

Abstract
CRISPR–Cas systems are bacterial adaptive immune systems, each typically composed of a locus of cas genes and a CRISPR array of spacers flanked by repeats. Processed transcripts of CRISPR arrays (crRNAs) play important roles in the interference process mediated by these systems, guiding targeted immunity. Here we developed computational approaches that allow us to characterize the expression of many CRISPRs in their natural environments, using community RNA-seq (metatranscriptomic) data. By exploiting public human gut metatranscriptomic data sets, we studied the expression of 56 repeat-sequence types of CRISPRs, revealing that most CRISPRs are transcribed in one direction (producing crRNAs). In rarer cases, including a type II system associated with Bacteroides fragilis, CRISPRs are transcribed in both directions. Type III CRISPR–Cas systems were found in the microbiomes, but metatranscriptomic reads were barely found for their CRISPRs. We observed individual-level variation of the crRNA transcription, and an even greater transcription of a CRISPR from the antisense strand than the crRNA strand in one sample. The orientations of CRISPR expression implicated by metatranscriptomic data are largely in agreement with prior predictions for CRISPRs, with exceptions. Our study shows the promise of exploiting community RNA-seq data for investigating the transcription of CRISPR–Cas systems.
Keywords: CRISPR–Cas systems, CRISPR RNA (crRNA), metatranscriptomics
INTRODUCTION
CRISPR–Cas systems are RNA-guided bacterial and archael adaptive immune systems against invasive nucleic acids (DNA or RNA molecules) (Barrangou et al. 2007; Carter and Wiedenheft 2015). These systems memorize the invasion history by incorporating pieces of the invader's genetic material into their so-called CRISPRs (clustered regularly interspaced short palindromic repeats), or arrays of repeat and spacer unit. The invader's segments become the spacers sandwiched between copies of a typically identical repeat. The cas loci, often found in the genomic neighborhood of the CRISPRs, contain CRISPR-associated genes (cas genes) which encode proteins involved in various steps of the defense procedure, including acquisition of the spacers, biogenesis of the RNA guides from the CRISPRs, and the interference step. The invaders (including viruses), on the other hand, feature various mechanisms to counter the defenses from the CRISPR–Cas systems, such as through the anti-CRISPR genes that were recently discovered (Bondy-Denomy et al. 2013, 2015).
In CRISPR–Cas systems, CRISPR arrays are transcribed and processed to generate small CRISPR RNAs (crRNAs). The short crRNAs assemble with Cas proteins (encoded by the cas genes) to form surveillance complexes in which crRNAs provide the guide for targeted immunity (Jackson et al. 2014; van der Oost et al. 2014). It has been shown that CRISPRs are transcribed first as precursor crRNA (pre-crRNA) molecules, which undergo maturation steps to generate short mature CRISPR RNAs (crRNA). The short, mature crRNAs guide Cas protein(s) to recognize and destroy invading DNAs/RNAs. There are three major types I–III (each has subtypes) of the CRISPR–Cas systems, classified mainly according to the composition of the companion cas genes (and the other two rarer, newly defined types IV and V) (Makarova et al. 2011, 2015). Previous studies have shown that the biosynthesis pathways of the guide RNAs are distinct for the different types of the CRISPR–Cas systems (Charpentier et al. 2015). Type I and III CRISPR–Cas systems use an endoribonuclease belonging to the Cas6 family to cleave the pre-crRNA within the repeat regions. Type II systems rely on dual-RNA complexes (of pre-crRNA and trans-acting small RNA, tracrRNA) for the processing of pre-crRNA molecules in which dual-RNA complexes are cleaved by the housekeeping RNase III. The tracrRNA genes contain an anti-pre-crRNA repeat (anti-repeat) such that tracrRNA and crRNA form dual tracrRNA–crRNA through the base-pairing between the anti-repeat and the repeat (Chylinski et al. 2013). RNA-seq has been used to study the mechanisms and functions of CRISPR RNA biogenesis (Heidrich et al. 2015). Various RNA-seq protocols coupled with different enrichment methods also have been developed with some targeting primary transcripts and others targeting for mature crRNAs (Deltcheva et al. 2011; Juranek et al. 2012; Dugar et al. 2013).
Antisense RNAs of crRNAs were detected in a few species, including Clostridium thermocellum (Richter et al. 2012), Sulfolobu acidocaldarius (Lillestol et al. 2009), and Pyrococcus furiosus (Juranek et al. 2012). In general, the abundance of antisense crRNAs is lower than their crRNA counterparts. In S. acidocaldarius (Lillestol et al. 2009), CRISPRs are found in both the genome and its plasmid (pKEF9): The crRNAs and antisense crRNAs in this genome are both transcribed with similar abundances but lead to spacer RNAs of different lengths. Hale and colleagues identified significant antisense transcription from a BRE/TATA promoter within CRISPR locus 1 in the P. furiosus genome, and the number of antisense RNA reads is about one-third the number of lead strand crRNA reads (Hale et al. 2012). Richter et al. (2012) identified antisense crRNA in C. thermocellum; although the amount of antisense crRNA transcripts is very small in comparison to the abundance of crRNAs, the authors reported that individual antisense crRNAs show a conserved processing pattern within the repeats. The discovery of antisense RNAs raised a question about functional significance of these antisense RNAs and has led to the speculation of regulatory functions by the antisense crRNAs (Zoephel and Randau 2013).
Experimental studies of crRNA biogenesis are still sparse compared to the large number of CRISPR–Cas systems in the reference genomes and metagenomes. However, knowledge of the crRNA biogenesis, including the strand encoding crRNA, is crucial for understanding the immunity process. It also has practical application to the characterization of leader regions (Wei et al. 2015) (a leader element typically locates between a cas locus and a CRISPR, and includes a promoter for the transcription of the CRISPR that follows the leader), protospacer-adjacent motifs (PAMs) found in invaders (Mojica et al. 2009), and tracrRNA. Computational methods have been developed to predict the transcription direction of CRISPRs. CRISPRDirection (Biswas et al. 2014) uses parameters (including secondary structure and AT-rich in the leader sequence) that are calculated from input CRISPR and flanking sequences, and combines them by weighted voting to reach a prediction. The second approach is CRISPRstrand (Alkhnbashi et al. 2014), which encodes and processes the repeat sequence and mutation information using a graph kernel to learn higher-order correlations. Both computational approaches were reported to have high prediction accuracy. However, both approaches were trained based on a small number of cases with experimental evidence. For example, although more than a thousand repeat consensus sequences (including 442 repeats in the REPEATSLange set, 419 repeats in the REPEATSKunin and 478 in the REPEATShah) were used to train and test CRISPRstrand (Alkhnbashi et al. 2014), only the repeats in the REPEATSLange (Lange et al. 2013) were based on 10 systems (associated with nine species) that had experimental evidence supporting the crRNA processing (Brouns et al. 2008; Haurwitz et al. 2010; Hatoum-Aslan et al. 2011; Garside et al. 2012; Juranek et al. 2012; Nam et al. 2012; Richter et al. 2012; Sternberg et al. 2012; Nickel et al. 2013; Scholz et al. 2013). It suggests that there is a demand for having more experimentally supported transcription for development and evaluation of such tools.
