. [DOI] [PubMed] [Google Scholar]
- Henry TC, Power JE, Kerwin CL, Mohammed A, Weissman JS, Cameron DM, Wykoff DD. 2011. Systematic screen of Schizosaccharomyces pombe deletion collection uncovers parallel evolution of the phosphate signal transduction pathway in yeasts. Eukaryot Cell 10: 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Kääriäinen S, Rosenström P, Schenkel A. 2008. Searching protein structure databases with DaliLite v.3. Bioinformatics 24: 2780–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797. [DOI] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, Grewal S. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bähler J, Bachand F. 2014. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol 21: 919–926. [DOI] [PubMed] [Google Scholar]
- Li F, Zhao D, Wu J, Shi Y. 2014. Structure of the YTH domain of human YTHDF2 in complex with an m6A mononucleotide reveals an aromatic cage for m6A recognition. Cell Res 24: 1490–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Tong L. 2014. Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc Natl Acad Sci 111: 13834–13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70: 583–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbogning J, Pagé V, Burston J, Schwenger E, Fisher RP, Schwer B, Shuman S, Tanny JC. 2015. Functional interaction of Rpb1 and Spt5 C-terminal domains in co-transcriptional histone modification. Nucleic Acids Res 43: 9766–9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas DA, Moreira-Ramos S, Zock-Emmenthal S, Urbina F, Contreras-Levicoy J, Käufer NF, Maldonado E. 2011. Rrn7 protein, an RNA polymerase I transcription factor, is required for RNA polymerase II-dependent transcription directed by core promoters with a HomolD box sequence. J Biol Chem 286: 26480–26486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell 43: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Sanchez AM, Shuman S. 2012. Punctuation and syntax of the RNA polymerase II CTD code in fission yeast. Proc Natl Acad Sci 109: 18024–18029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Bitton DA, Sanchez AM, Bähler J, Shuman S. 2014. Individual letters of the RNA polymerase II CTD code govern distinct gene expression programs in fission yeast. Proc Natl Acad Sci 111: 4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Sanchez AM, Shuman S. 2015. RNA polymerase II CTD phospho-sites Ser5 and Ser7 govern phosphate homeostasis in fission yeast. RNA 21: 1770–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Wittmann S, Kilchert C, Vasiljeva L. 2014. lncRNA recruits RNAi and the exosome to dynamically regulate pho1 expression in response to phosphate levels in fission yeast. Genes Dev 28: 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichino Y, Yamashita A, Yamamoto M. 2014. Meiotic long non-coding meiRNA accumulates as a dot at its genetic locus facilitated by Mmi1 and plays as a decoy to lure Mmi1. Open Biol 4: 140022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukleja M, Cuellar J, Siwaszek A, Kasprzak JM, Czarnocki-Cieciura M, Bujnicki JM, Dziembowski A, Valpuesta JM. 2016. The architecture of the Schizosaccharomyces pombe CCR4-NOT complex. Nat Commun 7: 10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Zhu Y, Bao H, Jiang Y, Xu C, Wu J, Shi Y. 2016. A novel RNA-binding mode of the YTH domain reveals the mechanism for recognition of determinant of selective removal by Mmi1. Nucleic Acids Res 44: 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt I, Straub N, Käufer NF, Gross T. 1993. The CAGTCACA box in the fission yeast Schizosaccharomyces pombe functions like a TATA element and binds a novel factor. EMBO J 12: 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt I, Kwart M, Gross T, Käufer NF. 1995. The tandem repeat AGGGTAGGGT is, in the fission yeast, a proximal activation sequence and activates basal transcription mediated by the sequence TGTGACTG. Nucleic Acids Res 23: 4296–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J. 2014. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10: 927–929. [DOI] [PubMed] [Google Scholar]
- Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. 2015. Structural basis for the discriminative recognition of N6-methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem 290: 24902–24913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Shichino Y, Tanaka H, Hiriart E, Touat-Todeschini L, Vavasseur A, Ding DQ, Hiraoka Y, Verdel A, Yamamoto M. 2012. Hexanucleotide motifs mediate recruitment of the RNA elimination machinery to silent meiotic genes. Open Biol 2: 120014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.