

Abstract
MicroRNAs (miRNAs) comprise a class of short noncoding RNA molecules that play diverse developmental and physiological roles by controlling mRNA abundance and protein output of the vast majority of transcripts. Despite the importance of miRNAs in regulating gene function, we still lack a complete understanding of how miRNAs themselves are transcriptionally regulated. To fill this gap, we predicted regulatory sequences by searching for abundant short motifs located upstream of miRNAs in eight species of Caenorhabditis nematodes. We identified three conserved motifs across the Caenorhabditis phylogeny that show clear signatures of purifying selection from comparative genomics, patterns of nucleotide changes in motifs of orthologous miRNAs, and correlation between motif incidence and miRNA expression. We then validated our predictions with transgenic green fluorescent protein reporters and site-directed mutagenesis for a subset of motifs located in an enhancer region upstream of let-7. We demonstrate that a CT-dinucleotide motif is sufficient for proper expression of GFP in the seam cells of adult C. elegans, and that two other motifs play incremental roles in combination with the CT-rich motif. Thus, functional tests of sequence motifs identified through analysis of molecular evolutionary signatures provide a powerful path for efficiently characterizing the transcriptional regulation of miRNA genes.
Keywords: miRNA, Caenorhabditis, gene regulation, cis-regulatory motifs, comparative genomics
INTRODUCTION
MicroRNAs (miRNAs) comprise a class of post-transcriptional negative regulators that target messenger RNAs (mRNAs) to fine-tune and buffer gene expression (Bartel and Chen 2004; Li et al. 2009; Wu et al. 2009; Herranz and Cohen 2010; Mukherji et al. 2011). Because they can regulate hundreds of target genes (Farh et al. 2005; Lewis et al. 2005), miRNAs have emerged as key modulators of gene function with links to human disease (Calin et al. 2004; Jazdzewski et al. 2009) and possible roles in phenotypic innovation (Heimberg et al. 2008; Iwama et al. 2013; Meunier et al. 2013). miRNAs were first identified in the nematode Caenorhabditis elegans through cloning of mutations affecting temporal control of cell fate determination during larval development (Lee et al. 1993; Reinhart et al. 2000). The first two miRNAs identified, lin-4 and let-7, were central to the discovery that miRNAs form a widespread class of regulatory molecules because of their deep phylogenetic origin (Mondol and Pasquinelli 2012). However, unlike their orthologs in other bilaterians, the C. elegans lin-4 and let-7 no longer cluster together in a cotranscribed unit. Instead, they occur on different chromosomes with sequential expression during larval development, suggesting that genomic rearrangement may enable individual miRNAs to acquire novel regulation, perhaps through modification of upstream regulatory control elements (Ambros 2004; Sokol 2012).
Despite the importance of miRNAs in transcriptome regulation, our understanding of the transcriptional regulation of miRNAs themselves remains far from complete. Most intergenic miRNAs undergo transcription by RNA polymerase II and have promoters similar to those of protein-coding genes, although C. elegans and human miRNAs seem to lack a TATA box (Zhou et al. 2007). In addition, intronic miRNAs are generally cotranscribed with their host gene when both occur in the same orientation, although intronic miRNAs sometimes also use distinct promoters (Martinez et al. 2008b; Isik et al. 2011; Ramalingam et al. 2013). Deeper insights into miRNA transcriptional regulation will inform both miRNA function and the trajectories of miRNA evolution. Indeed, while miRNA sequences experience strong selective constraints (Fahlgren et al. 2010; Meunier et al. 2013; Mohammed et al. 2013; Jovelin and Cutter 2014), functionally important phenotypic variation may derive from differences among individuals and species in the amount and pattern of miRNA expression (Ason et al. 2006; Huang et al. 2011; Arif et al. 2013; Yeh et al. 2014).
Systematic tests of interactions between known transcription factors and miRNA promoters have generated basic principles of regulatory network organization involving miRNA transcriptional regulation (Martinez et al. 2008a). A second strategy to reveal regulation of miRNAs involves identifying sequence motifs upstream of miRNAs, with the logic that these overrepresented and evolutionarily conserved motifs confer functionally important regulatory information (Ohler et al. 2004; Inouchi et al. 2007; Zhou et al. 2007, 2011; Heikkinen et al. 2008; Wang et al. 2008). This approach identified several shared and species-specific motifs for animal and plant miRNAs, although their function has not been characterized. For instance, motifs with consensus sequence CTCCGCCC and TTTCAAAA are overrepresented and conserved upstream of Caenorhabditis elegans and Caenorhabditis briggsae miRNAs, and a CT-rich motif occurs upstream of miRNA sequences from nematodes to human to plants (Ohler et al. 2004; Zhou et al. 2007; Heikkinen et al. 2008).
Despite these advances, it remains unclear whether convergent evolution generated the motifs shared between C. elegans and C. briggsae or whether these motifs are specific to a small subset of nematode miRNAs. Here, we take advantage of experimental miRNA annotation in four Caenorhabditis species and available genome sequences for eight species in order to (i) identify characteristic motifs in miRNA upstream sequences, (ii) quantify nucleotide changes on these putative regulatory motifs, and (iii) investigate the role of these motifs on miRNA expression. We found distinct motifs that occur upstream of many miRNAs in each species. We show that three motifs are conserved across the Caenorhabditis phylogeny and bear a clear signature of purifying selection to eliminate mutations to them. Finally, we demonstrate through site-directed mutagenesis and transgenic gfp reporters that a CT-rich motif is sufficient for let-7 expression. We also show that two CT-motifs and motif RKGCGGAGC interact to modulate let-7 expression. Consequently, our results provide experimental validation for functional roles of motifs in miRNA regulation that were identified through computational analysis of molecular evolutionary signatures.
RESULTS
Conserved motifs upstream of miRNAs in Caenorhabditis
We identified the five most overrepresented sequence motifs within 1-kb upstream of miRNA hairpins independently in each of eight Caenorhabditis species using MEME (Fig. 1), a probabilistic motif discovery tool (Bailey and Elkan 1994). We then quantified motif incidence in the miRNA upstream sequences with the position-weight matrices of each motif using the program MAST (Bailey and Gribskov 1998). A C. brenneri 7-bp motif with consensus sequence SAAAAAA did not pass the filtering threshold of the MAST search and was not investigated further. We did not find any differences in the proportion of miRNAs with and without motifs between intergenic and intronic miRNAs, suggesting that our search is unlikely to have identified splicing elements (not shown). To determine the specificity of the miRNA motifs, we compared the abundance of each motif upstream of miRNA hairpins and upstream of protein-coding genes. Motifs occur significantly more abundantly in miRNA upstream regions than in protein-coding upstream regions, with 1.38- to 20.15-fold enrichment, depending on motif and species (Table 1). Motif significance, the number of miRNAs with motifs, and the number of sites are summarized for each motif and each species in Table 1.