Microbiome studies have enabled the study of the diversity of CRISPR–Cas systems in bacterial communities, including those associated with human beings. Stern et al. (2012) reconstructed the content of the CRISPR bacterial immune system in the human gut microbiomes of European individuals and used it to identify a large catalog of phages targeted by CRISPR across all individuals, revealing a surprising, global sharing of gut phages among individuals. Gogleva et al. (2014) used human gut metagenomic data from three open projects to reconstruct CRISPR cassettes to track the dynamics of spacer content. Our group developed a few computational tools for identification of CRISPR–Cas systems from metagenomic sequences, and the application of our tools to the human microbiome project (HMP) data sets has resulted in the identification of a large collection of CRISPR–Cas systems and putative invaders in human-associated microbiomes (Rho et al. 2012; Zhang et al. 2013, 2014).
RNA-seq data of bacterial communities (metatranscriptomic data) provides information vital for elucidating functional characteristics of microbial communities and accurate annotations of genes and their regulation in their community—complementary to metagenomic sequencing (de Menezes et al. 2012; Giannoukos et al. 2012; Leimena et al. 2013; Jorth et al. 2014; Pearson et al. 2015). Here we explored the possibility of using metatranscriptomic data to characterize the transcription of CRISPRs and other components of the CRISPR–Cas systems including cas genes, leader sequences, and anti-repeats (for type II CRISPR–Cas systems). Using eight publicly available sets of human stool metatranscriptomic data sets (derived from eight human individuals, which were prepared using three different methods of sample preservation, including frozen, ethanol-fixed, and RNAlater-fixed) (Franzosa et al. 2014), we showed the promise of metatranscriptomics in studying the transcription of crRNAs while avoiding the limits of studying the biosynthesis of CRISPR transcript (crRNA) in single species.
RESULTS
We first show the testing of different assembly strategies for CRISPRs and then summarize the results of applying the chosen strategy to six gut microbiomes. We found that most CRISPR–Cas systems are transcribed from one strand with exceptions that CRISPRs are transcribed from both strands. We demonstrated that metatranscriptomic data could be utilized to provide transcription evidence to CRISPRs and other components in the CRISPR–Cas systems, including cas genes, leader sequences, and tracrRNA genes (in type II CRISPR–Cas systems).
Assembly of CRISPR arrays
CRISPRs in microbiomes are likely to contain unique spacers different from those found in reference bacterial genomes, so de novo assembly is necessary for the characterization of CRISPRs. Using the targeted assembly approach that we have developed for CRISPRs (Rho et al. 2012), given an input sequencing data set (metagenomic, metatranscriptomic, or combined), we fished out the reads that are likely to contain repeats (or part of the repeats) similar to the repeats found in 33 reference CRISPR–Cas systems (see Materials and Methods). We then de novo assembled the extracted pool of reads (usually a small fraction of the original data sets) using different k-mer sizes (the k-mer size has great impact on the performance of de novo assembly) and summarized the assembly results of the CRISPRs in Figure 1. The assembly results are compared in terms of the total number of spacers assembled and the length of the longest CRISPR array. Overall, k needs to be sufficiently large (e.g., >40) to achieve good assemblies of the arrays. However, when k gets too large, performance starts to degrade. We decided to use k-mer size of 53 nt for our targeted assembly, as well as the assembly of whole metagenome and combined metagenome and metatranscriptomics data sets.
FIGURE 1.

The impact of k-mer size on the assembly of CRISPRs. A and C show the total number of spacers identified when different k-mer sizes were used, and B and D show the longest CRISPR arrays (number of repeat-spacer units). A and B are based on the CRISPR arrays associated with 33 CRISPR repeats identified from reference genomes that were found to be highly expressed in the gut microbiome (Franzosa et al. 2014), while C and D show the results for the CRISPR arrays associated with B. fragilis (which has the longest repeat of 47 bp) only. The results for different data sets (from eight individuals) are shown in different colors.
Incorporating metatranscriptomic data set helps improve the assembly of CRISPRs
We compared the total number of spacers that can be identified from assembled contigs associated with the 33 reference CRISPRs (Fig. 2). Results show that for frozen samples, combining metagenomic and metatranscriptomic data sets resulted in, on average (across the eight individuals), 32% more spacers when compared to using metagenomic data sets alone (paired t-test; P-value = 2.66 × 10−5). The difference decreased when all data sets (derived from samples processed differently; see below) for each individual were combined for assembly, but still, the combined assembly approach that combines both metagenomic and metatranscriptomic sequencing reads resulted in an average of 16% more spacers (paired t-test; P-value = 0.00012), indicating the importance of using metatranscriptomic data sets for assembly of CRISPRs. We also compared the assembly results from data sets derived from the frozen samples, or combined the data sets derived using different experimental protocols (frozen, RNAlater-fixed, and ethanol-fixed). As shown in Figure 2, combining the different data sets greatly helped the assembly of the CRISPR arrays. On average, the total number of spacers was more than doubled when all data sets were used for the assembly.
FIGURE 2.

CRISPR assembly results using different assembly strategies. The strategies are frozen-WGS-targeted (“targeted” assembly of CRISPR using only metagenomic data sets from “frozen” samples); frozen-combined-targeted (targeted assembly using “combined” metagenomic and metatranscriptomic data sets from frozen samples); all-WGS-targeted (targeted assembly using only metagenomic data sets from all samples); all-combined-targeted (targeted assembly using combined metagenomic and metatranscriptomic data sets from all samples); and combined-soapdenovo (whole-metagenomic and metatranscriptomic assembly using data sets from all samples). Results from combined-soapdenovo were used in this study for downstream transcription analysis.
We therefore used the assembly results of the CRISPRs from combined data sets with all metagenomic and metatranscriptomic reads from all three experimental protocols for downstream transcription analysis. This way, we optimized the assembly of the CRISPRs, and at the same time, achieved assemblies of other components of the CRISPR–Cas systems including cas genes (which, however, may not be optimized). We note that the combined data sets were only used for the assembly of the CRISPRs. Considering the substantial differences among the different RNA-seq experimental protocols (Franzosa et al. 2014), we used individual metatranscriptomic data sets for transcriptional characterization.
New CRISPR–Cas systems are found in gut microbiomes. We used a conservative strategy to collect putative new CRISPR–Cas systems. First we collected contigs that contain both CRISPR and cas genes (the cas loci are most likely partial due to the fragmented nature of the metagenome assemblies). Starting from these contigs, we identified 1808 repeats that are not similar to the reference repeats. After clustering this set of repeats (at 90% sequence identity by CD-HIT-EST [Li and Godzik 2006]) and removing the singletons, we derived 104 representative repeat sequences. Only three of these repeats share similarity (based on BLASTN searches) with putative novel CRISPR repeats previously identified from the Human Microbiome Project (HMP) data sets (Rho et al. 2012). Therefore, the remaining 102 repeats are likely to represent new CRISPRs. We used the collection of a total of 137 CRISPR repeats (including 33 derived from reference genomes, and 104 putatively novel ones) for the following transcription studies.
Metatranscriptomic evidence for CRISPR transcription
We mapped metatranscriptomic data sets from all three experimental protocols against the contigs that contain CRISPRs and/or cas genes, and used the mapped reads to characterize the transcription of CRISPRs. The percentage of metatranscriptomic reads that can be mapped to CRISPR–Cas loci ranges from 0.07% to 0.28%. We focused on the CRISPRs that are supported by at least 10 (combined) RNA-seq reads for the analysis: 56 out of 137 representative CRISPRs satisfy this criterion. Notably, none of these 56 CRISPRs (and their associated species) have been previously studied experimentally, showing the promise of studying CRISPR–Cas systems (and their transcription) using metatranscriptomic data sets. Among the 56 CRISPRs with RNA-seq supports, 18 are from the reference collection of genomes (see Table 1), and the rest are putatively new ones. See Supplemental Table S1 for the information on the 56 CRISPRs with repeat sequences, the type of the associated systems (if type specific cas genes were found in the reference genomes or the contigs containing the CRISPRs), and their predicted transcription orientation. We only considered the transcription orientation of a CRISPR if most (at least 80%) of its metatranscriptomic reads (combined) was mapped to one strand (the dominant strand), and further checked the consistency across the samples.
TABLE 1.
Summary of the transcription of representative CRISPRs
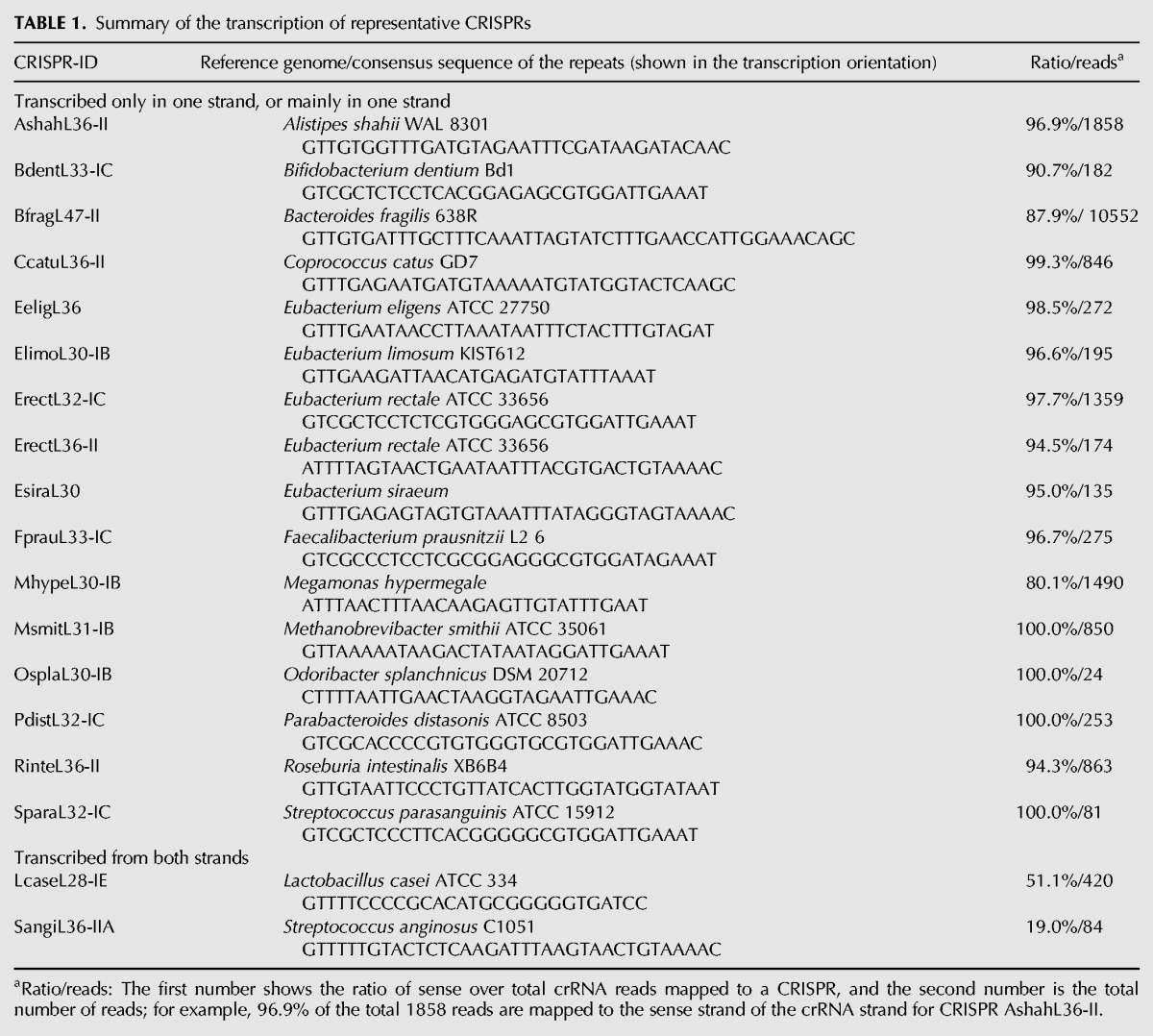
We analyzed an orphan CRISPR (EsiraL30), which was identified from the reference genome Eubacterium siraeum. CRISPRmap (Lange et al. 2013; Alkhnbashi et al. 2014) cannot predict orientation for this CRISPR (but it belongs to family 13 in CRISPRmap v2.1.3 at http://rna.informatik.uni-freiburg.de/CRISPRmap/Input.jsp). No contigs were identified from the gut microbiomes in which the genomic context can be used to infer the type of EsiraL30. Nevertheless, analysis of the assembled arrays shows that this CRISPR is likely to be active in the gut microbiomes with 28 spacers assembled, all unique (individuals do not share spacers). Metatranscriptomic analysis shows that this CRISPR was transcribed, mainly, in one direction: 100% (11 out of 11) of the reads in X316192082, 94.5% (74 out of 78) of the reads in X317802115, 100% (18 out of 18) of the reads in X317690558, and 85.7% (12 out of 14) of the reads in X317822438 can be mapped to one strand, which is therefore likely the lead strand of this CRISPR.