FIGURE 1.
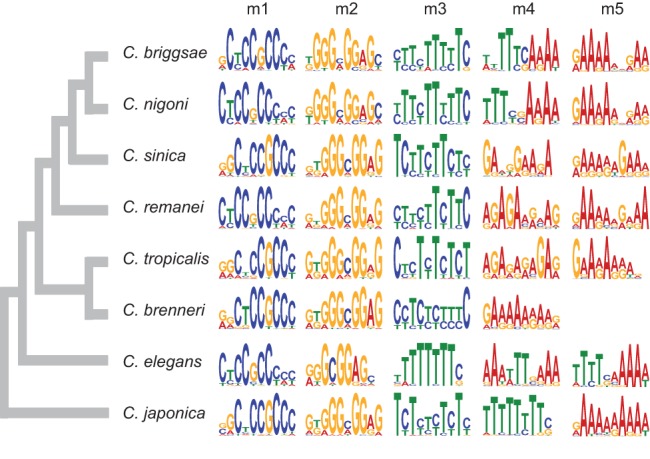
Logos of significantly overrepresented motifs in upstream miRNA sequences. Motifs are labeled m1 to m5 in each Caenorhabditis species, except for C. brenneri in which only four motifs are overrepresented. Each logo position includes the different nucleotides found at this position. The height of the nucleotide stack at a given position reflects conservation, and the height of a nucleotide is proportional to its frequency at that position. The phylogeny is from Kiontke et al. (2011).
TABLE 1.
Characteristics of overrepresented motifs located upstream of miRNA hairpins in Caenorhabditis

We generated sequence logos to summarize the information content of each motif (m1–m5) in the different Caenorhabditis species (Fig. 1). The C. elegans motif m1 (CYCCGCCYMY) was previously found to be conserved in C. briggsae and located preferentially within ∼200 bp of the miRNA hairpin (Ohler et al. 2004; Zhou et al. 2007; Heikkinen et al. 2008). Here, we demonstrate that this motif is conserved in eight Caenorhabditis species (Fig. 1). Moreover, the overrepresentation of motif m1 within 200 bp of the miRNA hairpins also is preserved across species (Fig. 2). Motif m2 (RKGCGGAGC) is the reverse complement of motif m1 and both the motif sequence and the motif distribution are also conserved across Caenorhabditis (Figs 1, 2). In contrast, motifs m3–m5 (BTTTTTTTY, AAAWTTSAAA, and TTTTSMAAAW) are distributed throughout the miRNA upstream sequence with no preferential position (Supplemental Fig. S1).
FIGURE 2.
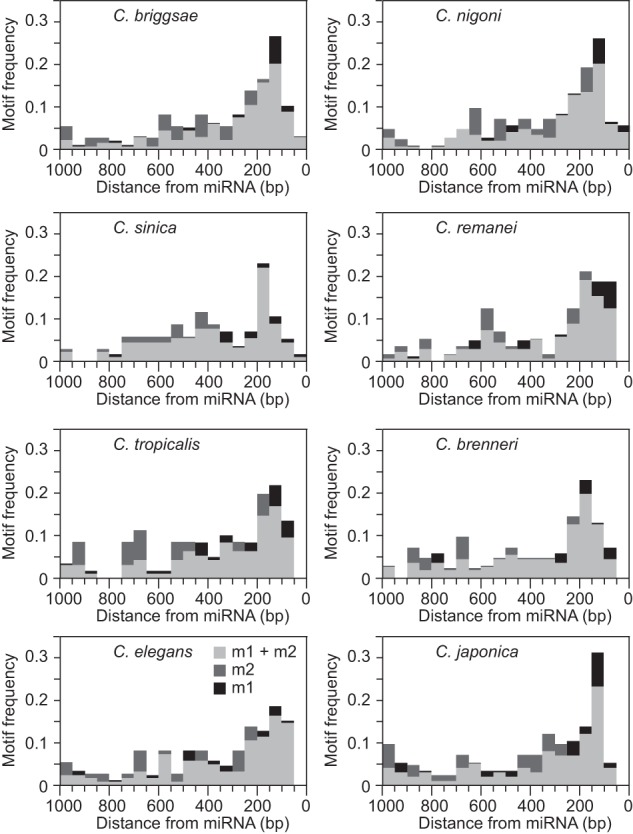
The frequency distributions of motifs m1, m2, and both motifs combined in 50-bp bins. Motifs m1 and m2 are more abundant within 200-bp upstream of miRNA hairpins in each Caenorhabditis species.
The most abundant motif in all Caenorhabditis species is the CT-rich microsatellite motif m3 located upstream of 62% of the C. elegans miRNAs with a total of 248 occurrences and in 91% of the C. japonica miRNAs with 136 occurrences (Table 1). All species except C. elegans also have at least one GA-rich motif, probably the reverse complement of the CT motif (Fig. 1). The C. elegans motif m5 with consensus sequence TTTTSMAAAW, previously found to be conserved in C. briggsae (Heikkinen et al. 2008), is also conserved in C. nigoni and is similar to C. elegans motif m4 AAAWTTSAAA (Fig. 1). Although C. elegans motifs m4 and m5 are not among the top five MEME motifs in C. sinica, C. remanei, C. tropicalis, and C. brenneri, we did find their instances in each species, albeit at lower abundance, using the motif's position-weight matrices (Supplemental Fig. S2). While differential motif abundance could suggest the presence of species-specific motifs in addition to broadly conserved motifs, we cannot rule out that those disparities might represent a byproduct of differences in the miRNA sets among species that we used during motif discovery.
Signatures of purifying selection in upstream miRNA motifs
Because DNA sequence conservation implicates fitness-associated functionality of the corresponding sequences, we quantified nucleotide divergence between species for the miRNA upstream motifs. First, we used a sliding window to compute nucleotide divergence between orthologous miRNA upstream regions in C. briggsae and C. nigoni, the two most closely related species available. We observed that sequence conservation declines with distance from the miRNA hairpin, similar to the pattern of conservation upstream of human and mouse miRNAs (Inouchi et al. 2007), and that sequence conservation is highest within the ∼200 bp of sequence immediately flanking miRNA hairpins (Fig. 3A).
FIGURE 3.

Nucleotide divergence in miRNA upstream regions and in each miRNA motif. (A) Sliding window of nucleotide divergence between C. briggsae and C. nigoni in miRNA upstream regions showing increased selective constraints in the proximity of the miRNA hairpins. The dark line indicates mean nucleotide divergence and the gray area shows the 95% confidence interval. (B) Nucleotide divergence between C. briggsae and C. nigoni for each C. briggsae motif. Motifs m1, m2, and m3 show signatures of purifying selection when compared with the random distribution. Means are represented ±1 standard error. Wilcoxon rank sum tests: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (ns) Not significant.
Second, we used pairwise alignments of orthologous miRNA upstream regions from C. briggsae and C. nigoni to quantify nucleotide differences with the C. briggsae motifs as reference. We generated the distribution of motif divergence under the model of no selection by computing nucleotide differences in 10,000 motifs of the same length drawn at random from the set of all miRNA upstream sequences. We found that sequence divergence for motif m1 is 2.2-fold lower than expected (P < 0.0001, Fig. 3B). We also detected purifying selection on motifs m2 and m3, with, respectively, 1.8-fold (P = 0.03) and 1.4-fold (P = 0.0010) reduction of nucleotide divergence relative to the random expectation (Fig. 3B). In contrast, nucleotide divergence is slightly higher than expected in motif m4 (P = 0.006) and does not deviate from the random expectation for motif m5 (Fig. 3B). To analyze motif sequence evolution in further detail, we counted the number of substitutions between C. briggsae and C. nigoni at each position of the instances of the C. briggsae motifs. Overall, we found that positions that contribute more to motif information content, that is, positions that are more conserved among motif instances, tend to be more preserved and accumulate fewer substitutions following species divergence (Supplemental Fig. S3).
Differences in miRNA expression level and miRNA sequence conservation
To further evaluate the relevance of the overrepresented motifs in miRNA upstream regions and their predicted role in miRNA regulation, we investigated the effect of the presence of motifs on the expression of the corresponding miRNAs in C. elegans. When we partitioned miRNAs according to the presence or absence of each motif and compared their expression level, we observed four- to fivefold higher expression for miRNAs associated with the m1 or m2 motif relative to miRNAs lacking these motifs (Fig. 4). We also found that miRNAs with both motifs m1 and m2 in their upstream sequence are expressed higher than miRNAs that have just a single motif or neither of these two motifs (Supplemental Fig. S4). Moreover, miRNA expression level correlates positively with the number of occurrence of motifs m1 or m2 (m1 Spearman's ρ = 0.431, P = 1.55 × 10−9; m2 ρ = 0.239, P = 0.0012). In contrast, the presence versus absence of motifs m3, m4, and m5 does not correlate significantly with any differences in miRNA expression (Fig. 4). The numerical incidence of motifs m3 and m4 also does not correlate with miRNA expression (m3 ρ = 0.058, P = 0.4383; m4 ρ = −0.140, P = 0.0611), whereas expression is somewhat lower for miRNAs with a greater abundance of motif m5 (ρ = −0.168, P = 0.0244).
FIGURE 4.
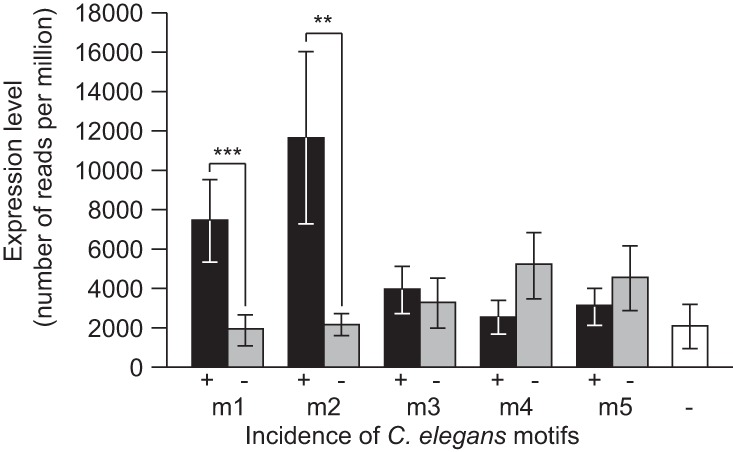
C. elegans miRNAs with motifs m1 or m2 are expressed at higher level than miRNAs lacking these motifs in their upstream sequence. miRNAs are partitioned based on the presence (+) or absence (−) of each motif, and normalized miRNA expression level (number of sequencing reads per million) is compared between each of the two groups. For comparison, mean expression levels of miRNAs lacking all motifs m1–m5 are indicated in white. Means are represented ±1 standard error. Wilcoxon two-sample tests: (**) P < 0.01, (***) P < 0.001.
Conserved miRNAs tend to be expressed at higher levels than those miRNAs that accumulate nucleotide changes or that are phylogenetically restricted (Liang and Li 2009; Shen et al. 2011; Jovelin 2013; Meunier et al. 2013; Jovelin and Cutter 2014). Therefore, we asked whether patterns of sequence evolution within the miRNAs themselves differ for miRNAs that have upstream motifs from those miRNAs that lack motifs in their upstream sequence. We found that miRNAs with motifs m1 and m2 tend to be more highly conserved at the nucleotide level than are miRNAs without either motif (Supplemental Fig. S5), although the difference is significant only for motif m2 (Km1+ = 0.0422, Km1− = 0.0563, P = 0.23; Km2+ = 0.0309, Km2− = 0.0560, P = 0.027).
Conserved motifs are sufficient for let-7 expression
The conservation of multiple motifs, most notably m1, m2, and m3, among miRNAs both within and across species strongly suggests that these motifs might play a role in miRNA biogenesis and/or in the regulation of miRNA expression. We then used miRNA transcription start site (TSS) annotations in C. elegans (Kruesi et al. 2013) to determine the context of motif occurrence and to gain further insight into motif function. We compared motif abundance upstream of TSS with abundance of motifs located between TSS and miRNA start. We found a significantly greater abundance of motifs located upstream of TSS, in the promoter, even after controlling motif counts by differences in sequence length between the two regions (Supplemental Table S1), suggesting that the motifs we identified may play a role in transcriptional regulation. Interestingly, C. elegans motifs m1/m2, m3, and m5 are very similar to known consensus sequences of binding sites, respectively, for transcription factors SP1 (Xi et al. 2007), HBL-1 (Inouchi et al. 2007), and EGL-5 (Niu et al. 2010), encouraging us to further explore the role of the motifs on miRNA expression.
We next sought to validate this hypothesis by investigating miRNA expression after manipulating motif incidence in transgenic lines. We explored this question with let-7 owing to the extensive study of its expression as one of the first discovered miRNAs and one of the few miRNAs to show a clear phenotypic effect when mutated (Reinhart et al. 2000; Johnson et al. 2003; Roush and Slack 2009; Kai et al. 2013). In particular, Johnson et al. (2003) used gfp reporters to identify a 116-nt-long enhancer required for the temporal regulation of let-7 expression that is located 1.2 Kb upstream of the let-7 mature sequence (pI16) (Supplemental Fig. S6). Interestingly, none of the motifs we identified are located within this minimum enhancer that has weak expression in the seam cells and low penetrance. However, another gfp reporter studied by Johnson et al. (2003) contained one instance of m2 and one instance of m3 (p2S1), and it is more strongly expressed in the seam cells and has high penetrance; a third reporter with an additional instance of m3 shows very bright expression with high penetrance (Supplemental Fig. S6, p1S1). Consequently, let-7 regulation provides an excellent system to study the function of the potential regulatory motifs in closer detail.