CRISPRs are dominantly transcribed in one direction
Most CRISPRs we identified in the gut microbiomes show transcription in one main direction. Figure 3 shows the fractions of reads from the main transcription strand over all reads for the 18 reference CRISPRs. Since most CRISPRs are mainly transcribed in one strand, the main direction therefore indicates the “sense” transcription of the corresponding CRISPRs (producing sense crRNAs). Using the gut metatranscriptomic data sets, we can assign “sense” strand for 16 out of 18 reference CRISPRs with metatranscriptomic supports (see Table 1; see Supplemental Table S1 for the results for all CRISPRs with metatranscriptomic support).
FIGURE 3.

A summary of the transcription orientations for the 18 reference CRISPRs, seen in eight sets of metatranscriptomic data sets from eight individuals. The transcription goes in one main orientation for most CRISPRs, except LcasL28-IE and SangiL36-IIA.
Figure 4 shows the genomic context and the predicted transcription orientation of the CRISPRs for a type II CRISPR–Cas system identified from the reference genome B. fragilis 638R (Fig. 4A), whose CRISPR orientation can be determined using metatranscriptomic data. We note the transcription orientation of the CRISPR array predicted by CRISPRDirection (Biswas et al. 2014) is the reverse strand (i.e., the same strand that encodes the cas genes). However, the metatranscriptomic data suggest the opposite direction (i.e., the CRISPR and cas genes are face to face; see an example in Fig. 4B), and all eight gut metatranscriptomic data sets support the same orientation—compelling evidence suggesting that the orientation prediction made by CRISPRDirection is wrong (although CRISPRDirection considered its prediction strong).
FIGURE 4.

Predicted transcription orientation for a CRISPR–Cas system associated with B. fragilis 638R, a type II-C CRISPR–Cas system, using metatranscriptomic data. As demonstrated in the reference genome (A), the transcription direction supported by metatranscriptomic sequencing data is opposite to the predicted orientation by CRISPRDirection. Repeat sequence in its predicted orientation is shown in Table 1. In this plot green arrows represent putative cas genes, and CRISPR arrays are shown in white hexagons with numbers inside the hexagons (followed by letter x) indicating the number of repeat-spacer units. (B) An example transcriptional profile for a contig (of 2278 bp; assembled from sample X317802115; id: 1280918) containing this CRISPR–Cas system. All metatranscriptomic reads mapped to the CRISPR in this contig (repeats are shown as red squares) are in one direction, which faces the cas genes, just as in the reference genome. We note no metatranscriptomic reads were mapped to the reverse strand of the contig (i.e., the sense strand of the cas genes).
We also analyzed AshaL36-II, the CRISPR associated with a type II CRISPR–Cas system identified from the reference genome Alistipes shahii. Querying the CRISPR repeat in the CRISPRmap server (v2.1.3) resulted in no annotation (i.e., it cannot be assigned to known structural/sequential families). We, however, found a wide spread of this CRISPR, potentially active, in the gut microbiomes we analyzed. We found CRISPR arrays associated with this CRISPR repeat in 69 contigs in the assemblies of the gut microbiomes, among which, six contain both CRISPR arrays and cas genes. Further, CRISPR arrays associated with AshahL36-II carry unique spacers (216 spacers can be extracted from the arrays assembled from the eight individual's microbiomes, and 196 of them are unique), indicating that this is an active CRISPR–Cas system with new spacers being captured in the CRISPR arrays. In all eight individuals, CRISPR arrays associated with this repeat are dominantly transcribed from one direction: Figure 5A shows the transcription of this CRISPR along with its cas loci found in a contig assembled from sample X316701492. It shows that both the cas genes (including cas9 and cas1) and the CRISPR were transcribed in this sample. It also shows that the transcription of the CRISPR starts in the leader sequence between the cas locus and the CRISPR array, which contains three putative transcription start sites (TSSs), including the most likely one closest to the cas locus. The TSSs were predicted by the BDGP neural network-based promoter prediction program, using the model for prokaryotes (http://www.fruitfly.org/seq_tools/promoter.html) (Reese 2001). For type II CRISPR–Cas systems, inferred transcription orientation of CRISPRs can also be applied to annotate the tracrRNA genes: An anti-repeat is predicted upstream of the putative cas9 gene in this contig (see Fig. 5A).
FIGURE 5.

Expression profiles for selective CRISPR–Cas systems. (A) A contig (of 15,939 bp; id: 1828570), which contains a putative type II CRISPR–Cas system sharing identical CRISPR repeats with A. shahii. Only part of the contig is shown for clarity. (B) A contig (of 20,551 bp; id: 1941627), which contains a type II CRISPR–Cas system associated with E. rectale. For each contig, putative elements of the CRISPR–Cas systems—including cas genes, the CRISPR, putative TSS(s) in the leader sequence, and an anti-repeat region—are shown in the plot with numbers indicating their genomic locations, and a black arrow above the CRISPR indicating the transcription orientation supported by metatranscriptomic reads. The read coverage curves are shown below the contig, with the coverage for forward and reverse strands shown in blue and green, respectively. Below the coverage plot shows the base-pairing between predicted anti-repeat region (Anti) and the CRISPR repeat (Repeat), with small circles indicating wobble base pairs (G–U).