To assess the contribution of the m2 and m3(a, b) motifs to let-7 expression and thus test their function in regulating the expression of miRNAs more generally, we made a transgenic construct similar to p1S1, described in Johnson et al. (2003). This plasmid contains a 670-bp fragment of the let-7 promoter (−1284- to −614-bp upstream of the mature region), including m2, m3a, and m3b sites, and is cloned in association with a minimal pes-10 promoter, which drives GFP expression. We used site-directed mutagenesis to create derivatives of this plasmid in which m2, m3a, m3b, m3a, and m3b, or all three sites together, were mutated (m3a: GTTTTTTTGT/gatatccgga; m2: GGGCGGAGTT/gttaacgcgt; m3b: ATTTTTTGGC/agcgctcgag). We then generated at least four independent worm strains per plasmid construct carrying each of these mutated reporter transgenes, as well as strains possessing the wild-type enhancer transgene as a positive control, and a transgene containing no let-7 enhancer sequences as a negative control. We then surveyed these strains for GFP expression.
Expression of let-7 was previously observed in the seam cells and in the pharynx using the wild-type enhancer transgene (Johnson et al. 2003). We recapitulated these results with our wild-type enhancer strains, examining fully developed adult worms. As anticipated, the transgene lacking a let-7 enhancer displayed no GFP expression in either tissue (Fig. 5 and data not shown). Next, we examined GFP expression in the seam cells of each individual site, double site, and triple site mutant strains, and found differential contributions of each element to GFP expression. We chose to examine seam cells instead of the pharynx because differences in GFP expression between strains were more easily quantifiable in the seam cells, making this a more sensitive assay. The GFP signal in the seam cells was variable amongst strains, thus we scored worms as seam cell GFP positive if they possessed any GFP signal in the appropriate cells (identified using DIC microscopy). First, when we mutated the m2 motif alone, seam cell GFP expression remained similar to strains possessing wild-type enhancer transgenes (Fig. 5) (P = 0.773). In contrast, loss of m3a severely compromised seam cell GFP expression, with a 104-fold reduction (P = 0.021) in the number of worms expressing GFP relative to the positive control (Fig. 5). Interestingly, m3b may act to repress let-7 expression because mutating m3b slightly increases, rather than decreases, GFP expression (Fig. 5). We observed 1.2-times more expression in the single m3b mutant than in the wild-type strain, albeit not significantly so (P = 0.564), and we recovered 4% GFP expression upon mutating m3b in the m3a mutant background (P = 0.021). However, further disruption of m2 in the m3a, m3b double mutant abolished the expression gain and resulted in an additional decrease in seam cell GFP expression with only 0.5% of worms showing GFP expression (P = 0.021) (Fig. 5). Collectively, our results demonstrate that m3a is sufficient to drive let-7 expression, while the m2 and m3b motifs appear to contribute more subtle modulations of expression in the adult seam cells.
FIGURE 5.
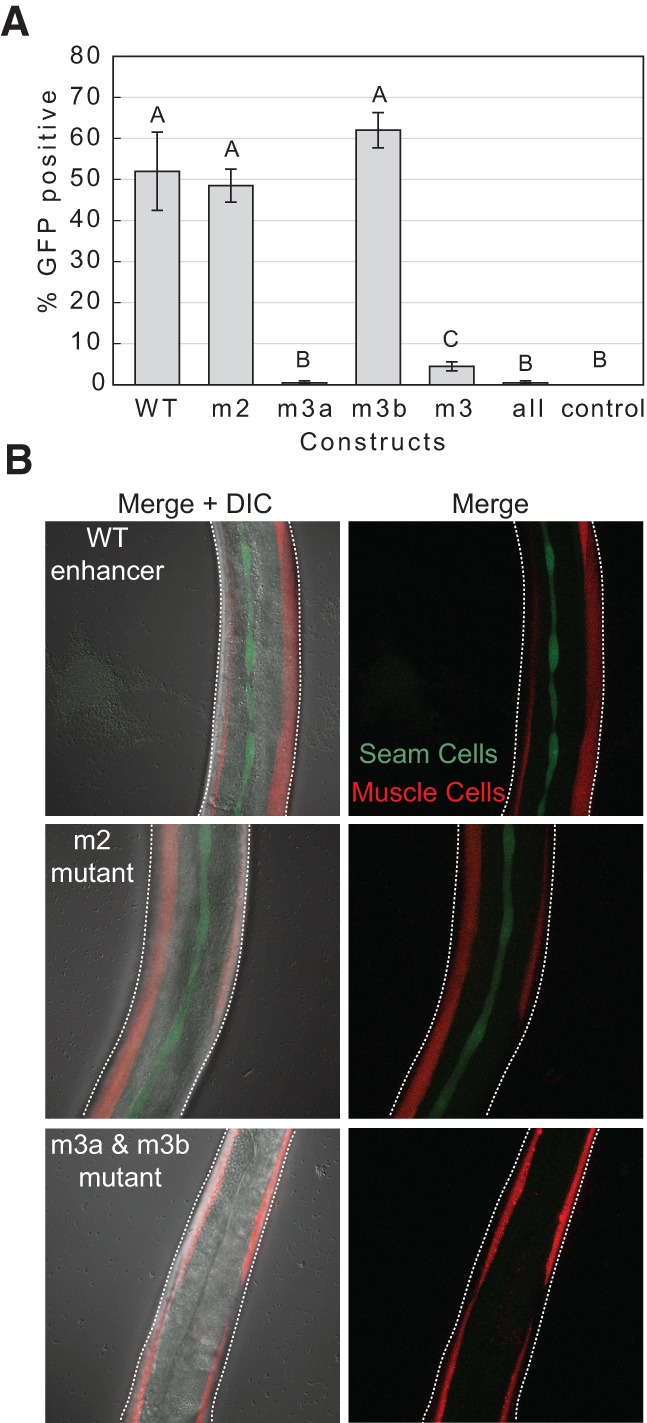
miRNAs motifs are sufficient for let-7 expression. (A) In vivo reporter assays demonstrate that m3a is sufficient for seam cell GFP reporter (and thus miRNA) expression in adult worms. Of note, 52% of adult worms with wild-type (WT) let-7 enhancer transgene show GFP expression in the seam cells whereas no expression is seen with a control plasmid possessing no enhancer. The wild-type enhancer transgene was mutated uniquely for motif m2, m3a, and m3b or for motifs m3a and m3b together or jointly for all three motifs. Means with different letters are significantly different from each other (Wilcoxon sum rank tests P < 0.05). Error bars indicate ±1 standard error. (B) Example images show positive seam cell GFP expression in the wild-type enhancer strain (top) and m2 mutant strain (center), and negative seam cell GFP expression in a triple mutant (m2, m3a, m3b) strain. Maximum intensity projections of a Z series of fluorescent images are shown with (left column) or without (right column) a single DIC slice overlay. Seam cell GFP is shown in green; muscle mCherry signal (a co-injection marker) is shown in red.
DISCUSSION
In the past decade, miRNAs have emerged as key regulators of gene function in a wide array of biological processes and are increasingly gaining attention for their potential role in disease, including cancer (Calin et al. 2004; Jazdzewski et al. 2009). Thus, insights into the regulation of miRNA biogenesis could reveal important aspects of miRNA function (Krol et al. 2010). miRNA loci are transcribed into primary transcripts with cap and poly(A) tail by RNA polymerase II (Lee et al. 2004), which are then sequentially cleaved by RNAse III Drosha and Dicer in the canonical miRNA biogenesis pathway (Yang and Lai 2011) to produce ∼22-nt-long mature miRNAs that regulate mRNA and protein abundance of their target genes (Bartel and Chen 2004; Baek et al. 2008; Mukherji et al. 2011). Promoters of miRNAs are similar to promoters of protein-coding genes in animals and plants (Zhou et al. 2007), suggesting that transcription factors (TFs) may be involved in regulating miRNA transcription. Indeed, modENCODE identified miRNAs that are heavily targeted by TFs in addition to a separate set of miRNAs that, in turn, preferentially regulate TFs (Gerstein et al. 2010).
To identify potential miRNA regulatory sequences, we applied the motif-finding algorithm MEME (Bailey and Elkan 1994) to miRNA upstream regions independently in eight Caenorhabditis species. Two motifs, termed here m1 and m2 and previously identified in C. elegans and C. briggsae (Ohler et al. 2004; Zhou et al. 2007; Heikkinen et al. 2008), are remarkably conserved across the Caenorhabditis phylogeny. A third CT-rich motif, termed here m3, also occurs in all species, along with GA-rich motifs, but with lower sequence conservation than motifs m1 and m2. Nevertheless, we revealed selective constraints preserving the sequences of motifs m1, m2, and m3 by comparing orthologous miRNAs between the two closest species C. briggsae and C. nigoni. We also showed that the incidence of motifs m1 and m2 upstream of miRNAs correlates with miRNA expression level and with miRNA sequence conservation.
It is possible that the motifs we identified play a role in miRNA biogenesis and processing rather than regulating miRNA expression. However, the motifs we identified here to be enriched upstream of miRNA genes also occur upstream of protein-coding genes, albeit at a significantly lower abundance, suggesting that they are not miRNA-specific motifs. In addition, we showed for C. elegans miRNAs with TSS annotations that all motifs were enriched upstream of the TSS, suggesting that they are part of the miRNA promoters. The average distance between TSS and miRNAs in C. elegans is 174 bp (Kruesi et al. 2013), and we showed that m1 and m2 SP1 motifs are preferentially located ∼200-bp upstream of miRNAs in all Caenorhabditis species, similar to the enrichment of SP1 motifs ∼50-bp upstream of TSS of protein-coding genes in humans (Xi et al. 2007). Analysis of TF target genes through chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq) revealed that most TFs regulate both protein-coding genes and noncoding RNAs, including miRNAs (Niu et al. 2010), implicating similar mechanisms of regulatory control. We showed that C. elegans motif m5 (TTTTSMAAAW), previously found to be conserved in C. briggsae (Heikkinen et al. 2008), is similar to C. elegans m4 (AAAWTTSAAA) and that both motifs are present in all species, even if the MEME search did not classify these two motifs among the top five overrepresented motifs in species other than C. briggsae and C. nigoni. We found that C. elegans motifs m4 and m5 are approximately twofold more abundant upstream of miRNAs than protein-coding genes. Interestingly, the binding sites of the homeobox gene egl-5 are associated with a motif highly similar to m5 (motif TSRDAAAA) both upstream of protein-coding genes and miRNAs, and the proportion of miRNAs regulated by egl-5 is twice the proportion of protein-coding genes (Niu et al. 2010). Whether motif TSRDAAAA is directly bound by EGL-5 or by a cofactor is unknown (Niu et al. 2010), but this suggests that our analysis of sequence motifs provides biologically relevant candidate elements to motivate further functional characterization.
To experimentally validate our approach, we performed functional tests of the conserved motifs m2 and m3 within the let-7 enhancer using GFP reporter transgenic C. elegans strains. Of note, we did not observe any significant enrichment of histone modifications (e.g. Histone H3 Lysine 21 acetylation) within the let-7 enhancer using data from the modENCODE Consortium (not shown), which is consistent with the idea that let-7 is primarily regulated through sequence-specific DNA binding proteins. We demonstrated through in vivo tests of motif function that motif m2 alone is not required for GFP reporter expression in seam cells, despite being highly conserved across species and well correlated with high levels of miRNA expression. However, it remains possible that m2 is required for expression in different tissues and/or at different developmental stages than the adult seam cells analyzed here, or that we did not sufficiently mutate the m2 site to fully abrogate its activity. In contrast, mutations in an instance of motif m3 (m3a) severely decreased GFP reporter expression. This result is consistent with the wide distribution of motif m3 upstream of numerous miRNAs across species, and points to a key role for this motif in regulating miRNA expression in general. Yet, mutation in a different instance of m3 (m3b) slightly increased GFP expression, suggesting that this instance may act as a repressor of gene expression.
These in vivo expression reporter analyses of m2 and m3 mutants are consistent with the notion that cooperative activity among multiple motifs is common within enhancers (Kazemian et al. 2013; Slattery et al. 2014) and that net expression is the result of multiple readouts encoded in regulatory sequences (Davidson 2006). Indeed, our analyses of single, double, and triple mutants illustrate how motifs may interact to regulate let-7 expression. Specifically, we observed that the strong reduction of GFP expression in m3a mutants is mitigated by mutation in m3b because GFP expression is still seen in 4.5% of adult worms on average in the double m3 mutant. In addition, disruption of m2 abolished this expression gain and further decreased GFP expression when paired with mutations in m3a and m3b. Interestingly, motif m3 is particularly T-rich in C. elegans compared with other species, and a T-rich (or A-rich) motif is a consensus of TF “hunchback” (Inouchi et al. 2007; Palsson et al. 2014). Roush and Slack (2009) identified three hunchback binding sites upstream of let-7, one of them corresponding to motif m3a, and provided strong support that HBL-1 binds these motifs to repress let-7 expression. This result suggests that cooperative binding among different TFs and cofactors through distinct binding sites can lead to different expression states. Indeed, deletion of m3a and the two other hunchback binding sites resulted in precocious GFP expression in larvae (Roush and Slack 2009), whereas our single, double, and triple m3a mutants strongly diminished GFP expression in adult.