Figure 5B shows the transcription profile of a contig that contains another type II CRISPR–Cas system associated with E. rectale. An anti-repeat is predicted between the putative cas9 gene and cas1 gene, and it is partially complementary to the corresponding CRISPR repeat as shown in Figure 5B. The RNA-seq coverage curve indicates that the anti-repeat and downstream cas genes were likely to be transcribed as a single unit. Only one putative TSS (and the predicted promoter is between 13,092 and 13,137 bp) was predicted in the leader sequence between the cas locus and the CRISPR array, and therefore it is likely to be the transcription start site of the downstream CRISPR array. We note that we did not find anti-repeats for BfragL47-II and CcatuL36-II; this is consistent with a previous study in which tracrRNA genes were not identified in the B. fragilis and C. catus reference genomes (Chylinski et al. 2013).
CRISPRs with bidirectional transcription
Some CRISPRs are transcribed in both directions. Using binomial testing (with P of 0.05, i.e., assuming the strand-specificity of the RNA-seq is 95%), we showed that among 1367 individual CRISPR arrays (associated with the 56 repeat-types) each having at least three copies of the associated repeat, 118 (8.6%) have detectable RNA-seq reads in both directions. The ratios vary if metatranscriptomic data sets derived using different experimental protocols were used. The ratios are 7.3%, 7.5%, and 11.3% for data sets derived from frozen, ethanol-fixed, and RNAlater-fixed samples, respectively.
We note that for the CRISPRs with bidirectional transcription, most still have one dominant transcription direction. Further, we found the bidirectional transcription for most CRISPRs is rather individual-specific. The CRISPRs are transcribed from one dominated direction in some individuals, whereas they are transcribed in both directions in others. For example, the expression of BfragL47-II is dominated by transcription in the sense strand (Fig. 6A). However, the relative abundance of antisense reads varies across individuals. In X6 (X319146421), a total of 2724 metatranscriptomic reads can be mapped to this CRISPR, among which only 1944 can be mapped to the sense strand (71.4%). In contrast, in X7 (X317690558), 476 out of 484 reads (98.3%) can be mapped to the sense strand.
FIGURE 6.
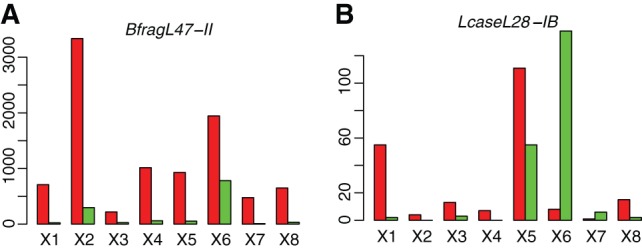
Transcription varies across individuals for CRISPRs. A and B show the number of sense reads (red bars) and antisense reads (green) that can be mapped to CRISPRs belonging to BfragL47-II (A) and LcaseL28-IB (B), respectively.
Strikingly, we found a case, CRISPRs associated with LcaseL28-IB, in which a significant portion of metatranscriptomic reads support the antisense transcription (Fig. 6B). Metatranscriptomic reads were found for 1, 2, 5, 1, 8, 9, 1, and 1 contig(s) containing this CRISPR, involving 11, 6, 18, 6, 123, 40, 6, and 16 repeat-spacer units in individuals X1, X2, X3, X4, X5, X6, X7, and X8, respectively. In six out of eight individuals, one orientation (so predicted to be the sense strand) dominates the transcription. However, in X5 (X316701492), there is a significant number of antisense reads (one-third of the total reads), and in X6 (sample ID: X319146421), reads from the antisense transcripts even dominated the total reads (94.5% of the total metatranscriptomic reads can be mapped to the antisense strand). The dominance of antisense transcription in individual X6 is unlikely to be an artificial result of experimental protocols as the metatranscriptomic data sets used here had strandedness >95% (Bao et al. 2015), and the antisense transcription is supported by the data sets derived from samples processed by the different experimental protocols: The fractions of antisense reads are 100% (all 32 reads) in the RNAlater-fixed samples, 96.6% (56 out of 58 reads) in the frozen samples, and 89.3% (50 out of 56 reads) in the ethanol-fixed samples.
Low transcription of type III CRISPR–Cas systems
It has been shown that type I CRISPR–Cas systems are more prevalent than the other two types of systems in microbial genomes: ∼60% or more of complete single-unit CRISPR–Cas loci are type I systems in both archaeal and bacterial genomes (Makarova et al. 2015). So it is not surprising that we observed more RNA-seq reads from CRISPR arrays associated with type I systems in the gut metatranscriptomic data sets than other types. Interestingly, although type II systems are less abundant than type I, their relative transcription levels appeared to be higher than type I counterparts. Type III systems are the least frequent in the gut data sets: We observed the existence of type III systems in the metagenomic data sets (at DNA level) but barely observed any RNA-seq reads from these arrays (only two arrays each had one RNA-seq read and others had none).