The CT-dinucleotide motif m3 represents the most abundant motif we found in Caenorhabditis, and a similar CT-rich motif was previously identified in plants and animals (Zhou et al. 2007), suggesting broad functional activity. Supporting this hypothesis, dinucleotide repeat motifs, including GA (and CT) motifs, are evolutionarily conserved and are enriched in enhancers active across multiple cell types, although they are also required in cell-specific enhancers (Yanez-Cuna et al. 2014). This could suggest that motif m3 may provide broad regulatory activity across multiple tissues and/or during development, while motifs such as m1 and m2, which do not occur upstream of all miRNAs despite their strong conservation, could provide additional regulatory information in a cell- or temporally specific manner. This hypothesis is consistent with our observation that a significant effect of mutating m2 becomes apparent only once m3 is already mutated.
In conclusion, our study demonstrates how an evolutionarily informed approach to identifying candidate regulatory sequence motifs, that includes functional tests of hypotheses generated by bioinformatic analysis, can efficiently expand our understanding of the transcriptional regulation of miRNA genes.
MATERIALS AND METHODS
miRNA data sets
We obtained from Wormbase WS241 the genomic sequences and genome annotations of C. elegans, C. brenneri, C. briggsae, C. remanei, C. japonica, C. tropicalis (formerly C. sp. 11), and C. sinica (formerly C. sp. 5) (Felix et al. 2014; Huang et al. 2014). We also obtained the genomic sequence of C. nigoni (formerly C. sp. 9) (Kumar et al. 2012; Felix et al. 2014), although no public genome annotation is available for this species. We downloaded the miRNA sequences of C. elegans, C. briggsae, C. remanei, and C. brenneri from miRBase v.20 (Kozomara and Griffiths-Jones 2011). We performed a BLAST search to identify miRNAs in C. japonica, C. tropicalis, C. nigoni, and C. sinica. Sequences around BLAST hits containing a seed match or conserved over a stretch of nucleotides longer than the mature sequence were examined for their ability to form a hairpin structure with RNAfold (Gruber et al. 2008) and were considered as miRNAs if their minimum folding energy (MFE) was lower than −15 kcal/mol.
For C. brenneri, 48% of the 214 miRNAs annotated in miRBase are present in two copies located on different contigs. These copies are most likely due to the residual heterozygosity in the genome assembly of C. brenneri rather than to segmental duplications (Barriere et al. 2009). Indeed, we computed nucleotide polymorphism with DnaSP v5 (Librado and Rozas 2009) using 3 kb of sequence upstream and downstream from each miRNA copy (using flanking sequences of the first and last clustered miRNA, see below) and aligned with T-Coffee (Notredame et al. 2000). We found pairwise estimates and average nucleotide diversity (N = 71, mean π = 12.1%) similar to polymorphism reported in this species (Jovelin 2009; Dey et al. 2013). Similarly, the two C. remanei mir-790 located on different contigs have highly similar flanking sequences and gene neighbors with identical protein sequences. Moreover, the C. elegans mir-790 ortholog is located on the chromosome with orthologous regions in C. remanei and in C. brenneri showing the highest propensity of heterozygosity in the genome assemblies (Barriere et al. 2009). We included only one copy of these allelic variants in our miRNA data set. Two C. brenneri miRNAs (Cbn-mir-41 and Cbn-mir-42) not deposited in miRBase v20 could be unambiguously identified based on sequence conservation and position within clusters. Cbn-mir-785 is already annotated in miRBase, but sequence conservation does not extend beyond the seed motif. Our BLAST search identified the C. brenneri mir-785 homolog, with sequence conservation extending over the entire hairpin, and we included this sequence in our analysis.
Motif discovery
We searched for overrepresented motifs within 1 kb of miRNA upstream sequence separately in each Caenorhabditis species using the motif discovery algorithm MEME (Bailey and Elkan 1994), thus providing independent lines of evidence for the biological importance of motifs. For this analysis, we first defined miRNA clusters for which the distance between two neighbors is <1 kb. Then we kept the most upstream clustered miRNA and excluded the other downstream miRNAs to prevent identical sequences from affecting the motif discovery step. We included intronic miRNAs because they may be expressed from their own promoters (Isik et al. 2011). The number of miRNA upstream sequences used for motif discovery were 190 for C. elegans, 131 for C. briggsae, 119 for C. remanei, 115 for C. nigoni, 113 for C. brenneri, 88 for C. sinica and for C. tropicalis, and 71 for C. japonica. Using MEME, we independently searched in each species the five most represented motifs with length between 5 and 10 nt, while allowing upstream sequences to contain 0 or 1 motif.
We then searched for motif occurrences in the miRNA upstream sequences with the position-weight matrices of each motif using the program MAST (Bailey and Gribskov 1998). To determine the specificity of the miRNA upstream motifs, we extracted 1 kb of upstream sequence for 1000 protein-coding genes randomly sampled in each species (except for C. nigoni which lacks a genome annotation), searched for motif occurrence using the position-weight matrices of each miRNA motif in the corresponding species using MAST, and compared site abundance between miRNAs and protein-coding genes.
C. elegans transcription start sites
To further understand the sequence context of the enriched motifs, we compared motif abundance upstream of transcription start sites (TSS) in the promoter and motif abundance downstream from the TSS but upstream of the miRNA start for C. elegans miRNAs with TSS annotations (Kruesi et al. 2013). We also divided motif counts by the length of the corresponding sequence to compare the number of motifs per nucleotide and to ensure that sequence length differences would not generate differences in motif abundance. We tested significance with paired Wilcoxon tests.
Sequence divergence
Upstream sequences of 115 orthologous miRNAs in C. briggsae and C. nigoni were aligned with T-coffee (Notredame et al. 2000). We computed sequence divergence (K) between species using a sliding window of 10-bp width and a 5-bp step between each orthologous pair, discarding windows with N < 50, and took the average nucleotide divergence per window to plot the distribution of K as a function of the distance from the miRNA hairpin. We also quantified K between C. briggsae and C. nigoni for each instance of the C. briggsae miRNA upstream motifs and counted the number of substitutions at each motif position. To infer selective constraints acting on motifs, we computed divergence in 10,000 motifs drawn at random from the set of 115 orthologous upstream sequences and compared mean K differences between motifs and the random distribution with Wilcoxon rank sum tests. We also aligned and computed nucleotide differences between the hairpin sequences of the 115 orthologous miRNAs in C. briggsae and C. nigoni and partitioned miRNAs based on the presence or absence of each motif. We used a p-distance to quantify sequence divergence between species and allowed at most three gaps and undefined nucleotides (Ns) per motif and window.