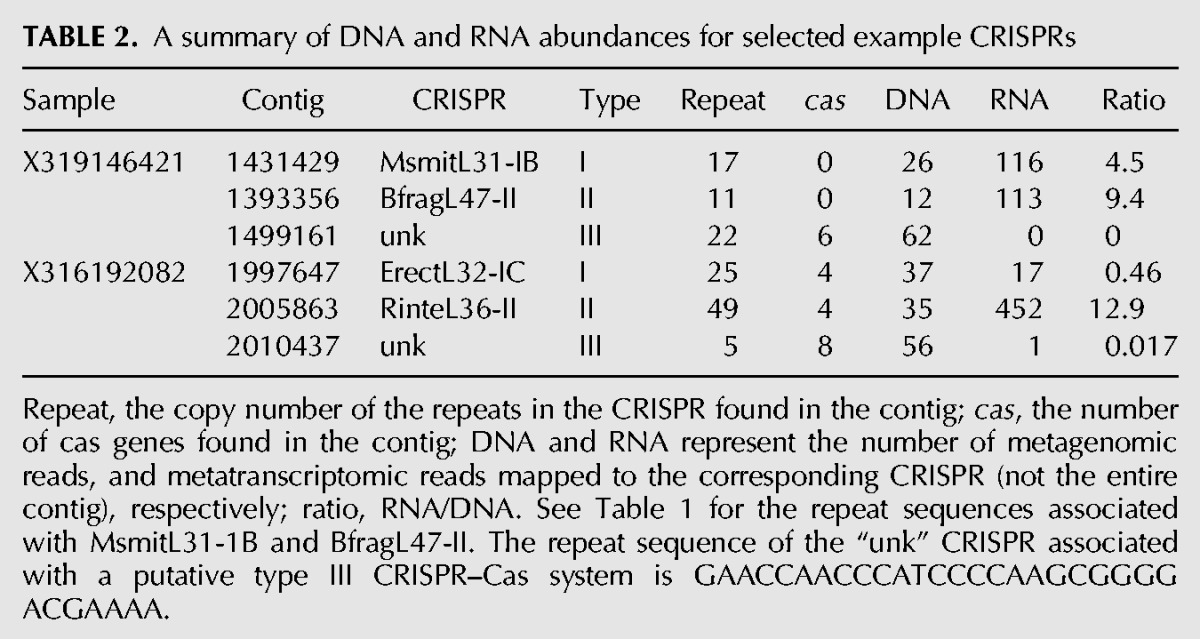
A lack of RNA-seq reads from a CRISPR can be the result of a low abundance of the associated genome (it was found that gene abundance and corresponding transcript abundance were well correlated [Franzosa et al. 2014]), or low transcription of the element, or both. To dissect the two confounding factors, we computed the ratio of the number of RNA-seq reads that can be mapped to the CRISPR array over the number of metagenomic reads that can be mapped to the same array (which approximates the RNA/DNA abundance ratios). The ratios are independent of the length of the CRISPRs (the length is canceled when computing the ratio). We used data sets derived from frozen samples for this calculation. We note that the values of the ratios do not indicate the expression levels of the corresponding CRISPRs, because they depend on the sequencing depth of the RNA-seq and the metagenome sequencing. But they can be used for comparing the relative expression levels of the different CRISPRs. Figure 7 shows that CRISPRs in type II systems appeared to have more transcripts (due to more transcription or other reasons) than CRISPRs belonging to type I and type III systems with t-test P-values of 0.000116 and <<0.0001, respectively. Arrays associated with type III systems have the lowest level of transcription, although some of them are of relatively high abundance at DNA level. Table 2 shows the comparison of a few CRISPR arrays. For example, a contig (ID: 1499161) assembled from the X319146421 data set contains a putative type III CRISPR array containing 22 repeats. A total of 62 metagenomic reads were mapped to the array, but no RNA-seq read was found for this array. In contrast, a total of 26 and 116 DNA and RNA reads were mapped to a MsmitL31-IB array (type I; associated with the archaeon Methanobrevibacter smithii) indicating a relative higher expression of MsmitL31-IB than the type III array in the sample. This result is consistent with a previous study (Franzosa et al. 2014) as well as our own (Bao et al. 2015), showing that M. smithii is abundant and highly transcriptionally active (supported by the huge numbers of RNA-seq reads) in the samples.
FIGURE 7.

Comparison of expression levels for the CRISPR arrays associated with different types of CRISPR–Cas systems (I, II, and III). In the boxplots, the y-axis represents the RNA/DNA abundance ratio (measured as the ratios of the number of RNA-seq reads divided by the number of metagenomic reads that can be mapped to the same array) for the arrays. (B) The zoomed-in view of A with the maximum ratio set to 2.
TABLE 2.
A summary of DNA and RNA abundances for selected example CRISPRs
DISCUSSION
We have developed a computational pipeline that allowed us to identify and characterize CRISPR transcription using metatranscriptomic data. Application of the pipeline to human gut metatranscriptomic data sets (combined with matched metagenomic data) revealed not only the transcription of many CRISPR–Cas systems but also the variation of the transcription of these CRISPRs in different human individuals. Metatranscriptomic data can be used to confirm the prediction of CRISPR transcription orientation, and, in some cases, can be used to correct wrong predictions as we show in the case of CRISPR associated with B. fragilis 638R (type II-C). Our analysis is limited in some aspects, however. Community RNA-seq captures both intact and fragmented transcripts, and degradation of transcripts is expected, so the results may be compounded by many factors. We cannot study mature crRNAs as they are likely to be filtered out in RNA-seq due to their small sizes. We also do not consider the different stability of crRNAs when we study the abundance of sense crRNA and antisense crRNA using reads count: Sense crRNAs are protected by other proteins within Cas protein interference complexes, whereas antisense reads do not benefit from this protection.
Using stranded RNA-seq reads, we were able to detect if transcription of a CRISPR goes in one direction or both. We proposed a statistical approach based on binomial testing for detecting CRISPR arrays with transcription in both directions (bidirectional) to avoid the artifact due to imperfect strandedness of the RNA-seq experiments. We emphasize that there could be other bias that may complicate the interpretation of the results. For example, we observed “bidirectional” transcription of similar levels in both directions (reads from one direction constitute 47.4%, 50.6%, 51.9%, 53.7%, 48.4%, 46.7%, 41.8%, and 54.0% of the total reads across eight individuals) for a CRISPR associated with Escherichia coli. However, the CRISPRs were not found in the matched metagenomic data sets. We believe this is a result of the DNA contamination in the RNA-seq experiments: The RNA-seq process was known to introduce 1%–2% E. coli genomic DNA into the final cDNA library, a result of E. coli-derived DNA polymerase I and ligase being used in the cDNA generation steps (Franzosa et al. 2014). We excluded the E. coli CRISPR in our study. On the other hand, this result indirectly shows that our pipeline produces accurate strand-specific expression levels for CRISPR.
Among the CRISPRs with metatranscriptomic evidence, there are type I and type II systems. Interestingly, type III CRISPR–Cas systems are found in the assemblies, but none of them show detectable transcription in metatranscriptomic data. The biological meaning of this observation remains to be explored. We observed that antisense transcription of CRISPRs varies among individuals, indicating that antisense crRNAs may play important regulatory functions (Zoephel and Randau 2013).
Although promising, using gut microbiome alone only surveyed a small number of CRISPRs compared to all known ones in the reference genomes and the new ones yet to be identified. Because different bacteria favor different environments, we believe with the increasing availability of metatranscriptomic data sets obtained from different environments and hosts it soon will become feasible to derive a comprehensive survey of the transcription of the CRISPRs of various types in their natural environments.
MATERIALS AND METHODS
Metagenomic and metatranscriptomic data sets
We used the human gut-associated strand-specific metatranscriptomic and matched metagenomic data from Franzosa et al. (2014). The data sets were downloaded from the SRA website (SRA accession: SRR769395–SRR769540). In total, we analyzed eight sets of metagenomic and metatranscriptomic data. Each set contains three metagenomic data sets, and three metatranscriptomic data sets derived from the same human individual but were prepared using three different methods of sample preservation (frozen, ethanol-fixed, or RNAlater-fixed) (Franzosa et al. 2014). The eight individuals are X310763260 (abbreviated as X1), X311245214 (X2), X316192082 (X3), X316701492 (X4), X317690558 (X5), X317802115 (X6), X317822438 (X7), and X319146421 (X8).
Assembly of CRISPR–Cas systems
It has been shown that some species are more transcriptionally active relative to their genomic abundance (Franzosa et al. 2014). Combining metatranscriptomic data sets with metagenomic data sets therefore has the chance of improving the assembly of some CRISPR–Cas systems from rare but highly expressed species. We compared the performance of the assembly of CRISPRs using metagenomic data sets only with the assembly using both metagenomic and metatranscriptomic data sets (combined assembly). Also, we applied both the targeted assembly of CRISPRs (Rho et al. 2012) we developed (Seq2CRISPR version 0.9 available at http://omics.informatics.indiana.edu/CRISPR) and “non-targeted” de novo assembly of the microbiomes using soapdenovo2 (Luo et al. 2012) using only metagenomic or combined metagenomic and metatranscriptomic data sets. Instead of using all the reads in a sequence data set, the targeted assembly approach first extracted the reads that contain segments similar to the repeats in reference CRISPRs and then assembled only the pooled reads (so significantly reduced the data set to be assembled).
Choice of assembler and k-mer size for the assembly
Similar to most de novo assemblers for short reads, the assembler we used, SOAPdenovo2, is based on de Bruijn graphs of k-mers (each is a short sequence of k nucleotides) (Compeau et al. 2011). It is therefore important to test the impact of choice of k-mer size on the assembly results of CRISPRs. In our previous targeted assembly, we used 43 as the k-mer size for targeted assembly for CRISPR arrays (Rho et al. 2012). Although this parameter works generally well in this study, we found that this parameter is less effective, especially for CRISPR arrays with long repeats such as the CRISPR associated with B. fragilis, which has the longest repeat of 47 bp. So in this study, we systematically tested the size of k-mers, and the results (see Results) show that k-mer size of 53 works generally well. We therefore applied this parameter for targeted assembly of CRISPR arrays and de novo assembly of the metagenomic, or combined metagenomic and metatranscriptomic data sets.
Reference collection of CRISPR repeats
We identified 33 CRISPR–Cas systems from 23 species that were shown to be highly expressed in the previous study (Franzosa et al. 2014). The repeats found in these CRISPR–Cas systems were used in our study as reference for the targeted assembly and characterization of CRISPRs in contigs by CRISPRAlign (Rho et al. 2012) (version 1.4 available at http://omics.informatics.indiana.edu/CRISPR/). We assigned IDs to the CRISPRs according to their associated species and other information: The ID uses five letters from the species name followed by the length of the repeats (length of 36 bp is shown as L36), and the type (subtype) information of the associated CRISPR–Cas system if it is available. For example, the CRISPR found in Lactobacillus casei ATCC 334 contains repeats of 28-bp long and is a type I-B system. It therefore is called LcaseL28-IB.
Identification of new CRISPR–Cas systems
From de novo assembly results, we can identify the contigs that contain both putative cas loci and CRISPR arrays, contigs that contain either cas loci or CRISPR arrays, and many more contigs that do not contain any. We focus on the contigs that have both putative cas loci and CRISPR arrays considering that they are more likely to represent true CRISPR–Cas systems than other contigs containing only one of the components (although it was shown that there are CRISPRs that are distant from any cas locus).
CRISPRs were predicted using CRISPAlign (Rho et al. 2012) against known CRISPR repeats (such that the predicted CRISPRs contain repeats sharing at least 90% sequence identity with one of the known repeats) for reference-based annotation, and metaCRT (Rho et al. 2012) (which we modified from CRT [Bland et al. 2007] to allow partial repeats at the ends of contigs) for de novo prediction. FragGeneScan (Rho et al. 2010) was applied to predict protein-coding genes from contigs, and the predicted proteins are used to annotate putative Cas proteins using hmmscan (Zhang et al. 2014) against the collection of 156 families of Cas proteins, including the known ones from a previous study (Makarova et al. 2011), and our newly defined Cas families from the human microbiomes (using a combination of context-based and similarity-search approaches). The type of CRISPR–Cas system was assigned using type signature cas genes (Makarova et al. 2011; Chylinski et al. 2014).
We then expanded the collection of CRISPR repeats from 33 reference repeats to 137 repeats. This expanded collection was used for identification of more CRISPR arrays, including those found in contigs that only contain the CRISPRs but no cas genes.
Quantification of CRISPR expression and detection of transcription direction
We mapped the RNA-seq reads to the assembled contigs that contain putative CRISPR (and cas genes) using Bowtie2 (Langmead and Salzberg 2012) and then summarized the expression of CRISPRs using mapped reads. Because strand-specific RNA-seq does not usually achieve 100% strand specificity (Sigurgeirsson et al. 2014), for a CRISPR with transcription only in one direction, we may find reads suggesting transcription from the other direction as well. Similar to the statistical approach we developed for detecting antisense transcripts to CDS (Bao et al. 2015), we applied binominal tests using a success rate of 0.05 to check if the observation of transcriptions from both directions is likely to be a consequence of the imperfect strandedness of the RNA-seq experiment or is more likely to represent the bidirectional transcription of the CRISPR. Specifically, we use binomial testing to detect CRISPRs with transcripts in both directions that are unlikely to result from such artifacts: Let P be the probability of having reads from one strand even though there is no real transcription in this strand (so real transcription occurs in the opposite strand). A total of c reads are sequenced from the CRISPR (c is approximated as the number of reads that can be mapped to the array), among which m reads represent transcripts from the strand opposite to the main direction. The null hypothesis is that there is no bidirectional transcription from this CRISPR. We use the binomial test in R (binom.test) to calculate the probability of having c reads (the number of successes) out of m trials (a total of m reads) with a success rate of P. If the probability is low (≤0.05 according to one-tailed binomial test), we consider that the CRISPR has bidirectional transcription (the alternative hypothesis). We used P = 0.05, since most of the metatranscriptomic data sets have less than this ratio of antisense reads (to protein-coding genes) (Bao et al. 2015), and it was shown that most library treatments in RNA-seq have a strandedness of >95% (Sigurgeirsson et al. 2014).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Haixu Tang and Kenneth Bikoff for reading the manuscript and the anonymous reviewers for their insightful comments. This work was supported by National Institutes of Health grant 1R01AI108888.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055988.116.