C. elegans miRNA expression level
We obtained the expression of C. elegans miRNAs from miRBase v20. For each motif, we then compared the average expression level between miRNAs with motifs in their upstream sequence and miRNAs lacking a given motif, and determined significance of the mean differences with Wilcoxon rank sum tests. In addition, we determined the average expression of miRNAs lacking any of the five motifs we identified with MEME. We also computed the correlation between miRNA expression levels with the number of occurrences of each motif with Spearman's rank correlation.
GFP reporter analysis
We cloned a fragment from −1284- to −614-bp upstream of the C. elegans let-7 mature miRNA into pPD97.78 (Andrew Fire, Stanford Univ., Addgene), to create a plasmid nearly identical to p1S1 (Johnson et al. 2003), which we named p1S1ADC. This 670-bp-long DNA fragment includes one instance of motif m2 and two instances (m3a and m3b) of motif m3 (Supplemental Fig. S6). pPD97.78 possesses a minimal pes-10 promoter, with little promoter activity on its own, making it useful for enhancer studies such as these. After sequence validating p1S1ADC, we performed site-directed mutagenesis to mutate m2, m3a, and m3b either singly or in combination using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to manufacturer's instructions and using the following primers (mutated sites in lower case). JMCo1835 (5′-AGTGCAAGTTGGTTCCGTGCAAACAATGAC-3′) is a reverse primer used in combination with JMCo1836, JMCo1837, or JMCo1838. JMCo1836 (5′-gatatccggaGGGCGGAGTTGAAAGCGAAAAAAAACTATCTAG-3′) is a forward primer used to mutate m3a. JMCo1837 (5′-GTTTTTTTGTgttaacgcgtGAAAGCGAAAAAAAACTATCTAGGAGGGAACTG-3′) is a forward primer used to mutate m2. JMCo1838 (5′-gatatccggagttaacgcgtGAAAGCGAAAAAAAACTATCTAGGAGGGAACTG-3′) is a forward primer used to mutate m3a and m2 together. JMCo1839 (5′-GATTTTGTATTTTTTTTATGGATGTTTAAATGTTTG-3′) is a reverse primer used in combination with JMCo1840. JMCo1840 (5′-agcgctcgagATCATTTCTCCTATTTTAAATAATTTTATTTTAAAAATTAGCTTCTC-3′) is a forward primer used to mutate m3b. All plasmids derived from site-directed mutagenesis were validated by sequencing. pJC028 possesses mutations in m2, m3a, and m3b; pJC029 possesses a confirmed mutation in the motif m2; pJC030 possesses confirmed mutations in both m3a and m3b motifs; pJC031 possesses a confirmed mutation in m3a; and pJC032 possesses a confirmed mutation in m3b. To generate transgenic strains, we injected a mixture of each reporter plasmid at 100 ng/µL with pCFJ104 at 10 ng/µL into Bristol N2 C. elegans young adult hermaphrodites. pCFJ104 is a muscle-specific mCherry co-injection marker from the laboratory of Eric Jorgensen at the University of Utah, obtained from Addgene. We identified transgenic strains using the muscle mCherry signal on an Olympus SZX16 fluorescent stereomicroscope. We established multiple independent transgenic lines with a maximum of one line per injected hermaphrodite to ensure that the lines were distinct. Worms were cultured according to standard protocols (Brenner 1974).
We scored and imaged seam cell GFP fluorescence for 50 worms per line and four lines per plasmid using a Nikon C2 confocal with a 60× oil immersion lens. We collected a Z series of images for these strains using a step size of 0.37 microns, and we have displayed these images (Fig. 5B) as a maximum intensity projection of fluorescence (muscle: mCherry, seam cell: GFP), overlaid with a single DIC slice. We also observed pharyngeal GFP expression much stronger than seam cell GFP expression, making it difficult to quantify accurately. Thus, the seam cells provided a more sensitive context for our quantification. Anatomical reference points were used to identify seam cells, including proximity to alae and a lack of muscle striations by DIC microscopy and lack of mCherry (muscle) signal by epifluorescence microscopy. Worms were scored as positive for seam cell expression if any GFP signal was detected in the seam cells regardless of intensity.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank three anonymous reviewers for constructive comments. This work was supported by grants from the Ontario Ministry of Research and Innovation to R.J. and the National Health Institutes (R01-GM096008) to P.C.P. and A.D.C. A.D.C. also is supported by a Canada Research Chair (CRC) in Evolutionary Genomics and the Natural Sciences and Engineering Research Council of Canada (NSERC). A.K., B.T., and J.M.C. are supported by NSERC (RGPIN-418), and J.M.C. is supported as a CRC in Small RNA Biology.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.055392.115.
REFERENCES
- Ambros V. 2004. The functions of animal microRNAs. Nature 431: 350–355. [DOI] [PubMed] [Google Scholar]
- Arif S, Murat S, Almudi I, Nunes MD, Bortolamiol-Becet D, McGregor NS, Currie JM, Hughes H, Ronshaugen M, Sucena E, et al. 2013. Evolution of mir-92a underlies natural morphological variation in Drosophila melanogaster. Curr Biol 23: 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ason B, Darnell DK, Wittbrodt B, Berezikov E, Kloosterman WP, Wittbrodt J, Antin PB, Plasterk RH. 2006. Differences in vertebrate microRNA expression. Proc Natl Acad Sci 103: 14385–14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14: 48–54. [DOI] [PubMed] [Google Scholar]
- Barriere A, Yang SP, Pekarek E, Thomas CG, Haag ES, Ruvinsky I. 2009. Detecting heterozygosity in shotgun genome assemblies: lessons from obligately outcrossing nematodes. Genome Res 19: 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. 2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5: 396–400. [DOI] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. 2004. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci 101: 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH. 2006. The regulatory genome. Elsevier, San Diego. [Google Scholar]
- Dey A, Chan CK, Thomas CG, Cutter AD. 2013. Molecular hyperdiversity defines populations of the nematode Caenorhabditis brenneri. Proc Natl Acad Sci 110: 11056–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, Smith LM, Dasenko M, Givan SA, Weigel D, et al. 2010. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22: 1074–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. 2005. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310: 1817–1821. [DOI] [PubMed] [Google Scholar]
- Felix MA, Braendle C, Cutter AD. 2014. A streamlined system for species diagnosis in Caenorhabditis (Nematoda: Rhabditidae) with name designations for 15 distinct biological species. PLoS One 9: e94723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. 2008. The Vienna RNA websuite. Nucleic Acids Res 36: W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen L, Asikainen S, Wong G. 2008. Identification of phylogenetically conserved sequence motifs in microRNA 5′ flanking sites from C. elegans and C. briggsae. BMC Mol Biol 9: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberg AM, Sempere LF, Moy VN, Donoghue PC, Peterson KJ. 2008. MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci 105: 2946–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz H, Cohen SM. 2010. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev 24: 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RS, Gamazon ER, Ziliak D, Wen Y, Im HK, Zhang W, Wing C, Duan S, Bleibel WK, Cox NJ, et al. 2011. Population differences in microRNA expression and biological implications. RNA Biol 8: 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RE, Ren X, Qiu Y, Zhao Z. 2014. Description of Caenorhabditis sinica sp. n. (Nematoda: Rhabditidae), a nematode species used in comparative biology for C. elegans. PLoS One 9: e110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouchi A, Shinohara S, Inoue H, Kita K, Itakura M. 2007. Identification of specific sequence motifs in the upstream region of 242 human miRNA genes. Comput Biol Chem 31: 207–214. [DOI] [PubMed] [Google Scholar]
- Isik M, Korswagen HC, Berezikov E. 2011. Expression patterns of intronic microRNAs in Caenorhabditis elegans. Silence 1: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama H, Kato K, Imachi H, Murao K, Masaki T. 2013. Human microRNAs originated from two periods at accelerated rates in mammalian evolution. Mol Biol Evol 30: 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazdzewski K, Liyanarachchi S, Swierniak M, Pachucki J, Ringel MD, Jarzab B, de la Chapelle A. 2009. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc Natl Acad Sci 106: 1502–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Lin SY, Slack FJ. 2003. The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev Biol 259: 364–379. [DOI] [PubMed] [Google Scholar]
- Jovelin R. 2009. Rapid sequence evolution of transcription factors controlling neuron differentiation in Caenorhabditis. Mol Biol Evol 26: 2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovelin R. 2013. Pleiotropic constraints, expression level, and the evolution of miRNA sequences. J Mol Evol 77: 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovelin R, Cutter AD. 2014. Microevolution of nematode miRNAs reveals diverse modes of selection. Genome Biol Evol 6: 3049–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai ZS, Finnegan EF, Huang S, Pasquinelli AE. 2013. Multiple cis-elements and trans-acting factors regulate dynamic spatio-temporal transcription of let-7 in Caenorhabditis elegans. Dev Biol 374: 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Pham H, Wolfe SA, Brodsky MH, Sinha S. 2013. Widespread evidence of cooperative DNA binding by transcription factors in Drosophila development. Nucleic Acids Res 41: 8237–8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiontke KC, Felix MA, Ailion M, Rockman MV, Braendle C, Penigault JB, Fitch DH. 2011. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol Biol 11: 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. 2010. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11: 597–610. [DOI] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. 2013. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife 2: e00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Koutsovoulos G, Kaur G, Blaxter M. 2012. Toward 959 nematode genomes. Worm 1: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. 2004. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20. [DOI] [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. 2009. A microRNA imparts robustness against environmental fluctuation during development. Cell 137: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Li WH. 2009. Lowly expressed human microRNA genes evolve rapidly. Mol Biol Evol 26: 1195–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- Martinez NJ, Ow MC, Barrasa MI, Hammell M, Sequerra R, Doucette-Stamm L, Roth FP, Ambros VR, Walhout AJ. 2008a. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev 22: 2535–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Ow MC, Reece-Hoyes JS, Barrasa MI, Ambros VR, Walhout AJ. 2008b. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res 18: 2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. 2013. Birth and expression evolution of mammalian microRNA genes. Genome Res 23: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Flynt AS, Siepel A, Lai EC. 2013. The impact of age, biogenesis, and genomic clustering on Drosophila microRNA evolution. RNA 19: 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondol V, Pasquinelli AE. 2012. Let's make it happen: the role of let-7 microRNA in development. Curr Top Dev Biol 99: 1–30. [DOI] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. 2010. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217. [DOI] [PubMed] [Google Scholar]
- Ohler U, Yekta S, Lim LP, Bartel DP, Burge CB. 2004. Patterns of flanking sequence conservation and a characteristic upstream motif for microRNA gene identification. RNA 10: 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson A, Wesolowska N, Reynisdottir S, Ludwig MZ, Kreitman M. 2014. Naturally occurring deletions of hunchback binding sites in the even-skipped stripe 3+7 enhancer. PLoS One 9: e91924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam P, Palanichamy JK, Singh A, Das P, Bhagat M, Kassab MA, Sinha S, Chattopadhyay P. 2013. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 20: 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906. [DOI] [PubMed] [Google Scholar]
- Roush SF, Slack FJ. 2009. Transcription of the C. elegans let-7 microRNA is temporally regulated by one of its targets, hbl-1. Dev Biol 334: 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Lv Y, Huang L, Liu W, Wen M, Tang T, Zhang R, Hungate E, Shi S, Wu CI. 2011. Testing hypotheses on the rate of molecular evolution in relation to gene expression using microRNAs. Proc Natl Acad Sci 108: 15942–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, Rohs R. 2014. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 39: 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol NS. 2012. Small temporal RNAs in animal development. Curr Opin Genet Dev 22: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gu J, Zhang MQ, Li Y. 2008. Identification of phylogenetically conserved microRNA cis-regulatory elements across 12 Drosophila species. Bioinformatics 24: 165–171. [DOI] [PubMed] [Google Scholar]
- Wu CI, Shen Y, Tang T. 2009. Evolution under canalization and the dual roles of microRNAs: a hypothesis. Genome Res 19: 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Yu Y, Fu Y, Foley J, Halees A, Weng Z. 2007. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res 17: 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Cuna JO, Arnold CD, Stampfel G, Boryn LM, Gerlach D, Rath M, Stark A. 2014. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res 24: 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lai EC. 2011. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43: 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SD, von Grotthuss M, Gandasetiawan KA, Jayasekera S, Xia XQ, Chan C, Jayaswal V, Ranz JM. 2014. Functional divergence of the miRNA transcriptome at the onset of Drosophila metamorphosis. Mol Biol Evol 31: 2557–2572. [DOI] [PubMed] [Google Scholar]
- Zhou X, Ruan J, Wang G, Zhang W. 2007. Characterization and identification of microRNA core promoters in four model species. PLoS Comput Biol 3: e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sun J, Wang QH, Song LQ, Zhao G, Wang HZ, Yang HX, Li X. 2011. Genome-wide analysis of clustering patterns and flanking characteristics for plant microRNA genes. FEBS J 278: 929–940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.