Freely available online through the RNA Open Access option.
REFERENCES
- Alkhnbashi OS, Costa F, Shah SA, Garrett RA, Saunders SJ, Backofen R. 2014. CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics 30: i489–i496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G, Wang M, Doak TG, Ye Y. 2015. Strand-specific community RNA-seq reveals prevalent and dynamic antisense transcription in human gut microbiota. Front Microbiol 6: 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712. [DOI] [PubMed] [Google Scholar]
- Biswas A, Fineran PC, Brown CM. 2014. Accurate computational prediction of the transcribed strand of CRISPR non-coding RNAs. Bioinformatics 30: 1805–1813. [DOI] [PubMed] [Google Scholar]
- Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. 2007. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493: 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, Davidson AR. 2015. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526: 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J, Wiedenheft B. 2015. SnapShot: CRISPR-RNA-guided adaptive immune systems. Cell 163: 260–260 e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, Richter H, van der Oost J, White MF. 2015. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev 39: 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Le Rhun A, Charpentier E. 2013. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol 10: 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Makarova KS, Charpentier E, Koonin EV. 2014. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res 42: 6091–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeau PEC, Pevzner PA, Tesler G. 2011. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol 29: 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Menezes A, Clipson N, Doyle E. 2012. Comparative metatranscriptomics reveals widespread community responses during phenanthrene degradation in soil. Environ Microbiol 14: 2577–2588. [DOI] [PubMed] [Google Scholar]
- Dugar G, Herbig A, Forstner KU, Heidrich N, Reinhardt R, Nieselt K, Sharma CM. 2013. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet 9: e1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. 2014. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci 111: E2329–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garside EL, Schellenberg MJ, Gesner EM, Bonanno JB, Sauder JM, Burley SK, Almo SC, Mehta G, MacMillan AM. 2012. Cas5d processes pre-crRNA and is a member of a larger family of CRISPR RNA endonucleases. RNA 18: 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, Levin JZ, Livny J, Earl AM, Gevers D, Ward DV, et al. 2012. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 13: R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogleva AA, Gelfand MS, Artamonova II. 2014. Comparative analysis of CRISPR cassettes from the human gut metagenomic contigs. BMC Genomics 15: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CVC, Graveley BR, Terns RM, et al. 2012. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A, Maniv I, Marraffini LA. 2011. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci 108: 21218–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329: 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich N, Dugar G, Vogel J, Sharma CM. 2015. Investigating CRISPR RNA biogenesis and function using RNA-seq. Methods Mol Biol 1311: 1–21. [DOI] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, van der Oost J, Terwilliger TC, Read RJ, Wiedenheft B. 2014. Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345: 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M. 2014. Metatranscriptomics of the human oral microbiome during health and disease. MBio 5: e01012–01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juranek S, Eban T, Altuvia Y, Brown M, Morozov P, Tuschl T, Margalit H. 2012. A genome-wide view of the expression and processing patterns of Thermus thermophilus HB8 CRISPR RNAs. RNA 18: 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. 2013. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res 41: 8034–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimena MM, Ramiro-Garcia J, Davids M, van den Bogert B, Smidt H, Smid EJ, Boekhorst J, Zoetendal EG, Schaap PJ, Kleerebezem M. 2013. A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets. BMC Genomics 14: 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Lillestol RK, Shah SA, Brugger K, Redder P, Phan H, Christiansen J, Garrett RA. 2009. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol 72: 259–272. [DOI] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155: 733–740. [DOI] [PubMed] [Google Scholar]
- Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, Ke A. 2012. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20: 1574–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel L, Weidenbach K, Jager D, Backofen R, Lange SJ, Heidrich N, Schmitz RA. 2013. Two CRISPR-Cas systems in Methanosarcina mazei strain Gö1 display common processing features despite belonging to different types I and III. RNA Biol 10: 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson GA, Lago-Leston A, Canovas F, Cox CJ, Verret F, Lasternas S, Duarte CM, Agusti S, Serrao EA. 2015. Metatranscriptomes reveal functional variation in diatom communities from the Antarctic Peninsula. ISME J 9: 2275–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG. 2001. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26: 51–56. [DOI] [PubMed] [Google Scholar]
- Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38: e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M, Wu YW, Tang H, Doak TG, Ye Y. 2012. Diverse CRISPRs evolving in human microbiomes. PLoS Genet 8: e1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. 2012. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res 40: 9887–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz I, Lange SJ, Hein S, Hess WR, Backofen R. 2013. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One 8: e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurgeirsson B, Emanuelsson O, Lundeberg J. 2014. Analysis of stranded information using an automated procedure for strand specific RNA sequencing. BMC Genomics 15: 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Mick E, Tirosh I, Sagy O, Sorek R. 2012. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22: 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Haurwitz RE, Doudna JA. 2012. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA 18: 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, Wiedenheft B. 2014. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol 12: 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chesne MT, Terns RM, Terns MP. 2015. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res 43: 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Rho M, Tang H, Doak TG, Ye Y. 2013. CRISPR-Cas systems target a diverse collection of invasive mobile genetic elements in human microbiomes. Genome Biol 14: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Doak TG, Ye Y. 2014. Expanding the catalog of cas genes with metagenomes. Nucleic Acids Res 42: 2448–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoephel J, Randau L. 2013. RNA-Seq analyses reveal CRISPR RNA processing and regulation patterns. Biochem Soc Trans 41: 1459–1463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.